


Abstract
Rho is a ring-shaped, ATP-fueled motor essential for remodeling transcriptional complexes and R-loops in bacteria. Despite years of research on this fundamental model helicase, key aspects of its mechanism of translocation remain largely unknown. Here, we used single-molecule manipulation and fluorescence methods to directly monitor the dynamics of RNA translocation by Rho. We show that the efficiency of Rho activation is strongly dependent on the force applied on the RNA but that, once active, Rho is able to translocate against a large opposing force (at least 7 pN) by a mechanism involving ‘tethered tracking’. Importantly, the ability to directly measure dynamics at the single-molecule level allowed us to determine essential motor properties of Rho. Hence, Rho translocates at a rate of ∼56 nt per second under our experimental conditions, which is 2–5 times faster than velocities measured for RNA polymerase under similar conditions. Moreover, the processivity of Rho (∼62 nt at a 7 pN opposing force) is large enough for Rho to reach termination sites without dissociating from its RNA loading site, potentially increasing the efficiency of transcription termination. Our findings unambiguously establish ‘tethered tracking’ as the main pathway for Rho translocation, support ‘kinetic coupling’ between Rho and RNA polymerase during Rho-dependent termination, and suggest that forces applied on the nascent RNA transcript by cellular substructures could have important implications for the regulation of transcription and its coupling to translation in vivo.
INTRODUCTION
Rho is a homo-hexameric molecular machine that couples the energy of ATP hydrolysis to translocation in the 5′→3′ direction on nascent single-stranded RNA (ssRNA) transcripts to terminate transcription. A rho open reading frame is present in the genomes of most (90%) of bacterial species (1), and Rho activity is essential for the general modulation of gene expression in Escherichia coli and other bacteria (2,3). In addition, Rho controls the expression of non-coding RNAs (small RNAs and antisense RNAs) (4), prevents the formation of toxic RNA-DNA structures (R-loops) during transcription (5), and silences foreign DNA (6,7).
Rho binds to single stranded RNA in a complex multi-step pathway that involves two distinct sites on the hexamer. The primary binding site (PBS), distributed on the N-terminal domains around the hexamer (Figure 1A, cyan), ensures initial anchoring of Rho to the transcript at a Rut (Rho utilization) site, a ∼70 nucleotides (nt) long, cytidine-rich and poorly-structured RNA sequence. Each Rho monomer contains a sub-site capable of binding specifically the base residues of a 5′-YC dimer (Y being a pyrimidine) (8). Biochemical and structural data suggest that Rho initially binds to RNA in an open, ‘lock-washer’ conformation that closes into a planar ring as RNA transfers to the central cavity (3,9–11). There, the ssRNA contacts an asymmetric secondary binding site (SBS) (Figure 1A, green), and this step, which presumably is rate-limiting for the overall reaction (11), leads to motor activation. Upon hydrolysis of ATP, the ssRNA is pulled upon conformational changes of the conserved Q and R loops of the SBS (10), leading to Rho translocation, and ultimately promoting RNA polymerase (RNAP) dissociation (3,12).
Figure 1.
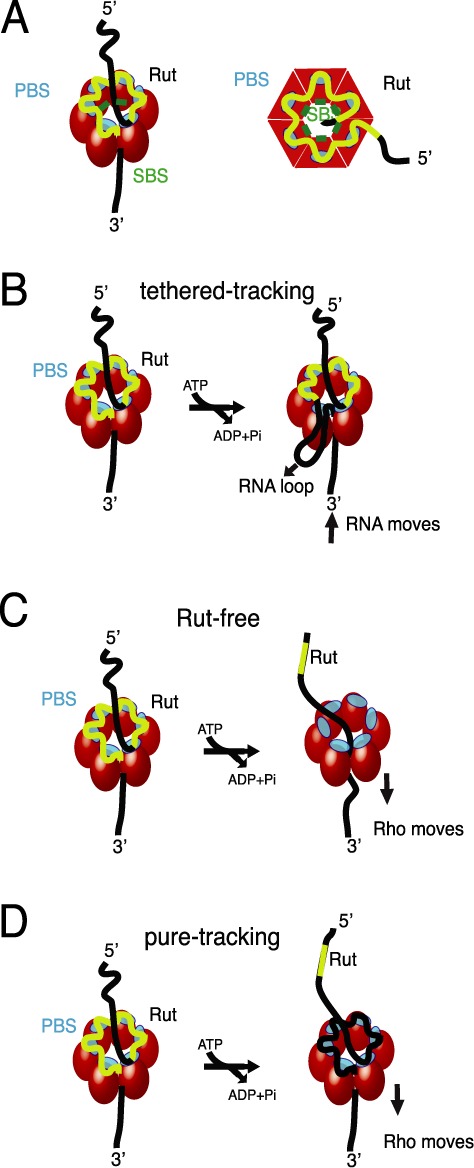
Schematic of Rho factor structure and mechanisms. (A) Rho assembles as a homo-hexameric ring (red spheres or tetragons), with RNA (black/yellow curve) binding to the primary binding sites (PBS, cyan) and the secondary binding sites inside the ring (SBS, green), where ATP-coupled translocation takes place. The Rut specific binding site is depicted in yellow. (B) The tethered-tracking model proposed that Rho translocates RNA while maintaining interactions between PBS and Rut. This model requires the formation of a loop that would shorten the extension of RNA upon translocation. (C) The Rut-free model posits that upon translocation Rho looses PBS/RNA contacts and only conserves contacts through the SBS. (D) In contrast, in the pure-tracking model, PBS/RNA contacts persist during translocation but are actively remodeled. Thus, this model does not predict changes in RNA extension during translocation.
The molecular mechanism of Rho translocation is still the subject of debate, and has important implications on the mechanism by which Rho dissociates RNAP. Over the years, three competing models accounting for Rho translocation have been proposed: The ‘tethered tracking’ model (13) postulates that Rho maintains its contacts between the PBS and the loading (Rut) site upon translocation (Figure 1B). This mechanism would allow Rho to maintain its high affinity interaction with Rut, and implies the growing of an RNA loop between the PBS and the SBS upon translocation. In contrast, for the two other proposed mechanisms, no RNA looping is expected. In the ‘Rut-free’ model (Figure 1C), Rho would completely disrupt its contacts with the RNA via its PBS upon translocation (10). Finally, the ‘Pure Tracking’ model involves a remodeling of the RNA/PBS contacts, and thus a disruption of the initial contacts with Rut (Figure 1D) (14,15).
The tethered tracking model recently received indirect support from single-molecule force spectroscopy measurements (16). In these experiments, Koslover et al. pulled on nascent RNA using optical tweezers, and showed that Rho binding to RNA induces a wrapping of 85 ± 2 nt, corresponding to Rho binding via its PBS + SBS, that is disrupted at a force of ∼14 pN. After incubation with ATP in the absence of force, the size of the RNA wrapped observed upon pulling increased (16). This observation is consistent with: (1) a model in which a distinct, single-step Rho oligomerization/conformational change promoted by ATP binding leads to additional RNA shortening; or (2) a mechanism in which a RNA loop is produced due to tethered tracking. These models cannot be discerned without direct, real-time measurement of Rho translocation.
Here, we implemented a single-molecule nano-manipulation assay combined with single-molecule fluorescence. These methodologies allowed us, for the first time, to monitor in real-time the translocation activity of Rho at the single-molecule level. Our data show that Rho progressively loops ssRNA at a constant rate during its ATP-dependent translocation, providing the first direct experimental evidence for the tethered tracking model. The application of opposing forces causes a strong inhibition in the overall efficiency of the helicase reaction by Rho, suggesting that no other force-independent translocation pathway competes with tethered-tracking. Finally, the rate of Rho translocation via tethered tracking is 2–5 times larger than the rate of RNA polymerase elongation (17), consistent with the regulation of transcription termination involving a kinetic competition between Rho and RNAP (18).
