


Abstract
Fanconi anemia (FA) is a fatal genetic disorder associated with pancytopenia and cancer. Cells lacking functional FA genes are hypersensitive to bifunctional alkylating agents, and are deficient in DNA double-strand break repair. Multiple genes with FA-causing mutations have been cloned, however, the molecular basis for FA remains obscure. The results presented herein indicate that a Rad50-dependent end-joining process is non-functional in diploid fibroblasts from FA patients. Introduction of anti-Rad50 antibody into normal fibroblasts sensitized them to DNA damaging agents, whereas this treatment had no effect on fibroblasts from FA patients. The DNA end-joining process deficient in FA cells also requires the Mre11, Nbs1 and DNA ligase IV proteins. These data reveal the existence of a previously uncharacterized Rad50-dependent DNA double-strand break repair pathway in mammalian somatic cells, and suggest that failure to activate this pathway is responsible, at least in part, for the defective DNA end-joining observed in FA cells.
INTRODUCTION
Fanconi anemia (FA) is a rare autosomal recessive disorder characterized by pancytopenia, developmental anomalies and cancer predisposition (1–4). Cells from FA patients exhibit hypersensitivity to bifunctional alkylating agents and chromosome instability. In addition, these cells display enhanced sensitivity to oxygen, and have subtle defects in the cell cycle and apoptosis (5–10). A number of investigators have also shown that FA cells are also more sensitive to ionizing radiation than are wild-type cells (11–13).
The disease is heterogeneous, with nine complementation groups having been identified thus far, referred to as FA-A, -B, -C, -D1, -D2, -E, -F, -G and -L (8,14). In the span of just over a decade, eight of the FA genes were cloned (14–22). However, examination of the sequences of the cloned FA genes has failed to provide significant insight into their function. Thus, despite these remarkable advances, there is as yet no clear evidence as to the function of the FA gene products.
A number of recent findings have led many to conclude that FA cells are defective in the repair of DNA double-strand breaks. It was shown some years ago that lymphoblast cell lines from patients suffering from FA possessed a subtle defect in the process of rejoining blunt-ended plasmid DNA molecules that had been introduced via electroporation (23,24). More recently, it was shown that nuclear protein extracts from a variety of FA cells have a profound defect in end-joining of linear plasmids (25). In addition, intact FA fibroblasts are deficient in DNA plasmid end-joining, and these cells are hypersensitive to restriction endonuclease-induced cell death (26).
DNA end-joining is a predominant form of DNA double-strand break repair (27). Studies in mammalian cells, and later in yeast, show that a mechanism minimally dependent on the concerted actions of a DNA end-binding heterodimer called Ku (28,29), DNA ligase IV (30) and its binding partner Xrcc4 (31), and the catalytic subunit of the DNA-dependent protein kinase (32) is essential for V(D)J recombination and is also capable of rejoining chromosomal DNA in somatic cells. The biochemical pathway responsible for this activity is referred to as non-homologous DNA end-joining (NHEJ). Interestingly, the available evidence suggests that FA cells are not deficient in NHEJ activity. Levels of the aforementioned proteins present in both lymphoblasts and fibroblasts from FA patients were indistinguishable from those seen in cells from normal donors (24,25). In addition, Ku-mediated end-joining activity in nuclear protein extracts from an FA fibroblast strain was similar to that seen in extracts from normal cells (25). FA cells are not as sensitive to ionizing radiation as are cells with defects in genes encoding the NHEJ proteins (33). Furthermore, lack of Ku-mediated NHEJ activity in vivo is associated with a severe form of immunodeficiency (34–36), which is not observed in FA patients. Thus, while it is possible that the end-joining defect in FA cells results from a deficiency in the Ku-mediated NHEJ pathway, it seems more likely that FA cells are deficient in an end-joining mechanism that is independent of the Ku-pathway.
This latter possibility is consistent with numerous findings indicating that both yeast and vertebrate cells possess DNA end-joining activities that are distinct from Ku-dependent NHEJ (25,28,29,37–39). A number of recent observations support the conclusion that the Rad50, Mre11 and Nbs1 proteins directly participate in DNA end-joining. Yeast clones lacking these proteins are deficient in plasmid end-joining activity (37). Reconstitution experiments using both yeast (40) and mammalian (41) proteins indicate that addition of these proteins enhances in vitro DNA end-joining activity. It was shown that addition of a fraction of mammalian cell extract enriched for Rad50, Mre11 and Nbs1 proteins stimulated in vitro end-joining (41). Finally, the presence of these proteins was necessary for proper in vitro DNA end-joining by extracts derived from human cells (42).
In addition to these findings, there is a growing body of evidence indicating that FA proteins function in a pathway that includes a complex of the Rad50, Mre11 and Nbs1 proteins, referred to as the RMN complex. First, it has been shown that subnuclear assembly of the RMN complex following cellular exposure to DNA crosslinking agents requires the Fancc protein (43). Second, the Fancd2 protein co-localizes with the Nbs1 protein following DNA damage (44). Third, cellular exposure to ionizing radiation results in phosphorylation of the Fancd2 protein in an Nbs1 and ATM-dependent manner (45). Finally, transgenic mice in which a hypomorphic allele of Rad50 called Rad50S has replaced the wild-type allele display hematopoetic failure and cancer predisposition that is similar to that seen in human FA patients (46). Taken together, these findings suggest that the deficiency in DNA double-strand break repair observed in FA cells may result from a failure to activate a DNA end-joining pathway dependent on the Rad50, Mre11, Nbs1 complex.
To gain insight into this question, we pursued an antibody inhibition-based strategy to examine the protein requirements for efficient plasmid end-joining in human diploid fibroblasts (HDFs). Based on findings presented herein, we conclude that the DNA end-joining defect associated with FA cells results from the inability of these cells to activate or properly regulate a previously uncharacterized Rad50-dependent DNA double-strand break repair activity.
MATERIALS AND METHODS
Cells and plasmid constructs
Cells were maintained in a humidified 5% CO2-containing atmosphere at 37°C. All cells were obtained from American Type Culture Collection cell repository unless otherwise noted. The immortalized cell line HT1080 was derived from a spontaneous human fibrosarcoma. Cell strains CCL-153, CCL-186 and PD.715.F are HDFs derived from normal donors. Cell strains PD.720.F, PD.551.F and PD.352.F (referred to as FA-A, FA-C and FA-G, respectively) are human diploid FA fibroblasts from patients of complementation groups A, C and G, respectively, and were obtained from Oregon Health Sciences University Fanconi Anemia Cell Repository.
Antibodies
All antibodies were used at a concentration of 0.4 μg per electroporation. Rabbit monoclonal anti-Ku86, rabbit monoclonal anti-DNA ligase IV and rabbit monoclonal anti-Xrcc4 antibodies were obtained from Serotec Ltd (Raleigh, NC). Mouse polyclonal anti-DNA ligase III, rabbit polyclonal anti-Fancd2, rabbit polyclonal anti-Rad50, rabbit polyclonal anti-Mre11 and rabbit polyclonal anti-Nbs1 antibodies were obtained from Novus Biologicals, Inc. (Littleton, CO). Goat polyclonal anti-Rad51 antibody was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Alkaline phosphatase-conjugated goat anti-rabbit IgG and rabbit anti-goat IgG were obtained from Sigma.
