


Abstract
Using an in vivo plasmid transformation method, we have determined the DNA sequences recognized by the KpnAI, StySEAI, StySENI and StySGI R-M systems from Klebsiella oxytoca strain M5a1, Salmonella eastbourne, Salmonella enteritidis and Salmonella gelsenkirchen, respectively. These type I restriction-modification systems were originally identified using traditional phage assay, and described here is the plasmid transformation test and computer program used to determine their DNA recognition sequences. For this test, we constructed two sets of plasmids, pL and pE, that contain phage lambda and Escherichia coli K-12 chromosomal DNA fragments, respectively. Further, using the methylation sensitivities of various known type II restriction enzymes, we identified the target adenines for methylation (listed in bold italics below as A or T in case of the complementary strand). The recognition sequence and methylation sites are GAA(6N)TGCC (KpnAI), ACA(6N)TYCA (StySEAI), CGA(6N)TACC (StySENI) and TAAC(7N)RTCG (StySGI). These DNA recognition sequences all have a typical type I bipartite pattern and represent three novel specificities and one isoschizomer (StySENI). For confirmation, oligonucleotides containing each of the predicted sequences were synthesized, cloned into plasmid pMECA and transformed into each strain, resulting in a large reduction in efficiency of transformation (EOT).
INTRODUCTION
Type I restriction-modification (R-M) systems consist of three genes (hsdR, hsdM and hsdS) and many have been allocated to one of four families (IA, IB, IC and ID), based on characteristics such as amino acid similarity and complementation (1,2). Type I restriction enzymes are multi-functional and are made up of three subunits that behave as either a restriction endonuclease or a methyltransferase depending upon the methylation status of the recognition sequence. The specificity subunit recognizes a bipartite DNA sequence, 13–15 bp in length, with a 3–4 bp component, a 6–7 bp random spacer sequence followed by a 4–5 bp component. Type I enzymes consistently methylate both strands of the recognition sequence at the N6 position of a specific adenine.
Currently there are over 60 known and 200 putative type I restriction and modification systems, but only 28 type I DNA recognition sequences have been identified (3). Recognition sequences for type I restriction enzymes have been previously found using several methods (2,4–6). An in vitro method, commonly used to find the recognition sequences for type II enzymes, involves cleaving DNA with a purified restriction enzyme and comparing the pattern of fragments with a set of patterns generated by a computer (7). Another in vitro method, requires a purified methylase to label substrate DNA of known sequence and then digesting DNA using several type II restriction enzymes to separate labeled fragments (4). Using computer analysis, the labeled fragments are then compared to find a common recognition sequence. An in vivo strategy (6) uses M13 phage vectors containing DNA fragments of known sequence (5). This method is based on the principle that a phage DNA fragment containing an unmodified target site plates with a reduced efficiency when transferred into a strain containing an R-M system (8,9). Similar observations were made regarding bacterial conjugation (10).
We have modified this in vivo approach and established a quantitative R-M system in Escherichia coli using a set of plasmids containing phage lambda DNA (11). Plasmids with unmodified recognition sites are cleaved, whereas modified plasmids and those lacking a given recognition sequence are not subject to restriction. Restriction activity is seen as a reduction in the efficiency of transformation (EOT) of 10−1 or less. The recognition sequences can be found using the program RM search developed for this purpose (12). Here we have applied this method to several restriction systems from genus Klebsiella and Salmonella. Because the determination of the recognition sequence for the KpnAI system was the first trial of this method, the process is described in detail.
KpnAI was discovered in Klebsiella oxytoca M5a1, a strain formerly recognized as Klebsiella pneumoniae (13,14). The R-M genes were cloned and KpnAI was characterized as a member of the newly defined type ID family (14,15). Using bacteriophage P1, Bullas et al. (16) identified 12 Salmonella strains with different restriction specificities in a collection of serotypes. However the recognition sequences for only four of these systems had been determined (3). We report the recognition sequences for StySEAI, StySENI and StySGI, three Salmonella systems found in Bullas' original study (16). In addition, the modified adenine in the each recognition sequence was determined using the methylation sensitivity of the various type II restriction enzymes (3,12).
MATERIALS AND METHODS
Reagents, strains and plasmids
Bacterial strains, phage and plasmids used in this study are listed in Table 1. Bacteria were grown in Luria–Bertani (LB) medium and incubated at 37°C with vigorous aeration. Ampicillin was added to a final concentration of 200 μg/ml (2 mg/ml in case of K.oxytoca). Growth was monitored using optical density measurements at 510 nm.
