


Abstract
The recent interest sparked due to the discovery of a variety of functions for non-coding RNA molecules has highlighted the need for suitable tools for the analysis and the comparison of RNA sequences. Many trans-acting non-coding RNA genes and cis-acting RNA regulatory elements present motifs, conserved both in structure and sequence, that can be hardly detected by primary sequence analysis alone. We present an algorithm that takes as input a set of unaligned RNA sequences expected to share a common motif, and outputs the regions that are most conserved throughout the sequences, according to a similarity measure that takes into account both the sequence of the regions and the secondary structure they can form according to base-pairing and thermodynamic rules. Only a single parameter is needed as input, which denotes the number of distinct hairpins the motif has to contain. No further constraints on the size, number and position of the single elements comprising the motif are required. The algorithm can be split into two parts: first, it extracts from each input sequence a set of candidate regions whose predicted optimal secondary structure contains the number of hairpins given as input. Then, the regions selected are compared with each other to find the groups of most similar ones, formed by a region taken from each sequence. To avoid exhaustive enumeration of the search space and to reduce the execution time, a greedy heuristic is introduced for this task. We present different experiments, which show that the algorithm is capable of characterizing and discovering known regulatory motifs in mRNA like the iron responsive element (IRE) and selenocysteine insertion sequence (SECIS) stem–loop structures. We also show how it can be applied to corrupted datasets in which a motif does not appear in all the input sequences, as well as to the discovery of more complex motifs in the non-coding RNA.
INTRODUCTION
Recent researchs have discovered a number of different functions for RNA molecules, which have been promoted from mere mediators of the genetic information encoded in the DNA to key players in a variety of cellular processes. Several databases solely devoted to different kinds of non-coding RNA are now publicly available [e.g. see (1–7)], and search tools especially tailored for non-coding RNA have been proposed, among others in (8,9). These facts have in turn highlighted the need for suitable algorithms and tools for the analysis and the comparison of non-coding RNA sequences. In fact, the function of many trans-acting non-coding RNA genes and cis-acting RNA regulatory elements depends on the presence of motifs that are conserved both in structure and (more loosely) in sequence. Examples of these functional motifs are the secondary structure signals present in the untranslated regions (UTRs) of mRNA that are involved in the post-transcriptional regulation of the gene expression (10,11). Other classes of RNAs, such as rRNAs, tRNAs, microRNAs and other non-coding small RNAs, as well as viral mRNAs, contain motifs with regulatory activity (12–14).
Many motifs of biological relevance can be described by using RNA secondary structure alone. Usually, in sequences sharing a functional motif similarity is limited to some parts of the sequences, like single hairpins, whereas rest of their overall structure is otherwise different, and methods for the prediction of consensus structures for two or more sequences cannot be applied. Moreover, although energy-based prediction algorithms can explore exhaustively the space of possible secondary structure conformations of a sequence picking one of the optimal (minimal) energy (15–18), RNA folding often involves intermediate states, and the study of RNA folding energy landscapes indicates that they present many different local minima (19). Thus, the actual structure of some functional RNA molecules does not correspond to the most thermodynamically stable one. If we just run a prediction algorithm and look for similar structural elements, we can miss a motif altogether, since it is not predicted in some (or most of the) sequences.
One possible approach is to align the sequences, and predict a consensus secondary structure for the most conserved parts of the alignment (20,21). Clearly, methods of this kind fail in the absence of significant sequence similarity and reliable alignments. Another idea is to align the sequences and predict a common secondary structure simultaneously, either locally or globally (22). The main drawback of this kind of approach lies in the high time complexity, exponential in the number of input sequences. In the FOLDALIGN and SLASH algorithms (23,24), the complexity is reduced, with the introduction of some heuristics, to about O(L4N4) for L sequences of N nucleotides, but this kind of approach remains usable on small sets of short sequences. Finally, if one has some hint about the structure and/or the sequence of a motif, pattern matching tools like RNAmot (25), PatSearch (26) and RNAMotif (27) can be used, to locate regions in the sequences that can fold into a given structure. The problem is that without substantial information, using strict constraints for the design of a descriptor may lead to miss some occurrences of a motif, while looser parameters yield in turn a very large number of false positives. A method used for post-processing the results of matching algorithms in order to extract the most similar hits has been proposed (28), whereas an approach based on evolutionary computation aimed instead at the optimization of the descriptor parameters is presented in (29). These methods are computationally expensive on large datasets. All in all, it is hardly a surprise that since the earlier works in this field, the problem has been considered to be as hard as ‘finding hairpins in a haystack’ (30).
METHODS
The method we present can be split into two separate parts. Our aim is to reduce, to the minimum possible, information concerning the motif needed by the algorithm and at the same time to keep a feasible computational complexity. Given a set of related RNA sequences expected to share a conserved motif, the algorithm first extracts a set of candidate regions from each one by using a very simple heuristic, associating with each region a potential secondary structure. The algorithm needs only one input parameter, which denotes the number of single hairpins contained in the structure that has to be associated with the regions. A priori, no further constraints are required for the size and number of the different elements comprising the structure (loops, stacks, connecting elements and so on) or any threshold for the folding energy. In the second part, candidate regions whose secondary structure contains the number of hairpins given as input are kept, and are compared with one another to build the group of ‘most similar’ ones, taking at most one candidate from each sequence. Candidate regions from the same sequence may overlap one another. The idea is to evaluate groups of regions by aligning them, and to find the group of regions that builds the best multiple alignment. The algorithm evaluates the alignments according to a scoring function that takes into account simultaneously the similarity to the sequence of the regions aligned as well as the similarity to the secondary structure it associated with them at the first step. Because building and evaluating all possible alignments is computationally unfeasible, we introduce a greedy heuristic for this task. The second part of the algorithm, however, could also be used independently from the first, e.g. to post-process the output generated by other motif finding programs.