Intracellular V(D)J recombination assay
V(D)J recombination was performed using a method similar to one that has been described previously (47,48). Briefly, 106 human cells were electroporated with 10 μg of the V(D)J plasmid recombination substrate pKSV(D)JSV40ori, 5 μg each of the plasmids pRAG1 and pRAG2 (gifts from Dr David Baltimore, California Institute of Technology, Pasadena, CA) that encode the RAG-1 and RAG-2 genes, respectively, 5 μg of the plasmid pRSVEdl884 that encodes the large T-antigen but does not have an SV40 origin of replication (49) and the indicated antibody. Following a 48 h incubation period, the plasmids were recovered from cells and digested with the restriction endonuclease DpnI. The frequency with which V(D)J recombination in the plasmid pKSV(D)JSV40ori occurred was subsequently determined by transforming this recovered DNA into DH10B electrocompetent reporter bacteria. The design of the recombination substrate is such that, upon V(D)J recombination, a portion of plasmid DNA flanked by recombination signal sequences (depicted by the white triangle and rectangle) is deleted (Figure 1A). As a consequence, a prokaryotic silencer element that is flanked by the recombination signal sequences (depicted in Figure 1A as an octagon) is deleted. As a consequence, transcription of the chloramphenicol acetyl transferase gene (depicted by shaded rectangle) is driven by the Escherichia coli promoter sequence (indicated as an arrowhead in Figure 1A), thereby conferring chloramphenicol resistance on bacteria transformed by the plasmid. Since both the non-recombinant and recombinant plasmids harbor a fully functional beta-lactamase gene (depicted in Figure 1A as a black rectangle), bacteria harboring either plasmid are resistant to ampicillin. Thus, the frequency of V(D)J recombination that occurs within the mammalian cells is calculated by dividing the number of chloramphenicol plus ampicillin-resistant bacterial colonies by the total number of ampicillin-resistant bacterial colonies obtained when the recovered plasmid DNA is transformed into sensitive bacteria. (None of the other plasmids used in this assay can confer either ampicillin or chloramphenicol resistance upon bacterial cells under the conditions utilized, so they do not interfere with this assay.)
Figure 1.
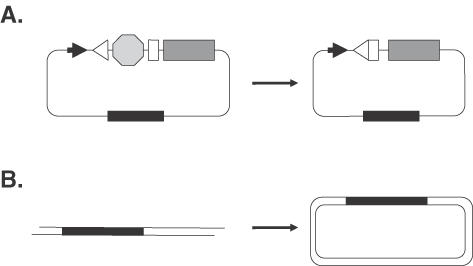
Schematic representations of intracellular DNA repair assays. (A) V(D)J recombination assay. Filled arrowhead, promoter sequence; open triangle and open rectangle, V(D)J recombination sequences; shaded octagon, prokaryotic transcriptional stop signal; shaded rectangle, chloramphenicol acetyltransferase gene; black rectangle, beta-lactamase gene. (B) DNA end-joining assay. Black rectangle, beta-lactamase gene.
Intracellular end-joining assay
DNA end-joining frequency was determined as described previously (23,26). Briefly, the plasmid pSV2Neo (50) was linearized by digestion with the restriction endonucleases EcoRI (to give cohesive ends) or SmaI (to give blunt ends), gel-purified and quantified. Human cells (106 per experiment) were then electroporated with 1.5 μg linearized pSV2Neo (or 1.5 μg pSV2Neo that was not digested), 5 μg of the plasmid pRSVEdl884 and antibody. After electroporation, cells were allowed to incubate for 48 h before plasmid DNA was recovered, digested with the restriction endonuclease DpnI, and introduced into DH10B electrocompetent reporter bacteria. Plasmids that had been rejoined while in the mammalian cells will transform bacteria and confer resistance to ampicillin due to the presence of the beta-lactamase gene (depicted as a black rectangle in Figure 1B), whereas plasmids that remained linear are unable to transform bacteria and will not result in the generation of bacterial colonies. Thus, the frequency of DNA end-joining is determined by comparing the number of ampicillin-resistant bacterial colonies obtained from electroporation with linearized pSV2Neo to the number of bacterial colonies obtained from a parallel experiment with circular pSV2Neo.
Electroporation of restriction endonucleases
Sensitivity to electroporated restriction endonucleases was determined as described previously (26). Briefly, ∼104 human cells were electroporated with restriction endonuclease PvuII diluted in its storage buffer to the indicated concentration and antibody. Subsequently, cells were incubated for 48–72 h before cell survival was determined using the sulforhodamine B assay (51).
Diepoxybutane sensitivity
Human cells (104) were mock electroporated or electroporated with the indicated antibody. Cells were allowed to recover for 6–12 h in drug-free media before media containing 200 ng/ml diepoxybutane was added. Cells were incubated in this drug-containing media for 96 h before cell survival was determined using the sulforhodamine B assay (51).
RESULTS
Human cells possess a DNA end-joining pathway that is independent of Ku-mediated NHEJ but dependent on DNA ligase IV
Previous results indicated that FA cells have a basic defect in DNA double-strand break repair (23–26). However, the identity of the DNA end-joining pathway that is non-functional in FA cells remains unknown. Since electroporation of specific antibodies has been used to block a variety of intracellular processes (52), we reasoned that an analogous approach could be used to address this issue as well. We pursued the following strategy: linear plasmid substrates were introduced into normal fibroblasts in the presence and absence of a number of specific antibodies, and the efficiency of plasmid end-joining was determined. Our aim was to identify antibodies that would inhibit DNA end-joining in wild-type cells, yet would not influence the efficiency of the residual DNA end-joining present in FA cells.
Before initiating this analysis, we tested the validity of the antibody inhibition strategy by examining a plasmid-based V(D)J assay through the co-electroporation of antibodies specific for known components of the V(D)J recombination machinery. Figure 1A depicts schematically the plasmid substrate utilized (for additional details, see Materials and methods). As the image depicts, when this plasmid is introduced into mammalian cells, site-specific recombination by the cellular V(D)J machinery (depicted by the arrow) generates a product that, when introduced into sensitive bacteria, confers resistance to the antibiotic chloramphenicol. It is noteworthy that while the non-recombinant substrate plasmid does not confer chloramphenicol resistance, it does render bacterial resistance to ampicillin. Therefore, the bacterial reporter system allows one to measure the efficiency with which V(D)J recombination occurs within a target mammalian cell population (for additional details, see Materials and methods).
We determined that plasmid V(D)J recombination occurs in the fibrosarcoma cell line HT1080 with a frequency of 0.18% (Table 1). The frequency of V(D)J recombination in a normal strain of HDFs, 0.21% was quite similar (Table 1, HDF). In both cases, V(D)J recombination frequency was measured in the presence of co-electroporated non-specific IgG. Additional control experiments revealed that similar frequencies of V(D)J recombination were obtained in the absence of antibody (data not shown). These V(D)J recombination frequencies, while lower than that observed when similar experiments are performed in human lymphoid cell lines (53), are similar to those seen by other investigators when V(D)J recombination was measured in a mammalian fibroblast background (47,48,54). In contrast, when antibodies specific to the essential V(D)J proteins Ku86, Xrcc4 or DNA ligase IV (30,31,55) were co-electroporated with the plasmid recombination substrate, a 18–45-fold reduction in the frequency of recombination was observed in both HT1080 and normal diploid fibroblast cells (Table 1). Control experiments showed that co-introduction of antibodies specific for DNA repair or recombination proteins that do not participate in the V(D)J recombination reaction, such as DNA ligase III (Table 1) and Rad51 (data not shown), had no effect on the efficiency of plasmid rearrangement.
Table 1. Antibody mediated inhibition of V(D)J recombination and rejoining of plasmid DNA.
| Antibody | V(D)J recombination frequency (%) | Plasmid DNA rejoining | ||
|---|---|---|---|---|
| Cells | Cells | |||
| HT1080 | HDF | HT1080 | HDF | |
| IgG | 0.18 | 0.21 | 27.1 | 27.1 |
| Ku86 | 0.0099* | <0.0052*,† | 29.9 | 29.2 |
| DNA ligase IV | 0.0081* | 0.0049* | 7.7* | 6.3* |
| Xrcc4 | 0.0040* | 0.0063* | 23.8 | 27.5 |
| DNA ligase III | 0.26 | 0.16 | 20.9 | 26.0 |
*P < 0.0001, χ2-test.
†No recombination events were detected. The frequency value depicted represents that which would have been calculated had a single V(D)J recombination event been detected.