Table 1. Bacterial strains and plasmids.
| Strain or plasmid | Relevant phenotype, genotype or description | Source or reference |
|---|---|---|
| K.oxytoca | ||
| M5a1 | R+KpnAI M+KpnAI | (13) |
| M5a1R | R−KpnAI M+KpnAI | (14) |
| Salmonella–E.coli hybrids | ||
| L4039 | R+SEAI M+SEAI | (16) |
| L4030 | R+SENI M+SENI | (16) |
| L4039 | R+SGI M+SGI | (16) |
| L4021 | R+STI M+STI | (16) |
| E.coli | ||
| DH5α | R−K M+K | Lab stock |
| Plasmids | ||
| Lambda subclonesa | ||
| pL1 (5.5 kb) | BamHI clone of phage lambda | (11) |
| pL2 (16.8 kb) | BamHI clone of phage lambda | (11) |
| pL3 (5.6 kb) | BamHI clone of phage lambda | (11) |
| pL4 (6.5 kb) | BamHI clone of phage lambda | (11) |
| pL5 (7.2 kb) | BamHI clone of phage lambda | (11) |
| pL6 (6.8 kb) | BamHI clone of phage lambda | (11) |
| pL7 (3.4 kb) | Subclone of pL1 | This study |
| pL8 (6.5 kb) | Subclone of pL2 | This study |
| pL9 (10.3 kb) | Subclone of pL2 | This study |
| pL10 (3.8 kb) | Subclone of pL4 | This study |
| pL11 (1.4 kb) | Subclone of pL7 | This study |
| pL12 (3.1 kb) | Subclone of pL8 | This study |
| pL13 (1.1 kb) | Subclone of pL9 | This study |
| pL14 (0.13 kb) | Subclone of pL11 | This study |
| pL15 (3.7 kb) | EcoRV clone of phage lambda | This study |
| pL16 (2.0 kb) | HindIII clone of phage lambda | This study |
| pL17 (2.3 kb) | HindIII clone of phage lambda | This study |
| pL18 (0.56 kb) | HindIII clone of phage lambda | This study |
| pL19 (6.6 kb) | HindIII clone of phage lambda | This study |
| pL20 (1.6 kb) | Subclone of pL19 | This study |
| pL21 (0.77 kb) | Subclone of pL20 | This study |
| pL26 (4.9 kb) | EcoRI clone of phage lambda | This study |
| pL27 (5.6 kb) | EcoRI clone of phage lambda | This study |
| pL28 (5.8 kb) | EcoRI clone of phage lambda | This study |
| pL29 (3.9 kb) | EcoRV clone of phage lambda | This study |
| pL30 (1.7 kb) | EcoRV clone of phage lambda | This study |
| E.coli subclonesa | E.coli map coordinate | (19) |
| pE2 (5.8 kb) | 1557431-1563188 | This study |
| pE3 (2.8 kb) | 1608569-1611375 | This study |
| pE4 (4.1 kb) | 1565001-1569077 | This study |
| pE5 (11.4 kb) | 1593721-1605160 | This study |
| pE6 (8.1 kb) | 1402492-1410639 | This study |
| pE8 (12.9 kb) | 1505286-1518231 | This study |
| pE9 (1.7 kb) | 1523044-1524690 | This study |
| pE10 (1.9 kb) | 1582185-1584064 | This study |
| pE11 (3.0 kb) | 1465811-1468761 | This study |
| pE12 (5.1 kb) | 1479912-1485078 | This study |
| pE14 (4.4 kb) | 1553017-1557430 | This study |
| pE15 (4.8 kb) | 1518232-1523043 | This study |
| pE16 (4.5 kb) | 1426716-1431168 | This study |
| pE17 (3.2 kb) | 1584065-1587223 | This study |
| pE18 (6.5 kb) | 1524691-1593720 | This study |
| pE19 (4.9 kb) | 1468762-1473696 | This study |
| pE22 (6.2 kb) | 1473697-1479911 | This study |
| pE23 (2.6 kb) | 1456295-1458942 | This study |
| pE24 (1.6 kb) | 1563189-1564830 | This study |
| pE26 (3.9 kb) | 1647821-1651757 | This study |
| pE28 (0.6 kb) | 1674657-1675295 | This study |
| pE29 (3.7 kb) | 1342364-1346037 | This study |
| pE31 (3.1 kb) | 1336254-1339389 | This study |
| pE33 (2.4 kb) | 1339984-1342363 | This study |
| pE38 (0.6 kb) | 1339390-1339983 | This study |
| pE44 (1.0 kb) | 1278264-1279262 | This study |
| pE45 (16.6 kb) | 1265543-1280035 | This study |
| Oligonucleotide clones | ||
| pKpnAI | Plasmid with KpnAI site | This study |
| pKpnAI-H1 | Plasmid with KpnAI/HindIII sites | This study |
| pKpnAI-H2 | Plasmid with KpnAI/HindIII sites | This study |
| pKpnAI-H3 | Plasmid with KpnAI/HindIII sites | This study |