MATERIALS AND METHODS
Protein and nucleic acid constructs
Wild-type Rho enzyme was overexpressed in BL21(DE3)pLysS cells harboring the pET28b-Rho plasmid (kindly provided by J. Berger, John Hopkins University) and purified as described previously (19). Purified Rho was stored at −20°C at micromolar concentration in 100 mM KCl, 10 mM Tris–HCl (pH 7.9), 0.1 mM ethylenediaminetetraacetic acid (EDTA), 0.1 mM Dithiothreitol (DTT) and 50% glycerol.
DNA oligonucleotides were purchased from Eurogentec and their sequences are listed in SI 8. Polymerase chain reaction (PCR) amplification reactions were performed with the KOD hot start polymerase (Novagen). R500 construct was produced by first PCR-amplifying (using primers 1 and 2) a 500-bp region of the phage λ DNA (Invitrogen) starting from the middle of the cro gene and containing the λtR1 terminator (SI 2, 9). The amplified fragment was sub-cloned between the unique EcoRI and HindIII restriction sites of the pBR322 plasmid in order to construct pBR322-λtR1. The pBR322-λtR1 plasmid was then used to PCR-generate (primers 3 and 4) a ∼1.7 kb DNA template for in vitro transcription by introducing the T7 RNA polymerase promoter. This template included the sub-cloned 500 bp λtR1 sequence flanked by two ∼600 bp, pBR322-native regions, up- and down-stream of λtR1 (corresponding, respectively, to the ‘handles’ A and B of the construct). The corresponding ssRNA was then produced by in vitro transcription by using the MEGAscript® T7 Kit (Ambion) according to the manufacturer's instructions. The produced RNA was purified by lithium chloride precipitation and the DNA template was removed by TURBO™ DNAse treatment (Ambion). The obtained RNA transcript was homogeneous and pure (see SI 10). The DNA handle to be hybridized at the 3′ end of the transcript (handle B) was generated by PCR amplification from pBR322 (primers 5 and 6) with the reverse primer being 5′-digoxigenin-labeled. The DNA handle to be hybridized at the 5′-end of the transcript (handle A) was produced by first PCR-amplifying a non-labeled fragment (primers 7 and 8). This fragment was subsequently 3′-biotinylated by using the 3′-5′ exonuclease activity coupled to the 5′-3′ polymerase activity of the T4 DNA polymerase (Invitrogen) in the presence of biotin-16-dUTP (Roche). The efficiency of the biotinylation reaction was assessed by incubating the biotinylated fragments with streptavidin-coated magnetic beads (Invitrogen). After several washing steps to remove the unbound DNA, the biotinylated handles were eluted and could be detected by gel electrophoresis. Finally, the ∼1700 nt RNA transcript was hybridized to the two DNA handles at a 1:1:1 molar ratio in an annealing buffer containing 1 mM EDTA (pH 8.0), 40 mM Pipes (pH 6.3), 0.4 M (NaCl) and 80% formamide in order to favor the RNA/DNA hybrid over the DNA/DNA pairing as previously reported (20). R500 was finally purified by ethanol precipitation and stored in 100 mM NaCl, 10 mM Tris (pH 8) buffer.
R500-Dcy3 was prepared as described for R500 but, in addition, a Cy3-labeled oligonucleotide (primer 13) was annealed at the 3′-end of the single-stranded λtR1 RNA region. R500-2 Dcy3 was prepared as described for R500 but with a longer handle A (generated with primers 7 and 16) covering the entire Rut (until the end of RutB, leaving a 294 nt ssRNA) and a Cy3-labeled oligonucleotide (primer 13) at the 3′ end of the λtR1 ssRNA region.
To generate RH120, first a 500-bp DNA fragment sequence was designed to contain 260 bp of the λtR1 sequence followed by a 240-bp inverted repeat coding for a 120-bp RNA hairpin (SI 9). This 500 bp fragment was produced by gene synthesis (DNA2.0, Menlo Park, CA, USA) and was subsequently sub-cloned between the unique EcoRI and HindIII restriction sites of the pBR322 plasmid in order to generate pBR322-H120. The RH120 construct was then produced as described for R500.
R500-long was produced by ligation of three precursor nucleic acid fragments. First, a large 15 721 bp DNA fragment was prepared by BamHI and EcoRI enzymatic digestion (Fast Digest, Thermo Scientific) of the phage λ DNA (Roche) and subsequent 0.7% agarose gel extraction (Qiagen kit) of the largest fragment. Next, a 440-bp digoxigenin-modified DNA linker was PCR-amplified by using the pUC19 plasmid as a template and primers flanking the multiple cloning site (primers 9 and 10) in the presence of a dNTP mixture containing a 1:4 molar ratio of digoxigenin-11-dUTP (Roche):dNTP. The amplified fragment was then purified (PCR purification kit, Qiagen), BamHI digested (Fast Digest, Thermo Scientific) and agarose gel extracted (Qiagen kit). Finally, the third fragment of the ligation is a ∼1.7 kb RNA/DNA Atto647N-labeled R500-5 construct. It was prepared similarly to the previously described R500 with, in addition, the annealing of an Atto647N-labeled oligonucleotide (primer 11) at the 3′-end of the single stranded λtR1 RNA region which was verified by agarose gel electrophoresis. Moreover, a phosphate group and an EcoRI overhang were added at the 5′ end of handle B through the reverse PCR amplification primer (primer 12) in order to anneal and ligate the construct to the corresponding EcoRI digested end of the λ DNA fragment. To produce the R500-long construct, the 440 bp digoxigenin-modified DNA linker, the 15721 bp λ DNA fragment and the ∼1.7 kb Atto647N-labeled R500-5 construct were ligated at a 1:1:1 molar ratio in the presence of T4 DNA Ligase (NEB). The ligation reaction was performed overnight at 16°C and the final product was heat inactivated (65°C) to ensure that the ligase did not remain associated with the nucleic acid construct.
Ensemble Rho kinetics
Fluorescence anisotropy experiments were performed using the R500 construct, hybridized to its DNA handles and bound to anti-digoxigenin-coated paramagnetic beads. A Cy3- or Atto647N-labeled fluorescent oligonucleotide (Eurogentec) was hybridized at the 3′-extremity of the ssRNA region, just before the beginning of the DNA/RNA handle. Fluorescence anisotropy measurements were performed at room temperature by using a Tecan Saphire II plate reader, by exciting at 530 nm, and detecting at 580 nm (bandwidth 20 nm) for Cy3 or exciting at 635 nm and detecting at 680 nm (bandwidth 20 nm) for Atto647N. The reaction was performed in helicase buffer (20 mM HEPES pH 7.5, 1 mM MgCl2, 0.1 mM DTT, 0.1 mM EDTA, 0.15 M potassium glutamate, 0.1 mg/ml bovine serum albumin (BSA)) supplemented with 1 mM adenosine diphosphate (ADP) or adenosine triphosphate (ATP). The concentration of the labeled construct was 1 nM and the concentration of Rho was 30 nM.
Magnetic tweezers
Single-molecule experiments were conducted on a magnetic tweezers instrument described elsewhere (21). Briefly, the optical setup is a home-made inverted microscope, equipped with high-power hybrid permanent magnets that can be translated to control precisely the degree of tension applied to single nucleic acid molecules. The high-power magnets enable the application of stretching forces up to ∼10 pN when using 1 μm-diameter paramagnetic beads (Dynal, My One Beads). Movies of fields of view up to 40 μm × 40 μm in size containing ∼10 beads were acquired at 34 Hz with a shutter time of 10 ms. A home-made offline analysis software was used to simultaneously track the 3D positions of the beads as in (21), which allowed to dynamically measure changes in tether extension in multiple beads at high time and spatial resolutions. To ensure accuracy of our extension measurements, height calibration was performed for each bead in the field of view, and the full extension and mechanical properties of each tether were individually tested. Instrument drift was calculated by using a stuck bead or by averaging the tether extensions of at least three beads in the field of view showing no looping event and subtracting it from the extension of the bead of interest (22). RNA extension was calculated from nm by assuming a 0.59 nm per nt (20).