These results indicated that we could use an analogous strategy to ask whether particular proteins are essential for intracellular plasmid DNA end-joining. Figure 1B outlines the nature of the plasmid end-joining strategy we utilized to address this question. As the figure indicates, the linear plasmid substrate encodes an ampicillin-resistance marker. Upon introduction into mammalian cells, cellular DNA repair machinery reseals this double-strand break (indicated by the arrow), regenerating a circular plasmid. Since linearized plasmids are unable to transform bacteria, the extent to which plasmid end-joining has occurred can be monitored by determining the number of ampicillin-resistant colonies obtained when plasmid DNA harvested from cells is used to transform drug-sensitive bacteria (see Materials and methods).
As Table 1 reveals, intracellular end-joining of linearized blunt-ended plasmid substrates in the presence of non-specific IgG occurs within both HT1080 and normal HDF cells with a frequency of 27.1%. Control experiments revealed that co-introduction of non-specific IgG had no effect on end-joining efficiency [data not shown, see also (26)]. We next asked whether introduction of antibodies specific for NHEJ proteins would inhibit DNA end-joining in these cells. Table 1 shows that co-introduction of antibodies specific for either Xrcc4 protein or Ku86 protein had no effect on DNA end-joining efficiency in either cell type. Conversely, co-introduction of an antibody specific for DNA ligase IV protein significantly reduced plasmid end-joining in both HT1080 cells as well as in the normal strain of HDFs. As indicated in Table 1, end-joining efficiency was reduced by between 3- and 4-fold when anti-DNA ligase IV antibody was present. When identical experiments were performed using substrates with cohesive ends, we observed that rejoining frequency was reduced by ∼4-fold by anti-DNA ligase IV antibody, whereas inclusion of anti-Ku86, anti-Xrcc4 or anti-DNA ligase III antibodies again had no effect on plasmid end-joining (data not shown). These results are reminiscent of our earlier finding that end-joining activity in mammalian nuclear protein extracts was dependent on DNA ligase IV, but functioned independently of Xrcc4 (25). They are also consistent with the finding that extrachromosomal end-joining occurs at wild-type levels in Chinese hamster ovary cells deficient in either Xrcc4 or Ku86 (56,57).
To confirm the results of the antibody-mediated inhibition experiments, we examined the frequencies of V(D)J recombination and DNA end-joining in HT1080 cells that stably expressed an antisense DNA ligase IV construct. Western-blot analysis revealed that these antisense expressing cells have a ∼50% reduction in DNA ligase IV protein expression compared with non-transgenic cells (data not shown). As Figure 2A reveals, V(D)J recombination in the DNA ligase IV antisense-expressing cells (AS) was significantly lower than that observed in non-transgenic HT1080 cells (HT). Figure 2A also shows that V(D)J recombination frequency in the antisense-expressing cells (AS) was indistinguishable from that observed in the cells that had been co-electroporated with anti-DNA ligase IV antibody (AB). Parallel experiments were performed to examine the efficiency of plasmid rejoining efficiency in these cells. As Figure 2B reveals, both antisense (AS) and antibody (AB) treated cells had significantly reduced levels of end-joining of both cohesive-ended substrates (black bars) and blunt-ended substrates (white bars), compared to non-treated (HT) cells. Again, we observed that the magnitude of the inhibitory effect was similar to both treatments. These findings convincingly demonstrate that introduction of specific antibodies into cells represents a viable method through which the molecular biology of DNA repair and recombination pathways can be examined.
Figure 2.
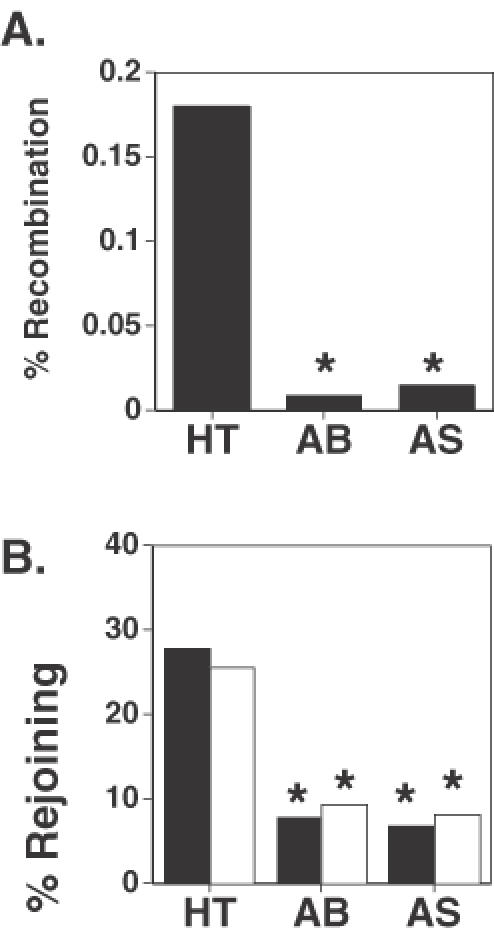
Expression of antisense DNA ligase IV reduces V(D)J recombination and DNA end-joining to levels similar to that of cells treated with anti-DNA ligase IV antibody. (A) V(D)J recombination was determined in unmodified HT1080 cells (HT), in HT1080 cells co-electroporated with anti-DNA ligase IV antibody (AB), and HT1080 cells stably expressing antisense DNA ligase IV (AS). *P < 0.0001, χ2-test. (B) DNA end-joining of cohesive-ended DNA (black bars) and blunt-ended DNA (white bars) was determined in the same cells as in part A. *P < 0.0001, χ2-test.
Fanconi anemia cells are deficient in a non-NHEJ-mediated DNA ligase IV-dependent end-joining pathway
The results presented above revealed that plasmid end-joining seen in cells in which DNA ligase IV function has been inhibited (∼7% rejoining efficiency) was similar to that seen in FA cells, which rejoin plasmid substrates with an efficiency of 4 and 6% (26). This observation raised the possibility that the DNA double-strand break repair defect in FA cells involves a DNA ligase IV-dependent repair pathway. One prediction of this model is that introducing an anti-DNA ligase IV antibody into FA cells would not influence plasmid end-joining efficiency. A series of experiments was therefore performed to test this hypothesis.
First, plasmid end-joining was evaluated in wild-type cells in the presence or absence of both anti-DNA ligase IV and anti-Fancd2 antibodies. It was shown previously that introduction of an anti-Fancd2 antibody reduced intracellular plasmid end-joining in wild-type cells to levels analogous to that of patient-derived FA cells but this treatment had no effect on end-joining in FA cells (26). Figure 3A reveals that introduction of antibodies specific to either anti-DNA ligase IV (L) or Fancd2 (D) reduced plasmid DNA end-joining efficiency in both HT1080 cells (black bars) and a normal strain of HDFs (white bars) by nearly 4-fold, compared to cells in which no antibody was introduced (N). It is noteworthy that each of the two antibodies exerted an equivalent inhibitory effect on plasmid end-joining in both the cells tested. Importantly, when both antibodies were co-introduced into the same cell (L + D), there was no further reduction in plasmid DNA end-joining frequency below that seen when either of the antibody was singly introduced into these cells (Figure 3A).
Figure 3.