Selecting initial candidates
It can be proved that the number of potential secondary structures that a RNA sequence of length n can form is exponential to the length of the sequence itself (31). Clearly, this is a number too large to be dealt with, even for short sequences. However, if we associate with each possible region of a sequence only one structure, this number becomes quadratic in the length of the sequence itself, since a string of length n has exactly n(n − 1)/2 substrings. Moreover, suppose that we are interested in stem–loop motifs. If we associate with each region the structure of minimal energy, and keep only those regions whose structure contains a single hairpin, the number is reduced further, and is estimated to be O(n). These considerations hold for any other type of secondary structure that comprises more than a single hairpin. Although perhaps not valid for each case, we expect this simple criterion to be reasonable for a large class of structural motifs, like regulatory signals in mRNA or in general conserved structure in the non-coding RNAs. In other words, we assume that a structural motif corresponds to a global-free energy optimum for the region forming it, and its formation has therefore to depend solely on local interactions among the nucleotides of the region and it is not the effect of the presence of other structures elsewhere in the sequence. This is also the reason why the algorithm processes every possible region instead of selecting a given region size and checking whether in some part it contains the structure required. Also, folding parameters used by energy-based RNA secondary prediction methods can be considered reliable enough when applied to small regions (i.e. a region folding into a structure with one or two hairpins usually ranges in size from 15 to 50 nt). Any available additional information concerning the motif (e.g. the number of its internal loops, the size of the hairpin loops, folding energy thresholds, sequence composition and so on) can be easily incorporated into the selection step so as to reduce further the number of initial candidates. For example, we might accept only hairpins whose loop has a given size (or is within a given size range) and/or that present a single internal loop or a bulge, and so on. Likewise, further constraints can be defined for more complex structures, e.g. disallowing multiloops accepting only series of hairpins (or vice versa).
Thus, given as input the number h of single hairpins that have to be contained in the secondary structure of the motif (and possible additional constraints), the algorithm selects from each input sequence the regions whose predicted optimal secondary structure contains exactly h hairpins. If this parameter is also not available, the algorithm can be simply iterated starting from a single hairpin, and increasing the number h at each run. In the absence of further constraints, the algorithm examines all the possible substrings of each input sequence. We also did not include any explicit energy threshold in the generation of the initial candidates. Although secondary structure motifs can be expected to be quite stable, deciding a priori a suitable threshold value is far from easy, and usually involves trying different values. Thus, we just chose to accept any region whose structure has energy lower than zero, i.e. lower than the unfolded state.
Finding similar regions
Finding motifs shared by nearly all the sequences
Once a set of candidate regions for each of the input sequences has been generated, if we assume that the motif appears in (nearly) all the sequences the problem arises for selecting one candidate for each sequence, in order to obtain a group of regions that, by considering their sequence and structure simultaneously, are similar enough to one another, and can be reasonably suspected to be instances of the same functional motif. Given a set of k input sequences of length n, however, an heuristic method has to be used to find an optimal solution, since finding the most similar group of candidates among the O(nk) possible solutions is computationally unfeasible for values of k > 3 or for values of n > 200–300 nt. For example, on a set of 10 sequences of length 200 with about 100 candidate regions for sequence, we have about 1011 possible region groups to evaluate. Therefore, we introduced a greedy heuristic to explore much more efficiently this large solution space.
More in detail, let S = {S1, S2, …, Sk} be the set of input sequences, and let {R1, R2, …, Rk} be the set of candidate regions associated with each sequence. First, the algorithm computes all the pairwise alignments between the regions of set R1 and R2, by considering sequence and structure information at the same time. Each alignment is described with a profile, scored with a suitable function S(M) that reflects the degree of sequence and structure similarity between the regions comprising it (further details can be found in the Supplementary Material). The p highest scoring profiles are kept, whereas the others are discarded. Next, the regions taken from S3 are aligned with each of the profiles saved. Each profile is scored again, and the p best alignments are saved. Then, the algorithm proceeds to the regions taken from sequence S4 and so on. The number p of profiles saved at each step can be chosen arbitrarily beforehand. Clearly, the larger is this number, the larger is the number of candidate solutions that are evaluated, and the more accurate (and slower) is the algorithm.
Thus, given as input a set of RNA sequences S = {S1, …, Sk}, and a secondary structure template defined by the number h of hairpins contained in it, the steps of the algorithm can be summarized as follows (Figure 1):
Figure 1.
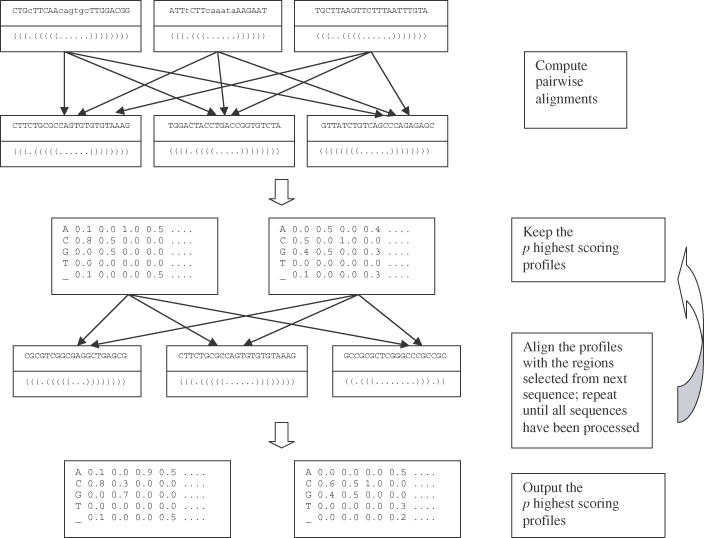
Schematic diagram of the structure of the algorithm.