Magnetic tweezers experiments were performed in a flow chamber built by drilling two holes on the opposite ends of a clean coverslip using a CO2 laser cutter (Thermoflan, France). The chamber was constructed by putting together a drilled coverslip, a non-drilled coverslip and a parafilm spacer (∼100 μm thickness). The chamber was sealed by heating on a heat block at 95°C, creating a two-channel microfluidics chamber. Polyethylene tubes (PE50, Harvard Apparatus, Holliston, USA) were connected to the holes through homemade adaptors. A flow was applied by gravity to the reaction chamber. More details on the flow chamber can be found elsewhere (23).
Single molecule assays
For magnetic tweezers experiments, the microfluidics chamber was first cleaned by extensive washing with water and ethanol and then coated with 5 mg/ml biotin–BSA (Invitrogen, France) in a home-made microfluidics device in phosphate-buffered saline (PBS). Streptavidin at 0.2 mg/ml (Invitrogen) in PBS was then bound to the biotin–BSA coated surface and incubated for 10 min at room temperature. Excess streptavidin was washed out by flowing >2 ml of PBS. Next, either the R500 or the RH120 nucleic acid constructs were incubated with α-digoxigenin antibody (Roche)-coated paramagnetic beads (MyOne beads, Dynal) in binding buffer (50 mM Tris–HCl pH 7.5, 500 mM NaCl, 5 mg/ml BSA) for 10 min. This solution was introduced into the chamber and beads were let to set. After 20 min of incubation at room temperature, unbound tethers were washed away by gently flowing binding buffer. The chamber was then thoroughly washed with Rho helicase buffer (20 mM HEPES (pH 7.5), 0.1 mM EDTA, 0.1 mM DTT, 0.15 M potassium glutamate, 1 mM MgCl2 and 1mM ATP or ADP). Rho protein was diluted in helicase buffer to a final hexamer concentration of 5–30 nM, and was introduced into the chamber. The distance between the magnets and the sample was held constant throughout each experiment, resulting in constant force during measurements (∼7 pN). To select for observations of single events, waiting times prior to the onset of activity were required to be five times longer than the time of single events. Individual events of Rho activity were fit using linear regression to obtain enzymatic rates and processivities (see Figure 3C). Due to resolution and drift limitations, we only considered events with processivities >30 nm.
Figure 3.
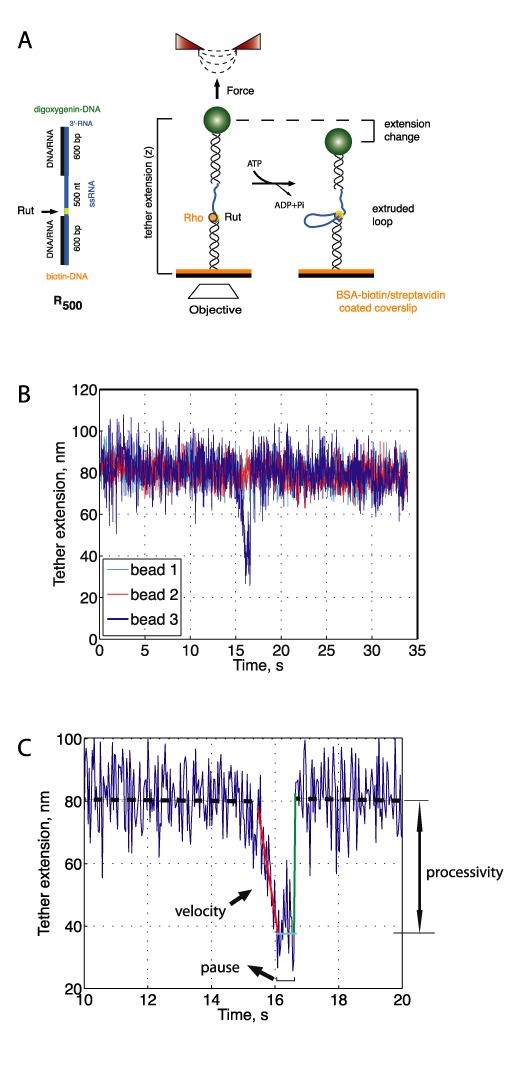
Observing real-time translocation by Rho at the single-molecule level. (A) A magnetic trap (right) consists of a magnetic bead (green sphere) tethered to a glass surface by one tether molecule (black helix and blue line). Magnets above the sample (red triangles) can be translated to exert tension on the DNA molecule. RNA looping by Rho (orange sphere) leads to the shortening of tether extension, monitored by the change in height of the magnetic particle detected through an inverted objective (white rhombus). Handles contain a biotin or a digoxygenin molecule for specific attachment to the BSA–biotin-coated coverslip surface (orange) or an anti-digoxygenin-coated bead. (B) Representative traces of the change in RNA extension due to single-molecule Rho activity on a tether molecule containing a Rut site. Activity of Rho on a single bead (bead 4, blue) in a field of view with several beads (beads 1–3, cyan, red and green) exhibiting no activity. Raw data are shown (34 Hz). Tether extensions represent relative, not absolute measurements of the tether length. Tethers extensions were vertically shifted for clarity. Events were observed at forces > 5 pN. (C) Single-molecule translocation event by Rho in the presence of ATP (raw data at 34 Hz). Forward and backward velocities for each event were obtained by fitting the linear regions (red and green lines represent linear fits) in the extension versus time curve. Forward (backward) velocities correspond to shortenings (recovery) of the RNA extension. Processivity was measured as the height difference between the baseline before the event (black dashed line) and the minimum extension in a event (cyan solid line). Pause length was measured by determining the time spent in a paused state after RNA shortening and before tether extension was fully recovered (cyan horizontal line). In this example, the forward translocation velocity was 90 ± 10 nt/s, the backward rate was 2100 ± 10 nt/s and the pause duration of 0.6 s. In the majority of events (>85%), the RNA extension recovered instantaneously, as shown in this panel.
For the magnetic tweezers experiments coupled with single-molecule fluorescence detection, a home-made epifluorescence setup based on a Zeiss Axiovert 200 inverted microscope equipped with a plan-apochromat 100×/1.4 NA objective (Zeiss, Le Peck, France) was used. The Atto647N dye of the oligonucleotide annealed to the ssRNA region of tethers was excited at 632 nm and its fluorescence passed through a dichroic filter (700 ± 75 nm, Chroma, USA). The camera exposure time used was 200 ms. Coverslips were functionalized with aminosilane (Abcys Laboratories, France) and a mixture of biotinylated and non-biotinylated polyethylene glycol (PEG, Abcys Laboratories). The flow chamber was prepared by first incubating with 0.2 mg/ml Streptavidin (Invitrogen) for 10 min, then washing excess streptavidin with 2 ml of TE buffer (20 mM Tris pH 7.5, 0.1 mM EDTA). 50 fmol of R500-long constructs, pre-incubated with anti-digoxigenin antibody (Roche)-coated beads (MyOne beads, Dynal), were introduced into the chamber in binding buffer. After 20 min incubation at room temperature to promote specific attachments, the unbound tethers were washed away. At this point, the chamber was thoroughly washed with Rho helicase buffer. Proper attachment of Atto647N-labeled oligonucleotides on R500-long tethers was assessed as described in SI 7. This procedure was also used to assess whether the activity of Rho led to the displacement of the fluorescently-labeled DNA oligonucleotide after the period of incubation.