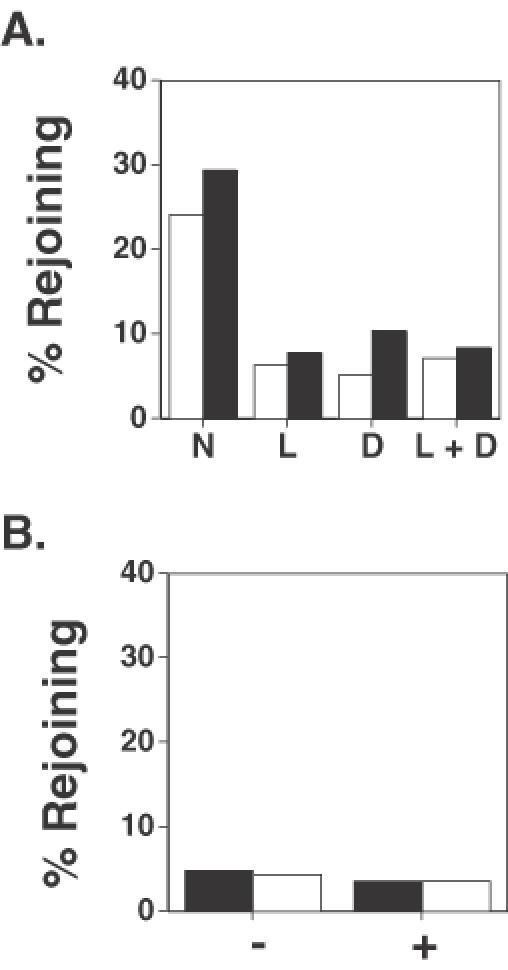
Co-introduction of anti-DNA ligase IV antibody reduces DNA end-joining frequency in normal diploid fibroblasts but does not affect FA cells. (A) End-joining frequency of cohesive-ended DNA was determined in HT1080 cells (black bars) and normal HDFs (white bars) in the presence of no antibody (N), in the presence of anti-DNA ligase IV antibody (L), in the presence of anti-Fancd2 antibody (D), and in the presence of both anti-DNA ligase IV and anti-Fancd2 antibodies (L + D). In all cases, antibody treatment significantly reduced plasmid end-joining levels compared to those observed in cells not treated with antibody, P < 0.0001, χ2-test. (B) End-joining frequency of cohesive-ended DNA (black bars) and blunt-ended DNA (white bars) was determined in patient-derived FA-C cells in the absence of antibody (−) and in the presence of anti-DNA ligase IV antibody (+).
The results shown in Figure 3A are consistent with the hypothesis that the plasmid end-joining defect in FA cells involves a DNA ligase IV-dependent pathway. A more direct test of this hypothesis was conducted by examining the influence of anti-DNA ligase IV antibody on plasmid DNA end-joining in a fibroblast strain derived from an FA patient. This analysis revealed that rejoining of introduced linear plasmid substrates with either cohesive ends (black bars) or blunt ends (white bars) occurred with an efficiency of ∼5% in patient-derived FA-C fibroblasts in both the absence (−) or presence (+) of anti-DNA ligase IV antibody (Figure 3B). Similar results were obtained when this experiment was performed on other FA fibroblast strains (data not shown).
Rad50, Mre11 and Nbs1 proteins are essential for the DNA ligase IV-dependent end-joining pathway that is deficient in Fanconi anemia cells
The results presented thus far indicate that DNA ligase IV is required for DNA end-joining in mammalian cells, and that this enzyme functions in a DNA end-joining pathway that is deficient in FA cells. The results presented above indicate that this DNA ligase IV-dependent pathway apparently functions independently of the other components of the Ku-dependent NHEJ pathway, namely Xrcc4 and the Ku heterodimer. Nevertheless, it seems likely that other protein factors are also involved in the DNA end-joining pathway that is defective in FA cells. Based on a number of recent findings (summarized in the Introduction), we asked whether the Rad50 protein represented one of these essential co-factors. To test this hypothesis, we examined the effect on plasmid DNA end-joining of co-electroporation of anti-Rad50 antibody. We found that when anti-Rad50 antibody was electroporated into a normal strain of HDFs along with linearized plasmids there was a nearly 3-fold reduction in the efficiency of end-joining of both cohesive- (Figure 4A) and blunt-ended (Figure 4B) DNA.
Figure 4.
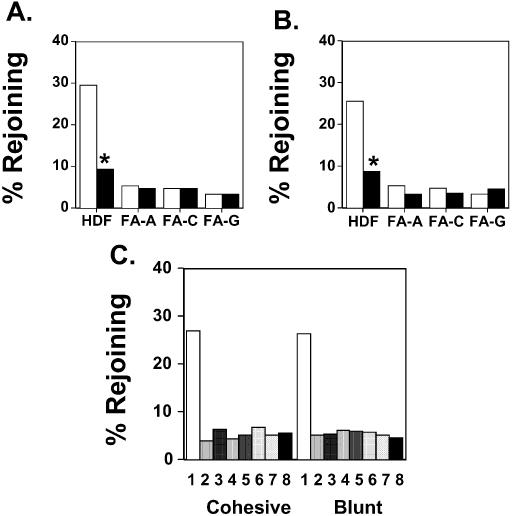
Effect of introduction of anti-Rad50, anti-Mre11 or anti-Nbs1 antibodies on DNA end-joining frequency in normal and FA fibroblasts. (A) The rejoining frequency of cohesive-ended DNA was determined in the HDFs in the absence of antibody (white bars) or in the presence of anti-Rad50 antibody (black bars). HDF, normal HDFs; FA-A, FA fibroblasts belonging to complementation group A; FA-C, FA fibroblasts belonging to complementation group C; FA-G, FA fibroblasts belonging to complementation group G. *P < 0.0001 compared to the same cell strain in the absence of antibody, χ2-test. (B) The rejoining frequency of blunt-ended DNA was determined in the same cells as in (A). *P < 0.0001 compared to the same cell type in the absence of antibody, χ2-test. (C) The rejoining frequency of cohesive- and blunt-ended DNA was determined in normal diploid fibroblasts co-electroporated with non-specific IgG (1), anti-Rad50 antibody (2), anti-Mre11 antibody (3), anti-Nbs1 antibody (4), both anti-Rad50 and anti-Mre11 antibodies (5), both anti-Rad50 and anti-Nbs1 antibodies (6), both anti-Mre11 and anti-Nbs1 antibodies (7), and anti-Rad50, anti-Mre11 and anti-Nbs1 antibodies (8). In all cases, antibody treatment significantly reduced plasmid end-joining levels compared to those observed in cells not treated with antibody, P < 0.0001, χ2-test.
The finding that the efficiency of plasmid DNA end-joining observed in normal diploid fibroblasts treated with the anti-Rad50 antibody is similar in magnitude to that seen in fibroblasts from FA patients (26) supports the hypothesis that a Rad50-dependent end-joining pathway is deficient in these latter cells. To test this hypothesis, we examined the effect on plasmid end-joining of electroporating anti-Rad50 antibody into FA fibroblasts belonging to several different complementation groups. Whereas co-introduction of anti-Rad50 antibody into wild-type cells reduced the efficiency with which a cohesive-ended plasmid substrate rejoined by ∼3-fold, we found that co-introduction of the anti-Rad50 antibody into FA fibroblasts belonging to complementation groups FA-A, FA-C and FA-G had no significant effect on the efficiency with which these cells re-joined either cohesive (Figure 4A) or blunt-ended (Figure 4B) plasmids. Interestingly, an additional series of experiments conducted in HT1080 and these same FA cell strains showed that co-introduction of both anti-Rad50 and anti-DNA ligase IV antibodies did not reduce DNA end-joining in any of the cell types beyond that of introduction of either antibody alone (data not shown). Based on these results, one can conclude that the DNA end-joining pathway that is deficient in FA cells is dependent on both the Rad50 and DNA ligase IV proteins.
Abundant evidence indicates that the Rad50 protein participates in DNA recombination/repair processes as part of a multi-component complex minimally comprised of Rad50, Mre11 and Nbs1 proteins (58–61). The findings presented in Figure 4A and B suggest that the introduction of antibodies specific for the Mre11 or Nbs1 proteins would also significantly reduce plasmid end-joining in normal fibroblasts. We tested this hypothesis by examining plasmid end-joining in a normal strain of diploid fibroblasts in the presence and absence of antibodies specific to the individual members of the RMN complex. Figure 4C indicates that introduction of these antibodies, either singly (samples 2–4), in pairwise combinations (samples 5–7), or all together (sample 8), resulted in a dramatic reduction of the frequency of end-joining of both cohesive-ended and blunt-ended plasmids. We performed a similar series of experiments in human fibrosarcoma HT1080 cells and observed that co-introduction of antibodies specific for any one of the members of the complex dramatically reduced end-joining efficiency. As was the case in the normal diploid fibroblasts, the degree of inhibition of plasmid end-joining seen when antibodies specific for two or more members of the complex were co-introduced did not exceed that seen when any of the antibodies was introduced separately (data not shown).