For each sequence Si, generate the set of candidate regions Ri. A candidate region is a region of Si whose structure of minimal folding energy contains exactly h hairpins.
Compute all pairwise alignments of the candidate regions of R1 with the candidates of R2, and compute the score S(M) of each profile.
Save the p highest scoring profiles.
- For each i ∈ {3,…, k}:
- Align each profile saved at the previous step with every region of Ri, and compute the score S(M) of the resulting profiles.
- Save the p highest scoring profiles.
End for
Output the p highest scoring profiles.
The algorithm is greedy, i.e. at each step i (each sequence processed) only the best p profiles [instead of all the potential O(ni)] are kept, hoping that at the end one of them will lead to the optimal solution (under the score function used). Thus, the final alignments, as well as their scores, can depend on the order in which the sequences are processed, i.e. the risk is to discard in the initial steps profiles corresponding to the real motif, and save only those corresponding to similar enough regions that at the end led to lower scoring solutions. One way to overcome these potential problems is to randomize the choice of the sequence processed at each step. That is, two sequences are picked at random (instead of the first two) at the first step, and in the same manner in the following steps a sequence that has not been used in the previous iterations is chosen at random. Then, the algorithm can be run a few times (with very high probability of choosing the sequences in different order at each try), and the best result is obtained by the comparison of the results output at each run. Also, well-conserved motifs should appear very frequently in the results, regardless of the order in which the sequences are processed.
One or more sequences in the input dataset might not contain an occurrence of the motif. However, the algorithm still picks the ‘best’ region from them trying to fit it into the alignment. In order to be able to tell which of the reported instances actually correspond to a real motif instance, the best p profiles found at the end undergo further processing before the final output. More in detail, a fitness value is associated with each of the regions comprising a profile, reflecting how well the region fits the profile itself (see Supplementary Material). The idea is that random regions not corresponding to actual motif instances should have a much lower (largely lower than zero) fitness value than the real ones, so as to permit to distinguish and discard them. As we demonstrate in the experiments, a positive or close to zero fitness value means that the instance reported fits well in the profile describing the motif, whereas a negative value indicates that the corresponding region should be suspected to come from a sequence not containing an instance of the motif. Finally, the profiles reported are analyzed further so as to give an idea on the degree of conservation both in sequence and in structure, expressed as a percentage value.
Finding motifs shared by a few sequences
The algorithm we described works well in practice if a conserved motif appears in all (or a significant fraction of) the input sequences. In case only a small subset of the input shares the same motif, we can employ a slightly different method for the progressive construction of the profiles. The regions are selected from each sequence in the same manner. Then, the regions of each sequence are compared with all the regions that have been selected from the other sequences. In other words, regions selected from sequence Si are aligned with all the regions of the sets Rj selected from the other sequences Sj, with j ≠ i. Also in this case, the highest scoring p profiles are saved. The difference is that, instead of saving the best alignment of regions coming from two sequences, the profiles saved correspond to the best pairwise alignments obtained by comparing all possible pairs of candidate regions. Then, at the second iteration each profile is aligned with all the candidate regions coming from the sequences that were not used to build it. That is, if profile M was built with two regions from sequences Si and Sj, then it is aligned with all the candidates from sequences Sl such that l ≠ i and l ≠ j. The result is a set of three-region alignments. Again, the p highest scoring profiles are kept, and at the following steps each profile is extended by aligning it to regions from sequences that had not been used at the previous steps. Also in this case, profiles can be extended until they contain exactly one region per sequence, or until they are formed q regions out of k input sequences. The algorithm can nevertheless output the best profiles obtained at each iteration (i.e. the best profiles comprised of two regions, three and so on up to q). This strategy is suitable for finding motifs appearing in q sequences out of k, where q is small (below k/2).
The advantage of this method is that, potentially, it is capable of finding motifs appearing in a small subset (of any size) of the input sequences. That is, if a motif is shared only by two sequences, then the corresponding regions should build the best alignment among the pairwise ones. The main drawback is that it is more prone to fall into local optima. If, for example, the input contains two nearly identical sequences, then the best pairwise alignments will turn out to be built from regions coming from these two sequences, and the profiles generated from them will influence the results of the successive iterations. Also, this approach is more expensive from the computational point of view. Although in the original algorithm, given k input sequences containing n regions the algorithm has to perform about n2 alignments in each of the k iterations [O(kn2) in all], in this case the number of alignments to be performed rises to about O(k2n2), with an increase in the execution time at each iteration roughly proportional to the number of input sequences. On the other hand, in this case the algorithm has to be run only once, since the results no longer depend on the order in which the sequences are processed. Thus, when little or no information are available concerning the actual number of sequences that could contain the motif, the best choice is to run the algorithm either in ‘full’ mode (looking for motifs comprised of regions taken from all the sequences), and then choosing the second strategy with a low threshold value for the number of regions (up to one-half of the sequences).
The second strategy can also be seen as a simple clustering method. In other words, given a set of candidate regions, the most similar pairs are merged into a cluster, represented by the alignment profile, and at the following iterations each cluster is expanded by adding to it one region. Although using the algorithm to process whole databases and genomes is not computationally feasible, it can be applied, e.g. in the post-processing of the results obtained from a genome-wide or database scan for regions fitting a motif descriptor (by saving each hit as a separate sequence), for which different programs can be used.
RESULTS
The algorithm has been implemented on a standard Pentium IV class desktop PC with 256 MB of RAM, running the Linux operating system. The routines for folding and free energy evaluation of the regions were taken from the RNAlib library, part of the Vienna RNA package publicly available at http://www.tbi.univie.ac.at/~ivo/RNA/, and are based on the energy parameters described in (16,17).