RESULTS
Direct observation of tethered tracking upon Rho translocation on ssRNA
To directly monitor the dynamics of ssRNA translocation by Rho, we implemented a single-molecule nano-manipulation assay based on magnetic tweezers to monitor the dynamics of Rho translocation on single ssRNA. The substrate was generated by annealing a 1700 nt ssRNA to two ssDNA molecules to obtain a 500 nt ssRNA region containing the Rho-dependent λ-tR1 terminator (SI 1 and 2) and two double stranded DNA/RNA hybrids acting as handles (R500, Figure 2A, ‘Materials and Methods’ section). The 3′-end of DNA handle A was labeled with biotin, while the 5′-end of handle B was labeled with digoxygenin (DIG). The ssRNA subtrate contained a natural Rut sequence located at ∼120 nt from the upstream RNA/DNA junction (handle A), and at 314 nt from the downstream RNA/DNA junction (handle B). First, we tested whether the presence of DNA/RNA hybrid regions in the tether, or the attachment of a bead to DIG affected the ensemble activity of Rho. To this aim, we implemented a fluorescence anisotropy-based helicase assay (24) to report on the ATP-dependent helicase activity of Rho (Figure 2B). A Cy3-labeled 20 nt DNA oligonucleotide (Dcy3) was hybridized at the 3′ end of the ssRNA region of R500, in the presence of anti-DIG antibody-coated magnetic beads that bind to handle B. Rho was able to bind, translocate, and displace the DNA oligonucleotide in R500 in an ATP-dependent manner, leading to a decrease in the fluorescence anisotropy signal (r) over time (Figure 2C). These data, obtained under multi-run helicase conditions (25), were fitted using the equation r = Ae−k1t−k2t + B, in which A and k1 represent the amplitude and the rate constant of the exponential (burst) phase, respectively, k2 is the rate constant of the steady-state linear phase, and B is the anisotropy of free Dcy3. The measured rate constant (k1 ∼ 0.18 min−1) is consistent with literature values using similar constructs (0.1–3 min−1 at 30°C (25,26)) and was not considerably affected when Atto647N was used (DA647N) instead of Cy3 (k1 ∼ 0.18 min−1 for Cy3, versus k1 ∼ 0.13 min−1 for Atto647N).
Figure 2.
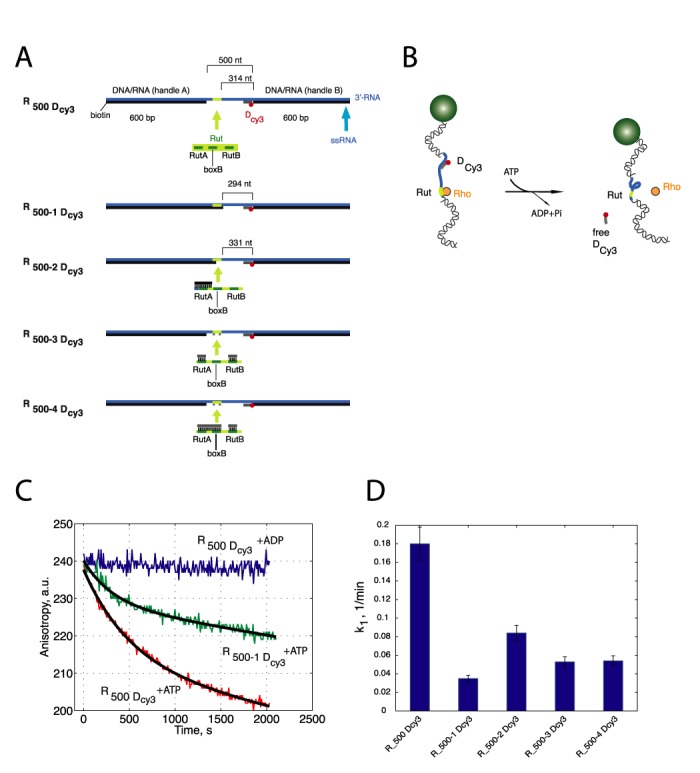
Rho activity is not affected by the presence of handles, or beads, but is stimulated by the presence of Rut. (A) Molecular constructs used for ensemble fluorescence anisotropy and single molecule fluorescence helicase experiments and optimized for single-molecule manipulation studies. R500 Dcy3 contains three segments: two, 600 bp DNA/RNA hybrids (black/blue) serving as handles and a ssRNA region derived from the λtR1 terminator (blue) containing a Rut site (yellow) located at 120 nt from the DNA/RNA handle A (5′ of the RNA) and 310 nt from the handle B (3′ of the RNA). A fluorescently labeled 21 bp oligonucleotide (Dcy3, grey) is hybridized next to handle B. Several variations of R500 Dcy3 were produced to investigate whether the activity of Rho was compromised by the inaccessibility of the Rut sequence in R500 Dcy3. In these substrates, the single-stranded region of the tether containing the Rut sequence was either partially or completely annealed to short oligonucleotide sequences or by extending handle A: R500-1 Dcy3, the DNA handle A is extended to render the entire Rut site inaccessible; in R500-2 Dcy3, extension of handle A covers only RutA; in R500-3 Dcy3, two short oligonucleotides block RutA and RutB; and in R500-4 Dcy3, two short oligonucleotides cover RutA-BoxB and RutB. (B) Schematic representation of an ensemble helicase activity assay on R500 Dcy3 substrates. Displacement of the oligonucleotide Dcy3 due to the helicase activity of Rho is monitored by a decrease in its fluorescence anisotropy signal due to an increased rotational mobility. (C) Typical helicase activity curves in which the fluorescence anisotropy signal decreases as a function of time. The kinetics of the observed helicase by Rho on the R500 Dcy3 construct (red) is strongly reduced when the Rut site is made unaccessible (R500-1 Dcy3 green), and abolished when ATP is replaced by ADP (blue). Data are fitted using Eq1 (exponential decay followed by a linear regime (see text for details)). (D) Helicase activity rates of Rho in substrates presented in panel (A). In all cases, reduction or abolishment of Rut accessibility leads to a significant reduction in the helicase activity of Rho.
Helicase activity experiments on R500 constructs where the Rut site was made completely inaccessible to Rho by extending handle A (R500–1, Figure 2A) resulted in a ∼5-fold reduction of the helicase kinetics (k1 = 0.035 min−1 from 0.18 min−1, Figure 2C). Similar results were obtained when Rut was covered by the annealing of complementary ssDNA oligonucleotides (D500-4 Dcy3, Figure 2A, D). When Rut was made only partially inaccessible to Rho, the rate of displacement was also significantly reduced (Figure 2A and D). This significant, but not complete inhibition of Rho activity can be explained by the use of potassium glutamate in the reaction buffer ([KGlu] = 150 mM), that has been reported to enhance Rho's activity significantly as compared to KCl (27,28). This effect has been explained by the ability of Rho to recognize and accommodate non canonical substrates in the presence of glutamate ions (29), thus lowering Rho discrimination against suboptimal (or Rut-less) substrates, a feature which may also have physiological significance (27,28,30). Overall, our results indicate that neither the presence of DNA/RNA hybrids in R500 nor the binding of beads to the 5′-end of handle B affect significantly the activity of Rho and are consistent with the Rut site in R500 being a preferred site for the initiation of Rho activity.
Next, we studied the dynamics of RNA translocation by Rho by implementing a magnetic tweezers instrument able to monitor the real-time activity of single motors in several single R500 molecules at the same time. The single R500 tethers were specifically attached to a streptavidin-coated surface at one end and to an anti-DIG antibody-coated magnetic bead at the other end (Figure 3A, ‘Materials and Methods’ section). Constant tensions were applied to the DNA/RNA molecule by translating a pair of permanent magnets parallel to the optical axis (21). A mid-throughput setup capable of simultaneously tracking several of beads per field-of-view was developed and used (‘Materials and Methods’ section). In this configuration, Rho translocation by tethered tracking would result in the progressive shortening of the tether extension, thus generating a measurable signal.