Based on the finding that introduction of any one of the antibodies dramatically reduced plasmid end-joining efficiency, and the observation that no further reductions in end-joining efficiency were associated with the introduction of combinations of the antibodies, we conclude that the individual components of the RMN complex function in the same end-joining pathway, most probably as a complex. The observation that the residual levels of end-joining seen in anti-RMN antibody-treated wild-type cells is nearly identical to that seen in FA cells further bolsters the view that the end-joining defect in these cells involves a pathway that is dependent on the RMN complex.
Deficient Rad50-mediated DNA repair renders cells sensitive to cytotoxicity induced by DNA damaging agents
The data presented above support the conclusion that the RMN complex plays an essential role in plasmid DNA end-joining within intact human fibroblasts. Furthermore, these data suggest that the RMN-dependent plasmid DNA end-joining pathway is defective in FA fibroblasts. We recently showed that FA fibroblasts are hypersensitive to cell death induced by restriction endonucleases, indicating that these cells are also deficient in chromosomal DNA double-strand break repair (26). We therefore asked whether the introduction of antibodies specific to the components of the RMN complex would sensitize a normal strain of HDFs to the cytotoxic effects of electroporated restriction endonuclease PvuII. Figure 5A shows a representative experiment. Electroporation of cells with 20 U of PvuII induced only a modest level (∼3%) of cell death. Control experiments revealed that co-introduction of non-specific IgG had no effect on PvuII-induced cell death (data not shown). In contrast, co-electroporation with antibodies specific to the individual members of the RMN complex (Figure 5A, samples 2–4) significantly enhanced the cytotoxic effect of electroporated PvuII. Interestingly, as in the case of plasmid end-joining (Figure 4), no additive effect was observed when pairwise combinations of antibodies were used to treat the cells (samples 5–7). Furthermore, the PvuII-induced cell death was no greater when a cocktail containing all three antibodies was introduced into the cells than when antibody specific for any one member of the RMN complex was introduced (Figure 5A, compare samples 8 with samples 2–5).
Figure 5.
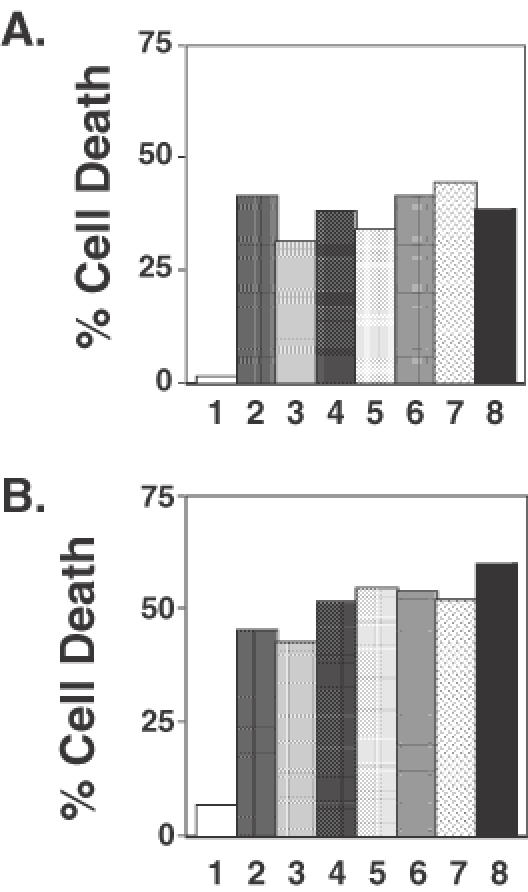
Introduction of antibodies directed against proteins in the RMN complex sensitizes normal diploid fibroblasts to agents that damage chromosomal DNA. (A) Cell death was determined following treatment with 20 U of the restriction endonuclease PvuII in cells co-electroporated with non-specific IgG (1), anti-Rad50 antibody (2), anti-Mre11 antibody (3), anti-Nbs1 antibody (4), both anti-Rad50 and anti-Mre11 antibodies (5), both anti-Rad50 and anti-Nbs1 antibodies (6), both anti-Mre11 and anti-Nbs1 antibodies (7), and anti-Rad50, anti-Mre11 and anti-Nbs1 antibodies (8). (B) Cell death was determined after treatment with 200 ng/ml diepoxybutane of cells electroporated with the same antibodies as in (A).
Sensitivity to the bifunctional alkylating agent diepoxybutane is the hallmark feature of FA cells (62). We therefore examined the influence of electroporation of anti-RMN complex antibodies on cellular sensitivity of wild-type cells to death induced by this drug. Figure 5B shows a representative graph of cell survival after exposure to 200 ng/ml diepoxybutane. Cells electroporated in the absence of antibody exhibited only modest levels of cell death (∼8%). This is indistinguishable to the cytotoxicity observed in cells electroporated with non-specific IgG (data not shown). In contrast, electroporation with antibodies specific for any of the RMN complex members rendered cells 5–7 times more sensitive to cell death induced by the drug diepoxybutane (Figure 5B, compare samples 2–4 with sample 1). We again observed that co-electroporation with antibodies specific for two or more members of the RMN complex failed to exert a greater effect on cellular sensitivity to diepoxybutane than did electroporation with antibody specific for any one member of the complex.
It is noteworthy that normal cells electroporated with antibodies specific for members of the RMN complex are as sensitive to the cytotoxic effects of diepoxybutane and PvuII as non-treated FA cells. These results support the conclusion that the RMN complex function is required to protect cells from death induced by DNA damaging agents, and that this RMN complex-dependent process is defective in FA cells.
If this interpretation was correct, whereas introduction of an anti-Rad50 antibody into normal fibroblasts dramatically sensitizes them to the cytotoxic effects of PvuII, as shown in Figure 5A, introducing this antibody into FA cells will have no effect on their sensitivity to PvuII-induced cell death. To test this hypothesis, we examined the effect on PvuII-induced cytotoxicity of introducing anti-Rad50 antibody into a diploid strain of fibroblasts belonging to complementation group FA-C. As Figure 6 reveals, introduction of anti-Rad50 antibody had no effect on the sensitivity of FA-C cells to PvuII-induced cell death [Figure 6, compare filled circles (antibody treated) with open circles (no antibody)]. As a control, we performed an identical experiment on FA-C fibroblasts that were transduced with a retrovirus expressing the Fancc protein. It has been previously shown that these retrovirally transduced cells are no longer sensitive to the cytotoxic effects of restriction endonucleases and have wild-type levels of plasmid end-joining (26). We observed that, consistent with our previous findings (26), retrovirally corrected FA-C fibroblasts were substantially more resistant to the cytotoxic effects of both 15 and 30 U of PvuII than were the non-corrected FA-C fibroblasts (Figure 6, compare open squares with open circles). We further observed that, as expected, treatment of the retrovirally corrected FA-C fibroblasts with anti-Rad50 antibody substantially enhanced their sensitivity to the cytotoxic effects of both 15 and 30 U of PvuII [Figure 6, compare open squares (no antibody) with filled squares (anti-Rad50 antibody treated)]. It was noteworthy that treatment of retrovirally corrected FA cells with anti-Rad50 antibody rendered them as sensitive to PvuII-induced cell death as were unmodified FA cells (Figure 5). Additional experiments revealed that introducing an anti-Rad50 antibody into FA-A fibroblasts had no effect on their sensitivity to PvuII-induced cell death, whereas similar treatment of retrovirally corrected FA-A fibroblasts substantially enhanced their sensitivity to death induced by PvuII (data not shown).
Figure 6.