There were many things to be verified in the experiments. First, to see whether the local optimum rule we introduced, to generate the starting set of candidates, is powerful enough to capture functional motifs, i.e. the latter actually correspond to a local minimum of the folding free energy. Second, we wanted to determine whether the greedy heuristic is capable of finding the highest scoring profile, and if the scores of profiles corresponding to conserved motifs are actually the highest, also including the possibility of having some sequences wrongly added to the datasets (not containing a motif instance). In all the experiments, the best p = 100 profiles were saved at each step (we kept this value as a default in every test). No explicit energy threshold was set for motif instances or any further constraint on the structure of the motifs. Moreover, the usual strategy for modeling and discovering motifs more complex than a single hairpin is either the prediction of a consensus secondary structure (in case the similarity covers most of the structure of the sequences considered), or to search for single hairpins first, and then combine them into more complex structures. Thus, we performed some tests also to determine whether the energy-based selection criterion applied to single hairpins works also in this case. In all the following experiments, we ran the algorithm on each dataset varying the number of hairpins h, starting from one. If the score of the best motif found with h + 1 hairpins remained stable (or was increased) when compared with h, we accepted the output of the last run as the motif, and proceeded by adding another hairpin. Otherwise, we stopped and reported the best motif found with h hairpins.
Iron responsive element (IRE)
This is a typical benchmark used in the tests of methods for the discovery of conserved structural elements in RNA. Among others, it has been studied in (24,28–30). The IRE is a conserved hairpin structure located in the 5′-UTR or in the 3′-UTR of various mRNAs coding for proteins involved in cellular iron metabolism (32,33). The interaction of the IRE with regulatory proteins modulates the translation of the mRNA according to the amount of iron present in the cell. Two alternative IRE consensus structures have been discovered (34). Some IREs present an unpaired nucleotide (usually cytosine) along the stem, whereas in others the cytosine nucleotide and two additional bases seem to oppose one free 3′ nucleotide, as shown in Figure 2. The lower stem are usually in the range 3-5 bp.
Figure 2.

The two canonical forms of the IRE.
First of all, the IRE motif provides a good example for the feasibility of our criterion for the selection of initial candidates. A search performed with the PatSearch algorithm (26) on the Untranslated Sequence Database (UTRdb) (7) using either of the motif templates shown above reported 65 candidate motifs in 5′-UTRs and 86 in 3′-UTRs. Among these (see also Supplementary Material), 16 (47 in 3′) had negative but not optimal energy associated with them and 9 (35 in 3′) had positive energy. Thus, according to the selection criterion of the algorithm, 25 (82 in 3′) non-optimal candidate IRE regions would be discarded a priori. The examination of the non-optimal candidates revealed that they had been located in the UTRs of mRNAs (or cDNAs) not annotated to be involved in iron metabolism, including a human ferritin pseudo-gene, thus very likely to be ‘false positives’. On the other hand, >90% of the optimal structures were located in mRNAs of proteins experimentally known to contain the IRE.
For the discovery test, we retrieved from GenBank the full mRNA sequences of human ferritin (light and heavy chain), and of aminolevulinate synthase 2, as well as their mouse homologs. To assess the performance and effectiveness of the algorithm also in the case of spurious sequence contamination, we added the mRNA sequences of three human ferritin pseudogenes to the dataset. Therefore, three sequences out of nine did not contain the IRE. The sequences ranged in size from ∼800-2000 nt. The results are shown in Figure 3. As we can see, the algorithm was capable of identifing the IRE in the correct sequences. Moreover, the motif instances reported in the pseudogenes have a much lower and negative fitness value, and thus can be reasonably suspected not to correspond to the functional motif.
Figure 3.

Highest scoring motif occurrences output by RNAProfile on the IRE dataset with their respective energy and fitness value. Note that the last three regions (reported in pseudogenes) have a much lower fitness value, thus very unlikely to be real IRE instances.
Atypical IREs
Although usually the IRE has one of the two forms described in the previous section, experimental observation has discovered some exceptions to the rule. We used these examples to test the ability of the algorithm also in the presence of instances much less conserved in sequence and structure with respect to the canonical forms. We built our dataset with the 5′-UTR of ferritin mRNA of fruit fly, bull frog, starfish and crayfish, in order to have the least degree of similarity in IRE instances. We also added to the input set the 5′-UTR of eight plant ferritin mRNA sequences, lacking the IRE. In this case, only one-third of the sequences contained the motif. We ran the algorithm to find motifs shared by a few sequences, saving the best groups up to size six (half of the sequences).
Figure 4 shows the result. The highest scoring four-region group contains the four IREs. As we can see, the bottom helix differs in length in the various instances. Also, both the starfish and crayfish IREs have a base pair missing at the bottom of the topmost helix. Moreover, crayfish does not contain the unpaired cytosine at the canonical position of the internal loop, usually considered the hallmark of the IRE. However, it has been experimentally proved that this IRE keeps its functional activity (35). Instead the frog and fruit fly IREs have the canonical form with a single unpaired C. Although structurally very different, all the correct IRE instances have been picked by the algorithm. When further regions are added, the highest scoring groups of five and six regions again contain the same four IREs, plus two additional regions with very low fitness values, that can be reasonably suspected not to be functional IREs.
Figure 4.

The results on the atypical IRE dataset. The first four instances, corresponding to the IREs, were included in the highest-scoring profile of four regions (and also the best two and three regions groups contained IRE instances). In the following two iterations, the best profile still contained the same four regions, plus the two shown at the bottom of the figure, unlikely to be IRE instances given the low fitness value.