| pSEAI-S1 | Plasmid with SEAI/ScaI sites | This study |
| pSEAI-S2 | Plasmid with SEAI/ScaI sites | This study |
| pSENI-S1 | Plasmid with SENI/ScaI sites | This study |
| pSENI-S2 | Plasmid with SEAI/ScaI sites | This study |
| pSGI-S1 | Plasmid with SGI/SpeI sites | This study |
| pSGI-T1 | Plasmid with SGI/Tth111I sites | This study |
aLocations of lambda and E.coli subclones are shown in Figure 1.
Chemical reagents were purchased from Fisher Scientific (Hanover Park, Ill,) and Sigma (St. Louis, MI). Restriction enzymes and T4 DNA ligase were obtained from New England Biolabs (Beverly, MA) and Promega (Madison, WI). DNA sequencing was done in the core facility of the Center for Molecular Biology and Gene Therapy at Loma Linda University (Loma Linda, CA).
Using pMECA(17) as the plasmid vector, a series of lambda subclones (pL series) were developed from both original lambda DNA and from the six lambda BamHI clones described previously (11). A second series of plasmids (pE series) were developed using the Kohara E.coli K-12 chromosomal library that was made in a lambda vector (18,19). These chromosomal fragments were subcloned into the EcoRI site of the pUC9 vector.
Restriction-modification (R-M) tests
R-M tests were performed using a plasmid transformation method (11). Results are described as a relative efficiency of transformation which is defined as the number of AmpR transformants divided by the number of AmpR transformants obtained from the control strain (11).
Cloning of oligonucleotides
Oligonucleotides were synthesized in the core facility of the Center for Molecular Biology and Gene Therapy (CMBGT) (Loma Linda, CA). Single-stranded DNA was heated to 90°C and then annealed at room temperature. Double-stranded oligonucleotides were ligated into the EcoRV site of pMECA and the mixture was transformed into E.coli DH5α using CaCl2-heat shock method (20). Clones were selected by plating the mixture on L-agar containing ampicillin and X-gal (5-bromo-4-chloro-3-indolyl-ß-D-galactoside) to a final concentration of 40 μg/ml. The resulting clones were sequenced at the CMBGT core facility.
RM search computer program
We developed a computer program ‘RM search’ to detect a unique DNA sequence present in all plasmids containing a recognition sequence (positive) but absent in the plasmids which do not contain the recognition sequence (negative) (12). RM search can be used to find both type I and type II recognition sequences as well as degenerate sequences.
RESULTS
The DNA recognition sequence of KpnAI
To confirm the presence of KpnAI recognition sequences in lambda DNA, plasmids were tested using a plasmid transformation method (11). Klebsiella strains M5a1(R+) and M5a1R(R− mutant) were used as recipients. When the plasmid vector, pMECA, was transformed into each strain, an equal number of AmpR transformants were obtained (EOT = 1.0) confirming that pMECA does not contain any KpnAI sites. Six lambda BamHI plasmids, pL1 to pL6, (11) were then tested and a strong restriction (EOT = 10−1–10−3) of plasmids pL1, pL2 and pL4 was observed indicating the presence of one or more KpnAI recognition sites. These plasmids were defined as ‘positive’(+). Two of the plasmids, pL3 and pL6, did not show any reduction in transformant numbers (EOT = 1.0) and were defined as ‘negative’ (−). Transfer of the plasmid pL5 into both M5a1 and M5a1R was unsuccessful. We assume that this is probably due to harmful lambda gene expression from the high copy number plasmid in K.oxytoca strains.