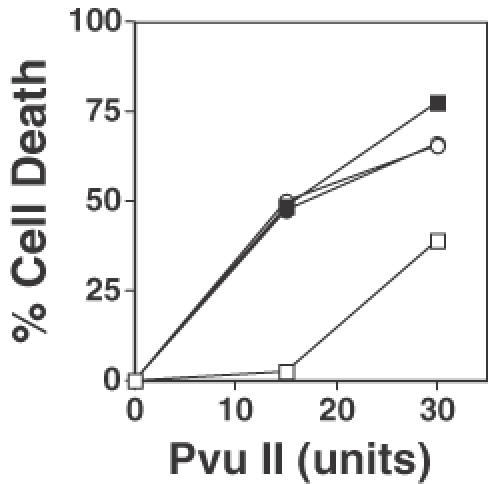
Anti-Rad50 antibody fails to sensitize FA-C cells to restriction endonuclease-induced cell death. Restriction endonuclease-induced cell death was measured in FA-C fibroblasts (circles), and in retrovirally-corrected FA-C fibroblasts (squares) in the absence (open symbols) and presence of anti-Rad50 antibody (filled symbols).
DISCUSSION
The results presented above provide compelling evidence that plasmid end-joining in human fibroblasts is dependent on DNA ligase IV and the Rad50/Mre11/Nbs1 (RMN) complex. Introduction of antibodies specific to DNA ligase IV or to any of the RMN complex proteins significantly diminished the efficiency with which both blunt-ended and cohesive-ended plasmid DNA substrates were rejoined in human fibroblast cells. Since co-introduction of antibodies to more than one of the RMN protein had no greater inhibitory effect on plasmid end-joining than did introduction of individual antibodies alone, we believe that these proteins function as a complex in the end-joining reaction. The data revealed that antibody-mediated inhibition of RMN complex function sensitizes fibroblasts to the cytotoxic effects of DNA-damaging agents, which support the conclusion that the RMN proteins are required for efficient repair of chromosomal DNA double-strand breaks as well.
The view that human fibroblasts possess an RMN-dependent end-joining pathway is consistent with a number of previous observations. For example, plasmid end-joining in the yeast Saccharomyces cerevisiae is dependent on the function of the Rad50, Mre11 and Xrs2 (the yeast homolog of Nbs1) genes (28,63). In addition, it was shown that expression of antisense mRNA specific for Rad50 significantly increased the sensitivity of human cells to DNA damaging agents (64). Furthermore, inclusion of an antibody specific for Rad50 significantly diminished the efficiency of plasmid end-joining in nuclear protein extracts prepared from a human cell line (42).
It is known that efficient end-joining of plasmid substrates in human fibroblasts and the yeast S.cerevisiae is dependent on DNA ligase IV and DNL4 proteins, respectively (65–67). However, while plasmid end-joining in yeast is entirely dependent on the DNL4-binding protein Lif1, a homolog of Xrcc4 (68), our results indicate that plasmid end-joining in human fibroblasts occurs with wild-type efficiency even in the presence of antibodies that neutralize Xrcc4 protein function. This is also consistent with the results showing that plasmid end-joining occurs at wild-type levels in a Chinese hamster ovary cell line deficient in Xrcc4 function (57). A further apparent difference between yeast and mammalian cells is that while Rad50/Mre11/Xrs2 (the yeast homolog of Nbs1)-dependent plasmid end-joining pathway in yeast is entirely dependent on the yeast homologs of Ku70/86, Hdf1/Hdf2 (28), we observed that the efficiency of plasmid end-joining in human fibroblasts is not dependent on Ku86. Again, experiments performed in Chinese hamster ovary cells found that plasmid end-joining occurred with essentially wild-type efficiencies in cells genetically deficient in the KU86 gene (57). Thus, it appears that while the RMN-dependent pathway of DNA end-joining has been conserved from yeast to mammals, there are some key differences in the molecular details of this pathway in the two species.
Our results suggest that an RMN-dependent end-joining pathway plays an important role in protecting the cells from induced chromosomal DNA damage. Our data show that interfering with a RMN-dependent DNA ligase IV-dependent process sensitizes wild-type cells to the cytotoxic effects of both introduced restriction endonucleases and the bifunctional alkylating agent diepoxybutane. Interestingly, antibody-mediated inhibition of the RMN proteins renders wild-type cells as sensitive to these agents as are FA fibroblasts. Conversely, inhibiting the RMN and DNA ligase IV proteins has no effect on plasmid end-joining efficiency in FA cells, or does it further sensitize these cells to the cytotoxic effects of introduced restriction endonuclease or diepoxybutane. These data support the conclusion that an RMN-dependent, DNA ligase IV-dependent pathway is non-functional in FA cells and that this defect is largely responsible for the hypersensitivity of these cells to DNA damaging agents.
This finding is consistent with a series of recent findings that have highlighted a link at the molecular level between the RMN complex and FA (see Introduction). Additionally, hypomorphic alleles of the Mre11 and Nbs1 genes are associated with the human genetic disorders Ataxia telangiectasia-like disorder (69) and Nijmegen breakage syndrome (70,71), respectively. Despite significant differences, all these disorders are characterized by cancer predisposition, and cells derived from these patients display genomic instability and hypersensitivity to agents, which cause lesions that are substrates for DNA double-strand break repair pathways. It is therefore conceivable that these disorders and FA may all result from subtly distinct defects within a common DNA repair pathway. Thus, more in-depth examination of the RMN-dependent DNA ligase IV-dependent DNA pathway described herein will provide significant insight into these connections.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr David Baltimore for kindly providing reagents used herein. This work was supported by the Leukemia Research Fund, the National Institutes for Health Grant AG16678 and Breast Cancer Research Programme Grant DAMD17-99-9299 from the United States Department of Defence.
REFERENCES
- 1.Auerbach A.D. and Allen,R.G. (1991) Leukemia and preleukemia in Fanconi anemia patients. A review of the literature and report of the International Fanconi anemia registry. Cancer Genet. Cytogenet., 51, 1–12. [DOI] [PubMed] [Google Scholar]
- 2.Dossantos C.C., Gavish,H. and Buchwald,M. (1994) Fanconi anemia revisited—old ideas and new advances. Stem Cells, 12, 142–153. [DOI] [PubMed] [Google Scholar]
- 3.Buchwald M. and Moustacchi,E. (1998) Is Fanconi anemia caused by a defect in the processing of DNA damage? Mutat. Res., 408, 75–90. [DOI] [PubMed] [Google Scholar]
- 4.Dapolito M., Zelante,L. and Savoia,A. (1998) Molecular basis of Fanconi anemia. Haematologica, 83, 533–542. [PubMed] [Google Scholar]
- 5.Dutrillaux B., Aurias,A., Dutrillaux,A.M., Buriot,D. and Prieur,M. (1982) The cell cycle of lymphoblasts in Fanconi anemia. Hum. Genet., 62, 327–332. [DOI] [PubMed] [Google Scholar]
- 6.Joenje H. and Oostra,A.B. (1983) Effect of oxygen tension on chromosomal aberrations in Fanconi anaemia. Hum. Genet., 65, 99–101. [DOI] [PubMed] [Google Scholar]
- 7.Joenje H., Arwert,F., Eriksson,A.W., de Koning,H. and Oostra,A.B. (1981) Oxygen-dependence of chromosomal aberrations in Fanconi's anaemia. Nature, 290, 142–143. [DOI] [PubMed] [Google Scholar]