Selenocysteine insertion sequence (SECIS)
Selenocysteine is the recently discovered 21st amino acid. The codon calling for this amino acid in mRNA is UGA, usually indicating the end of translation (36). To have UGA read as a selenocysteine codon instead of as a stop codon, and bound by the corresponding tRNA, mRNAs coding for proteins containing this amino acid (selenoproteins) present the SECIS element, a conserved stem–loop structure located in the 3′-UTR. The spacing between the UGA codon and the respective SECIS element determines whether the codon is actually translated as selenocysteine (37). Two forms of SECIS motif have been determined, respectively with long (12–14 nt, SECIS I) and short (3–6 nt) apical loops (SECIS II) (38) (Figure 5). SECIS I stems can be split into two parts, interspersed by an internal loop. The topmost helix contains non-canonical base pairings at the bottom, always with two G-A pairs. These non-canonical pairs are essential to mediate selenoprotein translation. In the second form, nucleotides forming the apical loop bind to one another, and as a result the upper helix is interrupted by another internal loop. Other than the non-canonical G-A pairs, SECIS hairpins present two or three consecutive unpaired adenine nucleotides, either in the apical loop (I) or inside an internal loop (II). Experimental evidence seems to support the fact that form II is found more often than the first (38). Moreover, recent discoveries have shown that in some cases the two unpaired adenines are replaced by two cytosines [selenoproteins M and O, (39,40)], making even harder the definition of a suitable unique descriptor for this motif, whose conservation seems to be mostly protein-specific. Several algorithms and programs have been developed for the discovery of potential SECIS elements in mRNA, and genome-wide scans have led to the discovery of new selenoproteins (41–44). Virtually all the methods rely on the presence of the non-canonical G-A pairs within a given distance range from an AA (or CC) dinucleotide.
Figure 5.
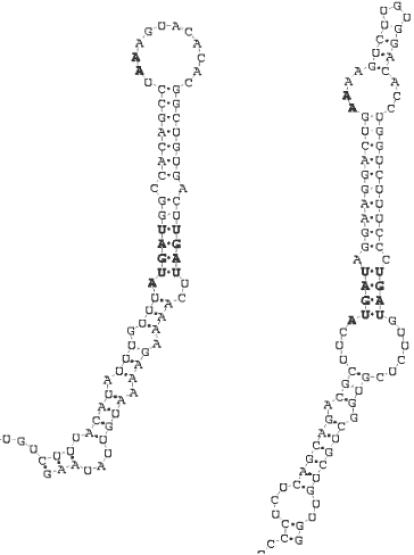
Secondary structure models of SECIS stem–loop elements: type I (left) and type II (right).
In our test, we did not include the possibility of non-canonical pairings in the generation of the initial candidates. Thus, the problem was whether the upper helix was conserved enough to be detected by the algorithm, and if it was possible to discriminate it against background spurious random motifs. We retrieved from GenBank the 3′-UTR of human, cow, mouse, rat and chicken mRNA sequences of the glutathione peroxidase 4 (GPX4). In addition, we added to the dataset the 3′-UTR of a glutathione peroxidase sequence reported not to contain the selenocysteine amino acid (thus, supposedly lacking the SECIS element). The results are shown in Figure 6. The algorithm was capable of detecting the upper part of the motif, just above the non-canonical pairs. As we can see, apart from the adenine nucleotides in the internal loop, there is very little conservation in unpaired nucleotides, differently from many other functional motifs, and the apical loop is variable in size. Moreover, the motif has been discovered without resorting to the non-canonical base pairings. By examining its fitness value, the non-selenocysteine sequence can be easily discriminated from the real instances of the SECIS motif.
Figure 6.

Highest scoring motif occurrences output by RNAProfile on the GPX4 dataset. The last region, with a significantly low fitness value, comes from a non-selenoprotein 3′-UTR.
Since the algorithm describes a motif with the alignment of a set of instances, a possible way to investigate new sequences suspected to contain a motif is to process them together with other sequences containing an instance of the motif itself. Selenoprotein M 3′-UTRs of man and mouse contain SECIS II elements whose AA dinucleotide is replaced by CC (39). Thus, if we just processed these sequences alone, we would have obtained a suspicious-looking motif that does not resemble the two known types of SECIS. We added to the GPX4 dataset the 3′-UTR sequences of human and mouse selenoprotein M. The results are shown in Figure 7. As we can see, the two instances reported for the new sequences contain two unpaired cytosines. If we inspect the fitness value associated with them, we can however see that they can be considered as instances of the motif, while the sequence not coding for a selenoprotein has an instance with a negative fitness value. Thus, despite the absence of the main hallmark of the SECIS II motif, the algorithm correctly identified them as possible new SECIS instances.
Figure 7.

Highest scoring motif occurrences in the GPX4 sequences combined with the selenoprotein M dataset.
Finding composite motifs
Drosophila Nanos mRNA 3′-UTR
The nanos protein in Drosophila is required for correct anterior/posterior patterning in the Drosophila embryo. Translation of the nanos mRNA is repressed in the bulk cytoplasm and activated in the posterior region. Repression is mediated by a translation control element (TCE) in the 3′-UTR of the mRNA. This element forms a Y-shaped structure, part of which is recognized by the Smaug protein leading to translational repression (45). In our experiment, we used the 3′-UTR of the nanos mRNA of Drosophila melanogaster, D.virilis and D.simulans, ranging in size from 200 to 400 nt and showing the least degree of similarity in their TCEs. In this case, the score of the best profile found remained stable in the two runs with one and two hairpins, and moreover, the best result of the first run was contained in the second. The latter, shown in Figure 8, had captured the two hairpins forming the Y structure.
Figure 8.

Highest scoring motif occurrences output by RNAProfile on the Drosophila nanos dataset.