Unique tether shortening events were indeed observed in presence of Rho and ATP at 7 ± 1 pN opposing force (Figure 3B). These events were observed only in a single bead in the field of view at any given time (Figure 3B and SI 3), ruling out any possible influence of axial stability or drift. Events were observed in a large proportion of single DNA-bead constructs (25% of 140) but rather infrequently (35 events, ∼85 min of tracking per bead on average). Similar tether shortening events were also observed using an alternative tether containing a Rut site and a 120 bp-long hairpin (RH120, see SI 1). Importantly, tether shortening events were never detected when ATP was replaced by ADP (N = 0 out of 60 beads, ∼153 min of tracking per bead on average). Rho is similarly saturated with ATP and ADP at the concentration of nucleotide used (1 mM; Kd ∼ 0.4 and 20 μM for ATP and ADP, respectively (31)), and both ATP and ADP favor Rho hexamerisation and RNA binding to similar extents (32). Importantly, at this concentration, ADP provides the same type of stabilization to Rho/RNA complexes (binding energy resulting mostly from PBS/RNA interactions) than does ATP or non-hydrolysable analogs (see SI 4 and (32)). Thus, the lack of significant RNA tether changes in the presence of ADP indicates that ATP hydrolysis is required to observe RNA shortening events, consistent with these events being due to active Rho translocation on RNA. Moreover, the occurrence of ATP-dependent shortening events strongly suggests that Rho translocates RNA using a tethered tracking mechanism (Figure 1B) rather than alternative mechanisms (Figure 1C and D).
The production of an active Rho/Rut complex able to translocate RNA arises from a sophisticated, rate-limiting process in which ssRNA sequentially binds to up to six different PBS clefts and enters the ring to contact the SBS (11,25,33). Single-molecule force-extension curve experiments showed that this multi-step activation pathway induces an overall ∼46 nm shortening of the RNA chain in the case of Rho binding to a λtR1-derived substrate (16). This process was reported to be rare, even in the absence of opposing loads (shortening events were observed in only ∼5% of the traces monitored), and independent from the supply of external energy (e.g. ATP hydrolysis) (16). A decrease in RNA extension induced by the formation of a Rho/Rut complex involves the production of work: W = x·F, where W is the amount of work, x is the distance change and F is the opposing force. A distance change of 46 nm at an external load of 7 pN represents 320 pN·nm of work, much larger than the energy that can be provided by the thermal bath (∼ 4 pN·nm). Thus, since RNA circulation on the PBS (and subsequent binding to SBS) is not fueled by external energy (derived from ATP hydrolysis), the efficiency of this process will be highly inhibited upon application of an external load on the RNA (see calculation on SI 5). This reduction explains the very low frequency of initiation of RNA translocation events observed in our study. Unfortunately, it was not possible to reduce the applied force on the RNA tether because this adjustment lowers dramatically the spatial resolution of our experiments (due to increased Brownian motion of the bead), precluding the detection of extension changes resulting from Rho translocation.
RNA shortening by ∼46 nm, which is expected to occur prior to the onset of translocation events (16), was not detected in our experiments. This observation is consistent with initial RNA binding being governed by several, kinetically-distinct transitions representing step-by-step associations of the RNA chain to each individual PBS cleft in the hexamer. A large RNA extension change occurring in a single kinetic step should have been detected and can thus be ruled out whereas step-by-step RNA binding transitions are not individually resolved in our conditions (SI 5). This is because each of these transitions produces a RNA extension change (∼5 nm) that is smaller than our instrument resolution (∼10 nm). Moreover, these small-scale transitions are expected to be sparsely spaced in time, since the RNA association rate for each PBS cleft is expected to be reduced by ∼4 orders of magnitude by the load applied on RNA (see above and SI 5). Thus, these transitions will appear separated in time and produce a change in tether extension below our instrument resolution and will therefore be very difficult to observe in our setup. Experimental traces displayed changes in extension that would be compatible with partial wrapping of RNA to PBS, but unfortunately we are unable to make an unambiguous attribution of these events due to instrumental limitations.
Tethered tracking apparent velocity and processivity are compatible with transcription termination
Tether looping events share a common structure. They start by a monotonous decrease in tether length, are followed by a pause, and finish by an abrupt recovery of tether length (Figure 3C). The monotonous nature of the tether shortening phase supports a progressive and ATP-dependent extrusion of a ssRNA loop by tethered tracking, instead of the instantaneous formation of an RNA loop without tracking. The extension decrease can be well fit by a linear function, and represents the velocity of Rho translocation by tethered tracking (Figure 3C). The average velocity of Rho is 56 ± 3 nt/s (mean ± SEM, N = 62, Figure 4C) independently of the substrate used (Figure 4A and B). This continuous decrease of tether extension is followed by a short pause with a mean pause length of 0.5 ± 0.2 s, and a rapid recovery of the original tether extension corresponding to the instantaneous release of the looped RNA (apparent rate of >1000 nt/s, Figure 3C). A summary of rates for the different substrates tested can be found in SI 6.
Figure 4.
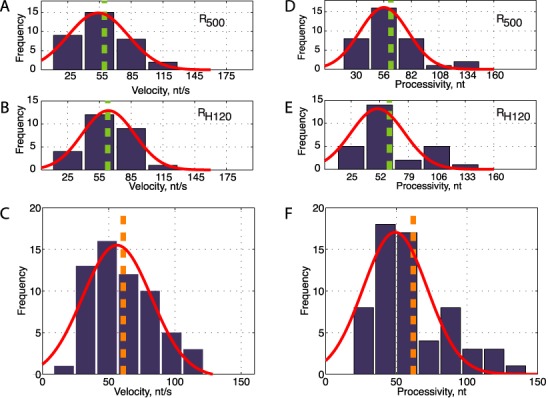
Rates and processivities of Rho translocation on single-stranded RNA. (A and B) Histograms of velocities of Rho translocation on R500 (panel A) and RH120 (panel B). Gaussian fits are represented in red (mean ± SEM: 50 ± 4 nt/s for R500 and 56 ± 4 nt/s for RH120), dashed green lines show unweighted averages (59.6 ± 4 nt/s for R500 and 62.8 ± 4 nt/s for RH120). (C) Histogram of compiled Rho single-molecule velocities (blue bars) fit to a Gaussian function (red line) indicates a mean rate of 56 ± 3 nt/s (mean ± SEM). Average velocity of 61 ± 3 nt/s (mean ± SEM) is indicated with a vertical dashed line. N = 62. (D and E) Histogram of Rho translocation processivities on R500 (panel A) and RH120 (panel B). Gaussian fits are represented in red (mean 56 ± 4 nt for R500 and 48 ± 4 nt for RH120), dashed green lines show the unweighted averages of all measurements (62 ± 4 nt for R500 and 61 ± 4 nt for RH120). (F) Histogram of compiled Rho single-molecule processivities (blue bars) fit to a Gaussian function (red line) showing a maximum at 49 ± 3 nt (mean ± SEM). Average processivity 62 ± 3 nt (mean ± SEM) is indicated with a vertical dashed line. Only events with processivities twice larger than the instrument noise (∼10 nm SD) were considered. N = 62.
RNA extensions before and after translocation are similar within measurement error, indicating that even if Rho has disrupted its RNA contacts via the SBS, it remained bound to the transcript, probably with RNA still engaged on the PBS. Indeed, previous single-molecule studies showed that complete Rho dissociation from RNA requires ∼14 pN of opposing force (16), twice the force applied here, and would have resulted in a ∼46 nm change in tether extension, that should have been detected under our experimental conditions. Persistence of Rho:λtR1-RNA complexes is consistent with very low dissociation rates (∼5 × 10−4 min−1 in 50 mM NaCl at 37°C and 0 pN) observed in the presence of ATP (32). This slow dissociation of Rho from RNA may lead to a sequential, step-wise loss of PBS–RNA contacts that would be undetectable in our experiments due the limitation in resolution as exposed above. We note, however, that we do not see consecutive translocation events to a significant extent in our single molecule traces, indicating that this step (reset of RNA/SBS interactions coupled to Rho activation) is rate-limiting in the overall helicase reaction, at least under high opposing forces (7 pN).