- 8.Joenje H., Oostra,A.B., Wijker,M., Disumma,F.M., Vanberkel,C.G.M., Rooimans,M.A., Ebell,W., Vanweel,M., Pronk,J.C., Buchwald,M. and Arwert,F. (1997) Evidence for at least eight Fanconi anemia genes. Am. J. Hum. Genet., 61, 940–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schindler D. and Hoehn,H. (1988) Fanconi anemia mutation causes cellular susceptibility to ambient oxygen. Am. J. Hum. Genet., 43, 429–435. [PMC free article] [PubMed] [Google Scholar]
- 10.Seyschab H., Sun,Y., Friedl,R., Schindler,D. and Hoehn,H. (1993) G2 phase cell cycle disturbance as a manifestation of genetic cell damage. Hum. Genet., 92, 61–68. [DOI] [PubMed] [Google Scholar]
- 11.Bigelow S.B., Rary,J.M. and Bender,M.A. (1979) G2 chromosomal radiosensitivity in Fanconi's anemia. Mutat. Res., 63, 189–199. [DOI] [PubMed] [Google Scholar]
- 12.Gluckman E. (1990) Radiosensitivity in Fanconi anemia: application to the conditioning for bone marrow transplantation. Radiother. Oncol., 1, 88–93. [DOI] [PubMed] [Google Scholar]
- 13.Noll M., Bateman,R.L., D'Andrea,A.D. and Grompe,M. (2001) Preclinical protocol for in vivo selection of hematopoietic stem cells corrected by gene therapy in Fanconi anemia group C. Mol. Therap., 3, 14–23. [DOI] [PubMed] [Google Scholar]
- 14.Meetei A.R., de Winter,J.P., Medhurst,A.L., Wallisch,M., Waisfisz,Q., van de Brugt,H.J., Oostra,A.B., Yan,Z., Ling,C., Bishop,C.E. et al. (2003) A novel ubiquitin ligase is deficient in Fanconi anemia. Nature Genet., 35, 165–170. [DOI] [PubMed] [Google Scholar]
- 15.de Winter J.P., Leveille,F., van Berkel,C.G.M., Rooimans,M.A., van der Weel,L., Steltenpool,J., Demuth,I., Morgan,N.V., Alon,N., Bosnoyan-Collins,L. et al. (2000) Isolation of a cDNA representing the Fanconi anemia complementation group E gene. Am. J. Hum. Genet., 67, 1306–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Winter J.P., Rooimans,M.A., van der Weel,L., van Berkel,C.G.M., Alon,N., Bosnoyan-Collins,L., de Groot,J., Zhi,Y., Waisfisz,Q., Pronk,J.C. et al. (2000) The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nature Genet., 24, 15–16. [DOI] [PubMed] [Google Scholar]
- 17.de Winter J.P., Waisfisz,Q., Rooimans,M.A., van Berkel,C.G.M., Bosnoyan-Collins,L., Alon,N., Carreau,M., Bender,O., Demuth,I., Schindler,D. et al. (1998) The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nature Genet., 20, 281–283. [DOI] [PubMed] [Google Scholar]
- 18.Foe J.R., Rooimans,M.A., Bosnoyan-Collins,L., Alon,N., Wijker,M., Parker,L., Lightfoot,J., Carreau,M., Callen,D.F., Savoia,A. et al. (1996) Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nature Genet., 14, 320–323. [DOI] [PubMed] [Google Scholar]
- 19.Howlett N.G., Taniguchi,T., Olson,S., Cox,B., Waisfisz,Q., De Die-Smulders,C., Persky,N., Grompe,M., Joenje,H., Pals,G. et al. (2002) Biallelic inactivation of Brca2 in Fanconi anemia. Science, 297, 606–609. [DOI] [PubMed] [Google Scholar]
- 20.Strathdee C.A., Gavish,H., Shannon,W.R. and Buchwald,M. (1992) Cloning of cDNAs for Fanconi's anaemia by functional complementation. Nature, 356, 763–767. [DOI] [PubMed] [Google Scholar]
- 21. The Fanconi anaemia/breast cancer consortium (1996) Positional cloning of the Fanconi anaemia group A gene. Nature Genet., 14, 324–328. [DOI] [PubMed] [Google Scholar]
- 22.Timmers C., Taniguchi,T., Hejna,J., Reifsteck,C., Lucas,L., Bruun,D., Thayer,M., Cox,B., Olson,S., D'Andrea,A.D., Moses,R. and Grompe,M. (2001) Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol. Cell, 7, 241–248. [DOI] [PubMed] [Google Scholar]
- 23.Escarceller M., Rousset,S., Moustacchi,E. and Papadopoulo,D. (1997) The fidelity of double strand breaks processing is impaired in complementation groups B and D of Fanconi anemia, a genetic instability syndrome. Somatic Cell Mol. Genet., 23, 401–411. [DOI] [PubMed] [Google Scholar]
- 24.Escarceller M., Buchwald,M., Singleton,B.K., Jeggo,P.A., Jackson,S.P., Moustacchi,E. and Papadopoulo,D. (1998) Fanconi anemia C gene product plays a role in the fidelity of blunt DNA end-joining. J. Mol. Biol., 279, 375–385. [DOI] [PubMed] [Google Scholar]
- 25.Lundberg R., Mavinakere,M. and Campbell,C. (2001) Deficient DNA end joining activity in extracts from Fanconi anemia fibroblasts. J. Biol. Chem., 276, 9543–9549. [DOI] [PubMed] [Google Scholar]
- 26.Donahue S.L. and Campbell,C. (2002) A DNA double strand break repair defect in Fanconi anemia fibroblasts. J. Biol. Chem., 277, 26243–26247. [DOI] [PubMed] [Google Scholar]
- 27.Valerie K. and Povirk,L.F.(2003) Regulation and mechanisms of mammalian double-strand break repair. Oncogene, 22, 5792–5812. [DOI] [PubMed] [Google Scholar]
- 28.Milne G.T., Jin,S., Shannon,K.B. and Weaver,D.T. (1996) Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 4189–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rathman W.K. and Chu,G. (1994) A DNA end-binding factor involved in double-strand break repair and V(D)J recombination. Mol. Cell. Biol., 14, 4741–4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frank K.M., Sekiguchi,J.M., Seidl,K.J., Swat,W., Rathbun,G.A., Cheng,H.L., Davidson,L., Kangaloo,L. and Alt,F.W. (1998) Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature, 396, 173–177. [DOI] [PubMed] [Google Scholar]
- 31.Li Z., Otevrel,T., Gao,Y., Cheng,H.L., Seed,B., Stamato,T.D., Taccioli,G.E. and Alt,F.W. (1995) The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell, 83, 1079–1089. [DOI] [PubMed] [Google Scholar]
- 32.Calsou P., Delteil,C., Frit,P., Drouet,J. and Salles,B. (2003) Coordinated assembly of Ku and p460 subunits of the DNA-dependent protein kinase on DNA ends is necessary for XRCC4-ligase IV recruitment. J. Mol. Biol., 326, 93–103. [DOI] [PubMed] [Google Scholar]
- 33.Karanjawala Z.E., Adachi,N., Irvine,R.A., Oh,E.K., Shibata,D., Schwarz,K., Hsieh,C.L. and Lieber,M.R. (2002) The embryonic lethality in DNA ligase IV-deficient mice is rescued by deletion of Ku: implications for unifying the heterogeneous phenotypes of NHEJ mutants. DNA Repair, 1, 1017–1026. [DOI] [PubMed] [Google Scholar]
- 34.Gu Y., Jin,S., Gao,Y., Weaver,D.T. and Alt,F.W. (1997) Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc. Natl Acad. Sci. USA, 94, 8076–8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nussenzweig A., Chen,C., da Costa,V., Soares,M., Sanchez,K., Sokol,K., Nussenzweig,M.C. and Li,G.C. (1996) Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature, 382, 551–555. [DOI] [PubMed] [Google Scholar]
- 36.Zhu C., Bogue,M.A., Lim,D.S., Hasty,P. and Roth,D.B. (1996) Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell, 86, 379–389. [DOI] [PubMed] [Google Scholar]
- 37.Boulton S.J. and Jackson,S.P. (1998) Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J., 17, 1819–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Labhart P. (1999) Ku-dependent non-homologous DNA end-joining in Xenopus egg extracts. Mol. Cell. Biol., 19, 2585–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verkaik N.S., Esveldt-van Lange,R.E.E., van Heemst,D., Bruggenwirth,H.T., Hoeijmakers,J.H.J., Zdzienicka,M.Z. and van Gent,D.C. (2002) Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur. J. Immunol., 32, 701–709. [DOI] [PubMed] [Google Scholar]