RNase P RNA
Ribonuclease P (RNase P) is a ubiquitous endoribonuclease, found in archaea, bacteria and eukaryotes as well as chloroplasts and mitochondria. Its best characterized activity is the generation of mature 5′ ends of tRNAs by cleaving the 5′-leader elements of precursor-tRNAs. Cellular RNase Ps are ribonucleoproteins. RNA from bacterial RNase Ps retains its catalytic activity in the absence of the protein subunit, i.e. it is a ribozyme. Isolated eukaryotic and archaeal RNase P RNA has not been shown to retain its catalytic function, but is still essential for the catalytic activity of the holoenzyme (46,47). Despite the many shared characteristics of RNase Ps, the structural and functional organization of diverse species of RNase P differs significantly across phylogenetic domains. The secondary structure of RNAse P RNA has been determined mainly by phylogenetic-comparative approaches. Eukaryotic RNAs display great variability in sequence and content of structural elements (48), and it is very difficult to align and compare sequences from even closely related groups like yeasts or vertebrates. In our test, we retrieved from the RNase P database (2) the eukaryotic RNA sequences of yeasts Arxiozyma telluris, Pichia mississippiensis and Kluyveromyces thermotolerans, as well as the sequences of toad, rat and human, having an average identity of 40%. In this case, the score of the two hair–pins run was the highest. The results are shown in Figure 9.
Figure 9.

Highest scoring motif occurrences output by RNAProfile on the RNAse P RNA dataset, corresponding to consensus helices 8 and 9 (see also Figure 10).
The profile describes two adjacent stem–loop structures, corresponding to helices 8 and 9 (see also Figure 10) in each of the input sequences. These two helices have been reported to be part of the core secondary structure of RNase P RNA, conserved from bacteria throughout eukaryotes. Although no functional role has been as yet assigned to this structure, perhaps the formation of the two adjacent helices serves as a catalyst for the overall folding of the sequence. To test this hypothesis, we folded the human sequence using mFold (49), first with no constraints, then constrained by the presence of the motif identified by RNAProfile. The structure described in the RNase P database (2) contains six stem–loop structures. While in the unconstrained case mFold reported a structure of optimal energy containing only three hairpins, the constrained fold contained seven hairpins, matching the ones described in (2), plus a small one formed in a region left unpaired in the structure of the database (between helices 11 and 19). Thus, as we have seen in this case, motifs reported by RNAProfile could be used as a constraint for the prediction of a consensus secondary structure, very difficult for RNase P RNA and other kinds of non-coding RNA, because compensatory mutations can hardly be detected, except in very close relatives (50).
Figure 10.
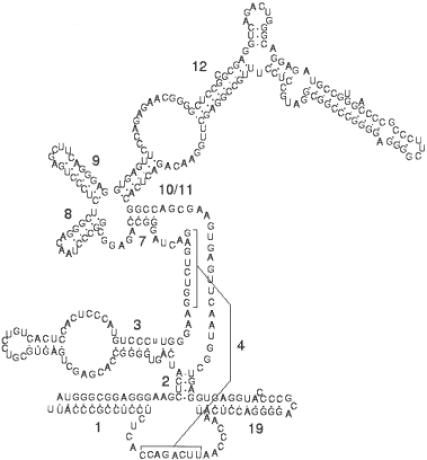
Secondary structure of human RNase P RNA [structure adapted from (4)]. Numbered helices form the consensus secondary structure of the molecule, conserved (despite no sequence conservation) in bacteria throughout eukaryotes.
DISCUSSION
The comprehensive identification and characterization of all encoded RNAs, the molecules they interact with, and the molecular structure of functional complexes is of utmost importance for a comprehensive understanding of the biology of a cell. Indeed, the important role played by non-coding RNAs in fundamental cellular processes is becoming even clearer, opening new avenues for a deeper understanding of life. Differently from DNA, whose double-stranded structure implies that the genetic information is fully encoded in the primary sequence of nucleotides, RNAs have a much higher structural flexibility and consequently may play a variety of crucial cellular functions whose specificity relies on both sequence and structural properties. This fact makes their functional annotation harder, particularly when dealing with single molecules. Comparative approaches, now made possible by the steady progress in the characterization of human and other organism transcriptomes, are certainly the most promising computational approaches for the prediction and the characterization of functional RNA motifs. Hence, the need for versatile computational methods for ‘comparative Rnomics’, able to detect functional RNAs to be then subjected to the suitable experimental validation.
The novel method presented here for the discovery of conserved functional and structural motifs from a set of related RNA sequences is simple, computationally efficient and showed to be effective in several test cases under a variety of conditions with loose conservation of sequence and/or structure. It outperforms the few existing methods both in terms of computation time and of prediction accuracy. In fact, methods for this task usually require a high degree of similarity in sequence (to align the sequences beforehand) and/or in structure (to predict consensus secondary structures), or substantial information regarding the structure of the motif to be found, whereas RNAprofile just needs a single parameter denoting how many hairpins a motif should contain. Moreover, the algorithm can be applied in different modes, to detect motifs globally conserved in a set of sequences, or motifs appearing only in a small subset of them. In addition, while not suitable for database and genome scans, it can be used to cluster the hits obtained with any other matching method. As we have shown in the experimental tests, our method can identify conserved motifs in RNA, comprised of a single hairpin or more than one, without aligning the sequences, or requiring any prior knowledge about the structure and/or the degree of conservation of a motif. In the absence of sequence similarity, the algorithm can anyway report motifs highlighting structural similarity alone. The heuristics the method is based on are also efficient enough to deal with quite long sequences. The simple criterion for the selection of candidate regions has proved itself to be feasible, also for the description of motifs comprised of more than a single hairpin. Finally, the algorithm does not need any input parameter other than a number of hairpins, and in any case can be simply iterated trying different numbers and comparing the results of each run. The implementation of the algorithm also proved to be quite efficient, taking a few (<3) minutes for each run on the first IRE dataset (that anyway included very long sequences), and always a few seconds in every other test.