From the change in total tether length, it is possible to extract the processivity of each single-molecule event (Figure 3C). The average processivity is 62 ± 3 nt (mean ± SEM, N = 62, Figure 4F) independently of the substrate used (Figure 4D and E, and SI 6). Importantly, many RNA shortening events exhibited a processivity considerably larger than the length of RNA sequestered during the formation of Rho–PBS complexes (∼46 nm), consistent with the observed shortening events corresponding to active RNA translocation by Rho. This mean processivity is larger than the distances between the end of Rut and transcription termination sites (∼ 13, 35 and 68 nt downstream in λtR1, SI 9), consistent with active translocation via tethered tracking being responsible for Rho-specific termination.
Tethered tracking as a major pathway for Rho translocation mechanism
To determine if tethered tracking is the dominant pathway, we investigated at the single-molecule level whether Rho is able to exert its helicase activity using pathways that do not involve RNA looping. To this aim, we coupled our magnetic tweezers setup to single-molecule fluorescence detection to monitor the displacement of DA647N upon Rho translocation at different applied forces. In order to decrease the background due to the bead auto-fluorescence, a new construct was generated (R500, long) by extending the DNA/RNA handle linking R500 to the bead by ligating it to a 14.7-kb dsDNA handle (Figure 5A). DA647N was annealed to the ssRNA region of the tether and used as a reporter for Rho's translocation activity. To verify the proper attachment of the fluorescently-labeled oligonucleotide in the absence of Rho, we imaged the fluorophore when applying flow force in the forward and backward directions and measured the distance between the two fluorophore positions at low tensions (∼ 2 pN, SI 7). Correctly attached fluorophores showed a distance of 1100 ± 150 nm (theoretical distance was 1200 nm, SI 7).
Figure 5.
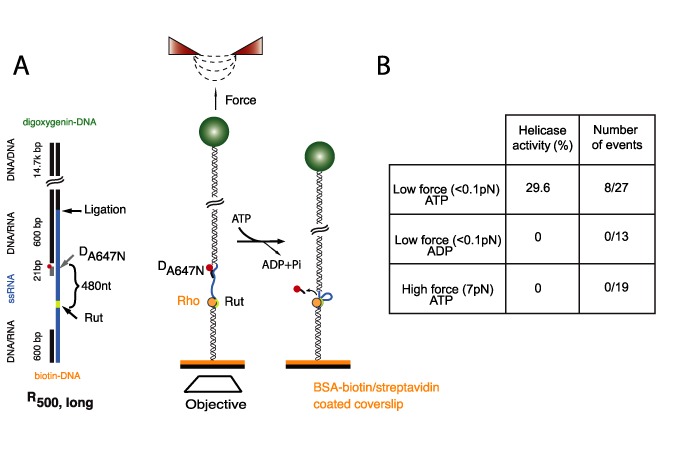
Helicase activity of Rho at low forces and proposed mechanism. (A) A fluorescent oligonucleotide labeled with a photo-stable fluorescent probe (Atto647N) is hybridized to the RNA next to the handle B, that has been extended by 14.7 kb to decrease the background fluorescence due to the bead. The disappearance of the fluorescence signal serves as reporter of Rho helicase activity, regardless of its RNA- looping action. At negligible forces, Rho displays a significant activity (on ∼30% of the constructs), while this helicase action is abolished by replacing ATP with ADP, or by applying an opposing force (see text). (B) Table displaying summary of single-molecule Rho helicase activity results under different experimental conditions.
The motor activity of Rho was tested under different conditions: (i) high force (7 ± 1 pN) in the presence of ATP, (ii) low force (<0.1 pN) in the presence of ATP and (iii) low force in the presence of ADP. After 30 min of incubation, and under imaging conditions where photobleaching is negligible, the number of displaced fluorophores was determined (results summarized in Figure 5B). In the presence of high forces, Rho was unable to displace the oligonucleotide (0 out of 19 molecules) located ∼300 nt away from the Rut site, consistent with the low processivity of tethered tracking by Rho under these conditions (62 ± 3 nt). Importantly, this tendency is reversed at low forces (<0.1 pN), in which case Rho was able to displace up to 30% of the oligonucleotides (8 out of 27 molecules, p-value versus high force <0.003). Critically, this ability of Rho to displace the oligonucleotide at low forces required the presence of ATP (0 out of 13 molecules displaced when ATP was substituted with ADP, p-value versus ATP <0.003). Overall, these results indicate that there is no other major, force-independent pathway accounting for the helicase activity of Rho. Thus, tethered tracking represents the major pathway for Rho translocation leading to transcription termination.
DISCUSSION
Several competing models for Rho translocation have been proposed in the literature (described in Figure 1), but direct determination of Rho translocation on RNA was necessary to refute or validate these mechanisms. A recent study used single-molecule force spectroscopy to measure changes in RNA extension due to Rho binding under different conditions (16). This study showed that RNA extension decreases upon Rho binding, and is further decreased in the presence of ATP. Although these results are consistent with a tethered tracking model for Rho translocation, the real-time gradual decrease of RNA extension was not detectable with the experimental setup. Thus, these data could also be conceivably explained by a conformational change in the Rho oligomer promoted by ATP binding, that would lead to a looping of the RNA transcript (looping without tracking). In this paper, we were able to follow the dynamics of Rho translocation at the single-molecule level, for the first time to our knowledge. Notably, we observed that ATP-fueled RNA translocation by Rho leads to a monotonous, constant-rate decrease in RNA extension, providing the first direct evidence for a mechanism of Rho translocation involving tethered tracking, and allowing for the first direct measurement of the velocity and processivity of this process (13). Critically, our results on the force-dependent Rho helicase activity and processivity monitored by single-molecule fluorescence coupled with nano-manipulation suggest that tethered tracking is the main mechanism for Rho translocation.
Overall, our data are consistent with a model in which:
RNA binding to Rho's PBS is a multi-step process. This initial, essential part of the activation pathway does not require the energy of ATP, but leads to a large change in RNA extension, and is thereby strongly inhibited by tension on the transcript. Due to the technical constrains of our experimental design (in terms of resolution, long-term mechanical drift, and importantly because of the opposing force applied to the transcript; see SI5), multi-step binding of RNA to Rho's PBS was not observed in our experiments.
After specific PBS-Rho contacts are formed, RNA binds the SBS and an active complex is generated. Importantly, the maintenance of PBS–Rho contacts leads to the formation of an RNA loop due to rapid, ATP-fueled Rho translocation at a velocity of 56 ± 3 nt/s. This velocity is consistent with the bulk ATP hydrolysis turnover number, that predicts a rate of ∼ 60 nt/s (12), providing additional evidence for tethered tracking being the main translocation mechanism. Importantly, the single-molecule rate of translocation is 2–6 times faster than the rate of transcription by RNAP (10–30 nt/s) (17), suggesting that, once properly loaded on RNA, Rho is able to quickly catch up with and inactivate RNAP. This is consistent with the ‘kinetic coupling’ model whereby slight variations in the relative rates of the two enzymes result in changes in termination efficiencies (termed the kinetic coupling model) (18).
The processivity of Rho translocation by tethered tracking is relatively modest for a ring-helicase (62 ± 3 nt), but seems optimally suited to bridge the average distance between Rut and termination sites (∼20–40 nt) ((12) and SI 2).
Rho remains bound to the transcript, even after the release of the RNA loop formed during tethered tracking. Such a mechanism might increase to a certain extent the overall efficiency of termination. Indeed, since Rho remains bound to the transcript, only the reset of RNA/SBS interactions and Rho activation are required so that translocation can restart. Translocation restart is, however, hardly detected in our conditions, probably because the transition from a RNA–PBS to a RNA–SBS complex also requires a significant shortening in RNA extension (∼29 nm) (16), which would produce a significant, force-induced inhibition in the kinetic rate of formation of SBS contacts (for the same reasons as for RNA binding to each PBS subsite; see results and SI 5).