- 40.Chen L., Trujilo,K., Ramos,W., Sung,P. and Tompkinson,A.E. (2001) Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol. Cell, 8, 1105–1115. [DOI] [PubMed] [Google Scholar]
- 41.Huang J. and Dynan,W.S. (2002) Reconstitution of the mammalian DNA double-strand break end-joining reaction reveals a requirement for an Mre11/Rad50/Nbs1-containing fraction. Nucleic Acids Res., 30, 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong Q., Boyer,T.G., Chen,P.L. and Lee,W.H. (2002) Deficient nonhomologous end-joining activity in cell-free extracts from Brca1-null fibroblasts. Cancer Res., 62, 3966–3970. [PubMed] [Google Scholar]
- 43.Picherierri P., Auerbeck,A.D. and Rosselli,F. (2002) DNA cross-link-dependent Rad50/Mre11/Nbs1 subnuclear assembly requires the Fanconi anemia C protein. Hum. Mol. Genet., 11, 2531–2546. [DOI] [PubMed] [Google Scholar]
- 44.Nakanishi K., Taniguchi,T., Ranganathan,V., New,H.V., Moreau,L.A., Stotsky,M., Mathew,C.G., Kastan,M.B., Weaver,D.T. and D'Andrea,A.D. (2002) Interaction of Fancd2 and Nbs1 in the DNA damage response. Nature Cell Biol., 4, 913–920. [DOI] [PubMed] [Google Scholar]
- 45.Taniguchi T., Garcia-Higuera,I., Xu,B., Andreassen,P.R., Gregory,R.C., Kim,S.T., Lane,W.S., Kastan,M.B. and D'Andrea,A.D. (2002) Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell, 109, 459–472. [DOI] [PubMed] [Google Scholar]
- 46.Bender C.F., Sikes,M.L., Sullivan,R., Huye,L.E., Le Beau,M.M., Roth,D.B., Mirzoeva,O.K., Oltz,E.M. and Petrini,J.H. (2002) Cancer predisposition and hematopoietic failure in Rad50(S/S) mice. Genes Dev., 16, 2237–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hesse J.E., Lieber,M.R., Gellert,M. and Mizuuchi,K. (1987) Extrachromosomal DNA substrates in pre-B cells undergo inversion or deletion at immunoglobulin V-(D)-J joining signals. Cell, 49, 775–783. [DOI] [PubMed] [Google Scholar]
- 48.Lieber M.R., Hesse,J.E., Mizuuchi,K. and Gellert,M. (1988) Studies of V(D)J recombination with extrachromosomal substrates. Curr. Top. Microbiol. Immunol., 137, 94–99. [DOI] [PubMed] [Google Scholar]
- 49.Ray F.A., Peabody,D.S., Cooper,J.L., Cram,L.S. and Kraemer,P.M. (1990) SV40 T antigen alone drives karyotype instability that precedes neoplastic transformation of human diploid fibroblasts. J. Cell Biochem., 42, 13–31. [DOI] [PubMed] [Google Scholar]
- 50.Southern P.J. and Berg,P. (1982) Transformation of mammalian cells to resistance with a bacterial gene under the control of the SV40 promoter. J. Mol. Appl. Genet., 1, 327–341. [PubMed] [Google Scholar]
- 51.Skehan P., Storeng,R., Scudiero,D., Monks,A., McMahon,J., Vistica,D., Warren,J.T., Bokesch,H., Kenney,S. and Boyd,M.R. (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl Cancer Inst., 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
- 52.Donahue S.L., Lundberg,R., Saplis,R. and Campbell,C. (2003) Deficient regulation of DNA double-strand break repair in Fanconi anemia fibroblasts. J. Biol. Chem., 278, 29487–29495. [DOI] [PubMed] [Google Scholar]
- 53.Lieber M.R., Hesse,J.E., Mizuuchi,K. and Gellert,M. (1988) Lymphoid V(D)J recombination: nucleotide insertion at signal joints as well as coding joints. Proc. Natl Acad. Sci. USA, 85, 8588–8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silver D.P., Spanopoulou,E., Mulligan,R.C. and Baltimore,D. (1993) Dispensable sequence motifs in the RAG-1 and RAG-2 genes for plasmid V(D)J recombination. Proc. Natl Acad. Sci. USA, 90, 6100–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taccioli G.E., Gottlieb,T.M., Blunt,T., Priestley,A., Demengeot,J., Mizuta,R., Lehmann,A.R., Alt,F.W., Jackson,S.P. and Jeggo,P.A. (1994) Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science, 265, 1442–1445. [DOI] [PubMed] [Google Scholar]
- 56.Liang F. and Jasin,M. (1996) Ku80-deficient cells exhibit excess degradation of extrachromosmal DNA. J. Biol. Chem., 271, 14405–14411. [DOI] [PubMed] [Google Scholar]
- 57.Kabotyanski E.B., Gomelsky,L., Han,J.O., Stamato,T.D. and Roth,D.B. (1998) Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res., 26, 5333–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Connelly J.C. and Leach,D.R.F. (2002) Tethering on the brink: the evolutionarily conserved Mre11-Rad50 complex. Trends Biochem. Sci., 27, 410–418. [DOI] [PubMed] [Google Scholar]
- 59.D'Amours D. and Jackson,S.P. (2002) The Mre11 complex: at the crossroads of DNA repair and checkpoint signaling. Nature Rev. Mol. Cell Biol., 3, 317–327. [DOI] [PubMed] [Google Scholar]
- 60.O'Driscoll M., Cerosaletti,K.M., Girard,P.M., Dai,Y., Stumm,M., Kysela,B., Hirsch,B., Gennery,A., Palmer,S.E., Seidel,J. et al. (2001) DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell, 8, 1175–1185. [DOI] [PubMed] [Google Scholar]
- 61.Paull T.T. (2001) New glimpses of an old machine. Cell, 107, 563–565. [DOI] [PubMed] [Google Scholar]
- 62.Sasaki M.S. and Tonomura,A. (1973) A high susceptibility of Fanconi's anemia to chromosome breakage by DNA cross-linking agents. Cancer Res., 33, 1829–1836. [PubMed] [Google Scholar]
- 63.Moore J.K. and Haber,J.E. (1996) Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 2164–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim Y.C., Koh,J.T., Shin,B.A., Ahn,K.Y., Choi,B.K., Kim,C.G. and Kim,K.K. (2002) An antisense construct of full-length human RAD50 cDNA confers sensitivity to ionizing radiation and alkylating agents on human cell lines. Radiat. Res., 157, 19–25. [DOI] [PubMed] [Google Scholar]
- 65.Grawunder U., Zimmer,D., Fugmann,S., Schwarz,K. and Lieber,M.R. (1998) DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol. Cell, 2, 477–484. [DOI] [PubMed] [Google Scholar]
- 66.Teo S.H. and Jackson,S.P. (1997) Identification of Saccharomyces cerevisiae DNA ligase IV: involvement in DNA double-strand break repair. EMBO J., 16, 4788–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilson T.E., Grawunder,U. and Lieber,M.R. (1997) Yeast DNA ligase IV mediates non-homologous DNA end-joining. Nature, 388, 495–498. [DOI] [PubMed] [Google Scholar]
- 68.Herrmann G., Lindahl,T. and Schar,P. (1998) Saccharomyces cerevisiae Lif1—a function involved in DNA double-strand break repair related to mammalian Xrcc4. EMBO J., 17, 4188–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stewart G.S., Maser,R.S., Stankovic,T., Bressan,D.A., Kaplan,M.I., Jaspers,N.G., Raams,A., Byrd,P.J., Petrini,J.H. and Taylor,A.M. (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell, 99, 577–587. [DOI] [PubMed] [Google Scholar]
- 70.Carney J.P., Maser,R.S., Olivares,H., Davis,E.M., Le Beau,M.M., YatesJ.R.,III, Morgan,W.F. and Petrini,J.H. (1998) The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell, 93, 477–486. [DOI] [PubMed] [Google Scholar]
- 71.Matsuura S., Tauchi,H., Nakamura,A., Kondo,N., Sakamoto,S., Endo, Smeets,D., Solder,B., Belohradsky,B.H., Der Kaloustian,V.M., Oshimura,M., Isomura,M. et al. (1998) Positional cloning of the gene for Nijmegen breakage syndrome. Nature Genet., 19, 179–181. [DOI] [PubMed] [Google Scholar]