PROGRAM AVAILABILITY
RNAProfile has been implemented and tested under various Linux/Unix platforms, and is available free of charge from ftp://www.pesolelab.it/pub/ (current version is 2.0). It includes the standard algorithm implemented in the two modes described in this paper, plus some additional features, like the possibility to post-process results obtained with different pattern matching algorithms and other methods for the generation of initial candidates and large-scale screening. Alternatively, the algorithm can be started with one or more initial profiles (that can be used as descriptors of known motifs), and regions taken from new sequences can be added to them so as to check whether the motif described by the profile appears in them.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by FIRB project ‘Bioinformatica per la Genomica e la Proteomica’ (Ministero dell'Istruzione e Ricerca Scientifica, Italy) and by Telethon.
REFERENCES
- 1.Bonnal S., Boutonnet,C., Prado-Lourenco,L. and Vagner,S. (2003) IRESdb: the Internal Ribosome Entry Site database. Nucleic Acids Res., 31, 427–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown J.W. (1999) The Ribonuclease P Database. Nucleic Acids Res., 27, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown J.W., Echeverria,M., Qu,L.H., Lowe,T.M., Bachellerie,J.P., Huttenhofer,A., Kastenmayer,J.P., Green,P.J., Shaw,P. and Marshall,D.F. (2003) Plant snoRNA database. Nucleic Acids Res., 31, 432–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffiths-Jones S., Bateman,A., Marshall,M., Khanna,A. and Eddy,S.R. (2003) Rfam: an RNA family database. Nucleic Acids Res., 31, 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenblad M.A., Gorodkin,J., Knudsen,B., Zwieb,C. and Samuelsson,T. (2003) SRPDB: Signal Recognition Particle Database. Nucleic Acids Res., 31, 363–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szymanski M., Erdmann,V.A. and Barciszewski,J. (2003) Noncoding regulatory RNAs database. Nucleic Acids Res., 31, 429–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pesole G., Liuni,S., Grillo,G., Licciulli,F., Mignone,F., Gissi,C. and Saccone,C. (2002) UTRdb and UTRsite: specialized databases of sequences and functional elements of 5′ and 3′ untranslated regions of eukaryotic mRNAs. Update 2002. Nucleic Acids Res., 30, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein R.J. and Eddy,S.R. (2003) RSEARCH: finding homologs of single structured RNA sequences. BMC Bioinformatics, 4, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rivas E. and Eddy,S.R. (2001) Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics, 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gray N.K. and Wickens,M. (1998) Control of translation initiation in animals. Annu. Rev. Cell Dev. Biol., 14, 399–458. [DOI] [PubMed] [Google Scholar]
- 11.Mignone F., Gissi,C., Liuni,S. and Pesole,G. (2002) Untranslated regions of mRNAs. Genome Biol., 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eddy S.R. (2001) Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet., 2, 919–929. [DOI] [PubMed] [Google Scholar]
- 13.Eddy S.R. (2002) Computational genomics of noncoding RNA genes. Cell, 109, 137–140. [DOI] [PubMed] [Google Scholar]
- 14.Huttenhofer A., Brosius,J. and Bachellerie,J.P. (2002) RNomics: identification and function of small, non-messenger RNAs. Curr. Opin. Chem. Biol., 6, 835–843. [DOI] [PubMed] [Google Scholar]
- 15.Zuker M. and Stiegler,P. (1981) Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res., 9, 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walter A.E., Turner,D.H., Kim,J., Lyttle,M.H., Muller,P., Mathews,D.H. and Zuker,M. (1994) Coaxial stacking of helixes enhances binding of oligoribonucleotides and improves predictions of RNA folding. Proc. Natl Acad. Sci. USA, 91, 9218–9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathews D.H., Sabina,J., Zuker,M. and Turner,D.H. (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol., 288, 911–940. [DOI] [PubMed] [Google Scholar]
- 18.Lyngso R.B., Zuker,M. and Pedersen,C.N. (1999) Fast evaluation of internal loops in RNA secondary structure prediction. Bioinformatics, 15, 440–445. [DOI] [PubMed] [Google Scholar]
- 19.Chen S.J. and Dill,K.A. (2000) RNA folding energy landscapes. Proc. Natl Acad. Sci. USA, 97, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Witwer C., Rauscher,S., Hofacker,I.L. and Stadler,P.F. (2001) Conserved RNA secondary structures in Picornaviridae genomes. Nucleic Acids Res., 29, 5079–5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hofacker I.L., Fekete,M., Flamm,C., Huynen,M.A., Rauscher,S., Stolorz,P.E. and Stadler,P.F. (1998) Automatic detection of conserved RNA structure elements in complete RNA virus genomes. Nucleic Acids Res., 26, 3825–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sankoff D. (1985) Simultaneous solution of the RNA folding, alignment and protosequence problems. SIAM J. Appl. Math., 45, 810–825. [Google Scholar]
- 23.Gorodkin J., Heyer,L.J. and Stormo,G.D. (1997) Finding the most significant common sequence and structure motifs in a set of RNA sequences. Nucleic Acids Res., 25, 3724–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorodkin J., Stricklin,S.L. and Stormo,G.D. (2001) Discovering common stem-loop motifs in unaligned RNA sequences. Nucleic Acids Res., 29, 2135–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laferriere A., Gautheret,D. and Cedergren,R. (1994) An RNA pattern matching program with enhanced performance and portability. Comput. Appl. Biosci., 10, 211–212. [DOI] [PubMed] [Google Scholar]