During the transcription of most E. coli genes, translation initiates soon after the ribosome binding site in the nascent transcript emerges from the RNA polymerase exit channel. Interestingly, the rate of translation is closely matched to that of transcription (34), and in conditions in which ribosomes slow down (e.g. due to amino-acid deficiency) the gap developing between the RNA polymerase and ribosome leads to premature transcription termination by Rho (35). Since the rate of Rho translocation (56 ± 3 nt/s) is at least twice the rates of translocation by the ribosome or RNA polymerase (17,36), tethered tracking by Rho (and the resultant development of an RNA loop between the PBS and SBS) may quickly generate tension on the RNA chain. This could be particularly true under in vivo conditions wherein the RNA polymerase and trailing ribosomes may provide dynamic anchor points (due to their respective anchoring to the bacterial chromosome and polysome complex). In this case, RNA tension may contribute to (or facilitate) termination. Alternatively, the developing RNA tension may put Rho–RNA interactions at risk during Rho translocation so that cofactors stabilizing the termination complex (e.g. NusG) are required under such conditions or when Rho–RNA interactions are suboptimal. Thus, tension on the nascent RNA may contribute to the regulation of Rho-dependent termination in vivo either by interfering with the formation of active Rho–Rut complexes (see above and SI 5) or by altering the stability of the transcription/termination complex during subsequent steps. These intriguing possibilities and their functional implications will require new, sophisticated experimental set-ups for rigorous testing.
Rho translocation via tethered tracking introduces an important topological constraint in the Rho hexamer, as this mechanism ensures that during active translocation a subset of subunits remain sequentially cross-linked to each other via the RNA chain running between the PBS sites. This mechanism may increase the processivity of the motor, as it decreases the likelihood of subunit dissociation or of conformational changes (e.g. into a ‘split-open’ hexamer) that would stop translocation. However, at least one inter-subunit interface should be devoid of such stabilizing contacts, introducing a clear asymmetry in the hexamer (Figure 1B). During the catalytic cycle, this ‘weak’ interface may be further destabilized periodically by the exchange of nucleotide cofactor (37). These features are compatible with Rho translocating with a 1 nt step-size (10), while the 7 nt periodicity observed in nucleotide analog interference mapping (NAIM) (38) would represent bursts of translocation regulated periodically by a special subunit (37) in a manner reminiscent to that observed for the Φ29 motor (39).
In brief, we provided the first direct experimental evidence that Rho translocates RNA mainly by a mechanism involving tethered tracking, and presented the first dynamical data at the single-molecule level. However, future single-molecule experiments at higher resolution (39,40) will be required to investigate the mechano-chemical cycle of this important model system. In future, higher-throughput setups may enable the study of the dependence of the initiation and translocation rates and processivity as a function of [ATP] to dissect the precise role of ATP in the mechano-chemical cycle of Rho. Recent technical developments in nano-patterning have been used in tethered-particle tracking applications to monitor hundreds of beads simultaneously (41). A similar development applied to magnetic tweezers could allow simultaneous tracking of a larger (10–100 times) number of beads, increasing considerably the efficiency of our assay and making these measurements possible. Alternatively, these measurements may be achieved using a different Rho/Rut combination less sensitive to tension on the tether. This combination could be searched by testing Rho homologs with distinct PBS and RNA requirements (e.g. Rho factor from GC-rich Micrococcus luteus; (42)) and/or by seeking force-resilient Rut sites from a randomized sequence pool using SELEX-derived approaches. Ideally, single-molecule experiments could be performed in the context of a transcription elongation complex, in order to assess the effect of auxiliary factors such as NusG. Finally, live single-molecule methods may be able to directly dissect the role of different partners (such as NusG) in the activity of Rho, the in vivo dynamics of transcription termination, and the mechanisms regulating Rho activity in the cell.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Diego Cattoni and Jeff Moffitt for critical reading of the manuscript and discussions.
FUNDING
Agence Nationale de la Recherche [ANR 2010 BLAN 1525 01 to M.B., E.M. and M.N.]; Human Frontiers Science Program through a Career Development Award (CDA-00017/2009 to M.N.); Ministère de la Recherche (to V.G.). Funding for open access charge: Agence Nationale de la Recherche [ANR 2010 BLAN 1525 01 to M.B., E.M. and M.N.].
Conflict of interest statement. None declared.
REFERENCES
- 1.D'Heygère F., Rabhi M., Boudvillain M. Phyletic distribution and conservation of the bacterial transcription termination factor Rho. Microbiology. 2013;159:1423–1436. doi: 10.1099/mic.0.067462-0. [DOI] [PubMed] [Google Scholar]
- 2.Boudvillain M., Figueroa-Bossi N., Bossi L. Terminator still moving forward: expanding roles for Rho factor. Curr. Opin. Microbiol. 2013;16:118–124. doi: 10.1016/j.mib.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Peters J.M., Vangeloff A.D., Landick R. Bacterial transcription terminators: the RNA 3′-end chronicles. J. Mol. Biol. 2011;412:793–813. doi: 10.1016/j.jmb.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peters J.M., Mooney R.A., Kuan P.F., Rowland J.L., Keles S., Landick R. Rho directs widespread termination of intragenic and stable RNA transcription. Proc. Natl. Acad. Sci. U.S.A. 2009;106:15406–15411. doi: 10.1073/pnas.0903846106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leela J.K., Syeda A.H., Anupama K., Gowrishankar J. Rho-dependent transcription termination is essential to prevent excessive genome-wide R-loops in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2013;110:258–263. doi: 10.1073/pnas.1213123110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cardinale C.J., Washburn R.S., Tadigotla V.R., Brown L.M., Gottesman M.E., Nudler E. Termination factor Rho and its cofactors NusA and NusG silence foreign DNA in E. coli. Science. 2008;320:935–938. doi: 10.1126/science.1152763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menouni R., Champ S., Espinosa L., Boudvillain M., Ansaldi M. Transcription termination controls prophage maintenance in Escherichia coli genomes. Proc. Natl. Acad. Sci. U.S.A. 2013;110:14414–14419. doi: 10.1073/pnas.1303400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bogden C.E., Fass D., Bergman N., Nichols M.D., Berger J.M. The structural basis for terminator recognition by the Rho transcription termination factor. Mol. Cell. 1999;3:487–493. doi: 10.1016/s1097-2765(00)80476-1. [DOI] [PubMed] [Google Scholar]
- 9.Skordalakes E., Berger J.M. Structure of the Rho transcription terminator: mechanism of mRNA recognition and helicase loading. Cell. 2003;114:135–146. doi: 10.1016/s0092-8674(03)00512-9. [DOI] [PubMed] [Google Scholar]
- 10.Thomsen N.D., Berger J.M. Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell. 2009;139:523–534. doi: 10.1016/j.cell.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim D.E., Patel S.S. The kinetic pathway of RNA binding to the Escherichia coli transcription termination factor Rho. J. Biol. Chem. 2001;276:13902–13910. doi: 10.1074/jbc.M011043200. [DOI] [PubMed] [Google Scholar]