- 26.Grillo G., Licciulli,F., Liuni,S., Sbisa,E. and Pesole,G. (2003) PatSearch: a program for the detection of patterns and structural motifs in nucleotide sequences. Nucleic Acids Res., 31, 3608–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macke T.J., Ecker,D.J., Gutell,R.R., Gautheret,D., Case,D.A. and Sampath,R. (2001) RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res., 29, 4724–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fogel G.B., Porto,V.W., Weekes,D.G., Fogel,D.B., Griffey,R.H., McNeil,J.A., Lesnik,E., Ecker,D.J. and Sampath,R. (2002) Discovery of RNA structural elements using evolutionary computation. Nucleic Acids Res., 30, 5310–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Y.J. (2002) Prediction of consensus structural motifs in a family of coregulated RNA sequences. Nucleic Acids Res., 30, 3886–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dandekar T. and Hentze,M.W. (1995) Finding the hairpin in the haystack: searching for RNA motifs. Trends Genet., 11, 45–50. [DOI] [PubMed] [Google Scholar]
- 31.Waterman M.S. (1995) Introduction to Computational Biology. Chapman & Hall, London, UK. [Google Scholar]
- 32.Ke Y., Sierzputowska-Gracz,H., Gdaniec,Z. and Theil,E.C. (2000) Internal loop/bulge and hairpin loop of the iron-responsive element of ferritin mRNA contribute to maximal iron regulatory protein 2 binding and translational regulation in the iso-iron-responsive element/iso-iron regulatory protein family. Biochemistry, 39, 6235–6242. [DOI] [PubMed] [Google Scholar]
- 33.Theil E.C. (1998) The iron responsive element (IRE) family of mRNA regulators. Regulation of iron transport and uptake compared in animals, plants, and microorganisms. Met. Ions Biol. Syst., 35, 403–434. [PubMed] [Google Scholar]
- 34.Gdaniec Z., Sierzputowska-Gracz,H. and Theil,E.C. (1999) Iron regulatory element and internal Loop/Bulge structure for ferritin mRNA studied by Cobalt(III) hexammine binding, molecular modeling, and NMR spectroscopy. Biochemistry, 38, 5676. [DOI] [PubMed] [Google Scholar]
- 35.Huang T.S., Melefors,O., Lind,M.I. and Soderhall,K. (1999) An atypical iron-responsive element (IRE) within crayfish ferritin mRNA and an iron regulatory protein 1 (IRP1)-like protein from crayfish hepatopancreas. Insect Biochem. Mol. Biol., 29, 1–9. [DOI] [PubMed] [Google Scholar]
- 36.Low S.C. and Berry,M.J. (1996) Knowing when not to stop: selenocysteine incorporation in eukaryotes. Trends Biochem. Sci., 21, 203–208. [PubMed] [Google Scholar]
- 37.Martin G.W., Harney,J.W. and Berry,M.J. (1996) Selenocysteine incorporation in eukaryotes: insights into mechanism and efficiency from sequence, structure, and spacing proximity studies of the type 1 deiodinase SECIS element. RNA, 2, 171–182. [PMC free article] [PubMed] [Google Scholar]
- 38.Fagegaltier D., Lescure,A., Walczak,R., Carbon,P. and Krol,A. (2000) Structural analysis of new local features in SECIS RNA hairpins. Nucleic Acids Res., 28, 2679–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korotkov K.V., Novoselov,S.V., Hatfield,D.L. and Gladyshev,V.N. (2002) Mammalian selenoprotein in which selenocysteine (Sec) incorporation is supported by a new form of Sec insertion sequence element. Mol. Cell. Biol., 22, 1402–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kryukov G.V., Castellano,S., Novoselov,S.V., Lobanov,A.V., Zehtab,O., Guigo,R. and Gladyshev,V.N. (2003) Characterization of mammalian selenoproteomes. Science, 300, 1439–1443. [DOI] [PubMed] [Google Scholar]
- 41.Castellano S., Morozova,N., Morey,M., Berry,M.J., Serras,F., Corominas,M. and Guigo,R. (2001) In silico identification of novel selenoproteins in the Drosophila melanogaster genome. EMBO Rep., 2, 697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lescure A., Gautheret,D., Carbon,P. and Krol,A. (1999) Novel selenoproteins identified in silico and in vivo by using a conserved RNA structural motif. J. Biol. Chem., 274, 38147–38154. [DOI] [PubMed] [Google Scholar]
- 43.Kryukov G.V., Kryukov,V.M. and Gladyshev,V.N. (1999) New mammalian selenocysteine-containing proteins identified with an algorithm that searches for selenocysteine insertion sequence elements. J. Biol. Chem., 274, 33888–33897. [DOI] [PubMed] [Google Scholar]
- 44.Driscoll D.M. and Chavatte,L. (2004) Finding needles in a haystack. EMBO Rep., 5, 140–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crucs S., Chatterjee,S. and Gavis,E.R. (2000) Overlapping but distinct RNA elements control repression and activation of nanos translation. Mol. Cell, 5, 457–467. [DOI] [PubMed] [Google Scholar]
- 46.Frank D.N. and Pace,N.R. (1998) Ribonuclease P: unity and diversity in a tRNA processing ribozyme. Annu. Rev. Biochem., 67, 153–180. [DOI] [PubMed] [Google Scholar]
- 47.Xiao S., Scott,F., Fierke,C.A. and Engelke,D.R. (2002) Eukaryotic ribonuclease P: a plurality of ribonucleoprotein enzymes. Annu. Rev. Biochem., 71, 165–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frank D.N., Adamidi,C., Ehringer,M.A., Pitulle,C. and Pace,N.R. (2000) Phylogenetic-comparative analysis of the eukaryal ribonuclease P RNA. RNA, 6, 1895–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res., 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perriquet O., Touzet,H. and Dauchet,M. (2003) Finding the common structure shared by two homologous RNAs. Bioinformatics, 19, 108–116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.