- 12.Richardson J.P. Rho-dependent termination and ATPases in transcript termination. Biochim. Biophys. Acta. 2002;1577:251–260. doi: 10.1016/s0167-4781(02)00456-6. [DOI] [PubMed] [Google Scholar]
- 13.Steinmetz E.J., Platt T. Evidence supporting a tethered tracking model for helicase activity of Escherichia coli Rho factor. Proc. Natl. Acad. Sci. U.S.A. 1994;91:1401–1405. doi: 10.1073/pnas.91.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walstrom K.M., Dozono J.M., Robic S., Hippel von, P.H. Kinetics of the RNA-DNA helicase activity of Escherichia coli transcription termination factor rho. 1. Characterization and analysis of the reaction. Biochemistry. 1997;36:7980–7992. doi: 10.1021/bi963179s. [DOI] [PubMed] [Google Scholar]
- 15.Walstrom K.M., Dozono J.M., von Hippel P.H. Kinetics of the RNA-DNA helicase activity of Escherichia coli transcription termination factor rho. 2. Processivity, ATP consumption, and RNA binding. Biochemistry. 1997;36:7993–8004. doi: 10.1021/bi963180r. [DOI] [PubMed] [Google Scholar]
- 16.Koslover D.J., Fazal F.M., Mooney R.A., Landick R., Block S.M. Binding and translocation of termination factor rho studied at the single-molecule level. J. Mol. Biol. 2012;423:664–676. doi: 10.1016/j.jmb.2012.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larson M.H., Landick R., Block S.M. Single-molecule studies of RNA polymerase: one singular sensation, every little step it takes. Mol. Cell. 2011;41:249–262. doi: 10.1016/j.molcel.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin D.J., Burgess R.R., Richardson J.P., Gross C.A. Termination efficiency at rho-dependent terminators depends on kinetic coupling between RNA polymerase and rho. Proc. Natl. Acad. Sci. U.S.A. 1992;89:1453–1457. doi: 10.1073/pnas.89.4.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boudvillain M., Walmacq C., Schwartz A., Jacquinot F. Simple enzymatic assays for the in vitro motor activity of transcription termination factor Rho from Escherichia coli. Methods Mol. Biol. 2010;587:137–154. doi: 10.1007/978-1-60327-355-8_10. [DOI] [PubMed] [Google Scholar]
- 20.Liphardt J., Onoa B., Smith S.B., Tinoco I.J., Bustamante C. Reversible unfolding of single RNA molecules by mechanical force. Science. 2001;292:733–737. doi: 10.1126/science.1058498. [DOI] [PubMed] [Google Scholar]
- 21.Nollmann M., Stone M.D., Bryant Z., Gore J., Crisona N.J., Hong S.-C., Mitelheiser S., Maxwell A., Bustamante C., Cozzarelli N.R. Multiple modes of Escherichia coli DNA gyrase activity revealed by force and torque. Nat. Struct. Mol. Biol. 2007;14:264–271. doi: 10.1038/nsmb1213. [DOI] [PubMed] [Google Scholar]
- 22.Ribeck N., Saleh O.A. Multiplexed single-molecule measurements with magnetic tweezers. Rev. Sci. Instrum. 2008;79:094301. doi: 10.1063/1.2981687. [DOI] [PubMed] [Google Scholar]
- 23.Cattoni D.I., Fiche J.-B., Valeri A., Mignot T., Nollmann M. Super-resolution imaging of bacteria in a microfluidics device. PLoS ONE. 2013;8:e76268. doi: 10.1371/journal.pone.0076268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu H.Q., Zhang A.H., Auclair C., Xi X.G. Simultaneously monitoring DNA binding and helicase-catalyzed DNA unwinding by fluorescence polarization. Nucleic Acids Res. 2003;31:e70. doi: 10.1093/nar/gng070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walmacq C., Rahmouni A.R., Boudvillain M. Influence of substrate composition on the helicase activity of transcription termination factor Rho: reduced processivity of Rho hexamers during unwinding of RNA-DNA hybrid regions. J. Mol. Biol. 2004;342:403–420. doi: 10.1016/j.jmb.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 26.Rabhi M., Gocheva V., Jacquinot F., Lee A., Margeat E., Boudvillain M. Mutagenesis-based evidence for an asymmetric configuration of the ring-shaped transcription termination factor Rho. J. Mol. Biol. 2011;405:497–518. doi: 10.1016/j.jmb.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 27.Zou L.L., Richardson J.P. Enhancement of transcription termination factor rho activity with potassium glutamate. J. Biol. Chem. 1991;266:10201–10209. [PubMed] [Google Scholar]
- 28.Walstrom K.M., Dozono J.M., von Hippel P.H. Effects of reaction conditions on RNA secondary structure and on the helicase activity of Escherichia coli transcription termination factor Rho. J. Mol. Biol. 1998;279:713–726. doi: 10.1006/jmbi.1998.1814. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz A., Walmacq C., Rahmouni A.R., Boudvillain M. Noncanonical interactions in the management of RNA structural blocks by the transcription termination rho helicase. Biochemistry. 2007;46:9366–9379. doi: 10.1021/bi700493m. [DOI] [PubMed] [Google Scholar]
- 30.Shashni R., Qayyum M.Z., Vishalini V., Dey D., Sen R. Redundancy of primary RNA-binding functions of the bacterial transcription terminator Rho. Nucleic Acids Res. 2014;42:9677–9690. doi: 10.1093/nar/gku690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balasubramanian K., Stitt B.L. Evidence for amino acid roles in the chemistry of ATP hydrolysis in Escherichia coli Rho. J. Mol. Biol. 2010;404:587–599. doi: 10.1016/j.jmb.2010.09.039. [DOI] [PubMed] [Google Scholar]
- 32.Gan E., Richardson J.P. ATP and other nucleotides stabilize the Rho-mRNA complex. Biochemistry. 1999;38:16882–16888. doi: 10.1021/bi991559i. [DOI] [PubMed] [Google Scholar]
- 33.Walmacq C., Rahmouni A.R., Boudvillain M. Testing the steric exclusion model for hexameric helicases: substrate features that alter RNA-DNA unwinding by the transcription termination factor Rho. Biochemistry. 2006;45:5885–5895. doi: 10.1021/bi0600648. [DOI] [PubMed] [Google Scholar]
- 34.Proshkin S., Rahmouni A.R., Mironov A., Nudler E. Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science. 2010;328:504–508. doi: 10.1126/science.1184939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson J.P. Preventing the synthesis of unused transcripts by Rho factor. Cell. 1991;64:1047–1049. doi: 10.1016/0092-8674(91)90257-y. [DOI] [PubMed] [Google Scholar]
- 36.Wen J.-D., Lancaster L., Hodges C., Zeri A.-C., Yoshimura S.H., Noller H.F., Bustamante C., Tinoco I. Following translation by single ribosomes one codon at a time. Nature. 2008;452:598–603. doi: 10.1038/nature06716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soares E., Schwartz A., Nollmann M., Margeat E., Boudvillain M. The RNA-mediated, asymmetric ring regulatory mechanism of the transcription termination Rho helicase decrypted by time-resolved nucleotide analog interference probing (trNAIP) Nucleic Acids Res. 2014;42:9270–9284. doi: 10.1093/nar/gku595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartz A., Rabhi M., Jacquinot F., Margeat E., Rahmouni A.R., Boudvillain M. A stepwise 5′-hydroxyl activation mechanism for the bacterial transcription termination factor Rho helicase. Nat. Struct. Mol. Biol. 2009;16:1309–1316. doi: 10.1038/nsmb.1711. [DOI] [PubMed] [Google Scholar]
- 39.Moffitt J.R., Chemla Y.R., Aathavan K., Grimes S., Jardine P.J., Anderson D.L., Bustamante C. Intersubunit coordination in a homomeric ring ATPase. Nature. 2009;457:446–450. doi: 10.1038/nature07637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abbondanzieri E.A., Greenleaf W.J., Shaevitz J.W., Landick R., Block S.M. Direct observation of base-pair stepping by RNA polymerase. Nature. 2005;438:460–465. doi: 10.1038/nature04268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plénat T., Tardin C., Rousseau P., Salomé L. High-throughput single-molecule analysis of DNA-protein interactions by tethered particle motion. Nucleic Acids Res. 2012;40:e89. doi: 10.1093/nar/gks250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nowatzke W.L., Burns C.M., Richardson J.P. Function of the novel subdomain in the RNA binding domain of transcription termination factor Rho from Micrococcus luteus. J. Biol. Chem. 1997;272:2207–2211. doi: 10.1074/jbc.272.4.2207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.