When all positive and negative DNA sequence data were entered into the RM search program (12) more than one hundred candidate type I recognition sequences were found. To limit the number of candidate sequences, small positive clones or large negative clones were necessary. Thus, we constructed a number of additional subclones from the original ‘positive’ pL series plasmids. Lambda DNA was also digested with EcoRI, EcoRV or HindIII to obtain fragments for subcloning. This series of subclones, pL, are described in Figure 1A. Plasmid restriction tests were performed and the EOT values are described in Figure 2A. The presence or absence of the KpnAI site was obvious (Figure 2A) since EOT values were close to 1.0 when the plasmid had no sites (negative plasmids) and less than 10−1 when the plasmid contained at least one site (positive plasmids). A computer search using this additional data narrowed the search to seven type I sequences.
Figure 1.
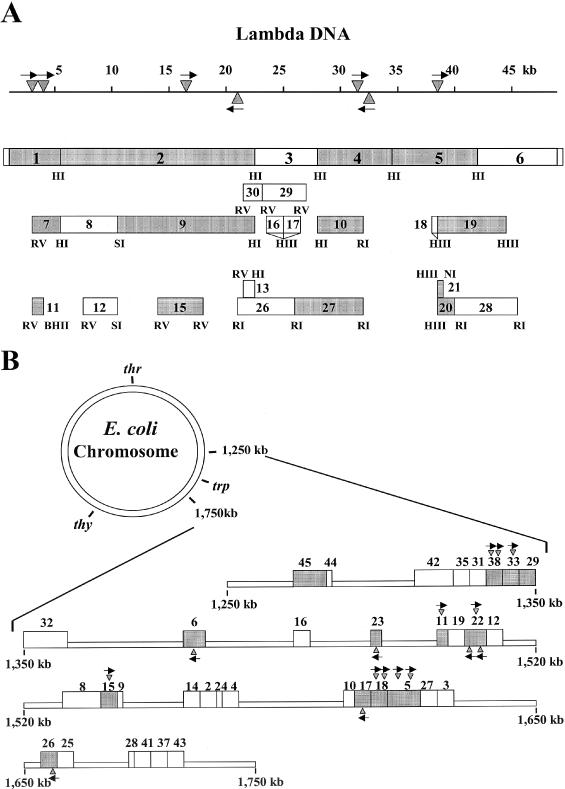
pL series plasmid subclones derived from phage lambda DNA (A) and pE series plasmids derived from fragments of E.coli K-12 chromosomal DNA (B). Each number and the corresponding box represent the plasmid inserts. Shaded boxes contain KpnAI recognition site(s) (data from Figure 2). The triangles and arrows show the location and direction of the KpnAI sites predicted from this study. Restriction sites are also shown: HI, BamHI; RI, EcoRI; RV, EcoRV; HIII, HindIII; PI, PstI; NI, NdeI; BHII, BssHII; and SI, SphI. All chromosomal fragments in the pE series plasmids were cloned into the EcoRI site.
Figure 2.
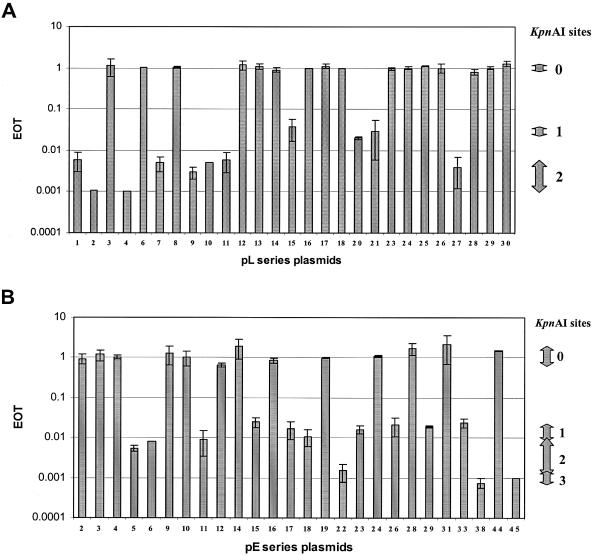
EOT values obtained from pL series plasmids (A) and pE series plasmids (B). Each plasmid was transformed into both strains M5a1 and M5a1R (control). Relative transformant numbers (EOT) were calculated. The number of KpnAI sites varies from 0 to 3. The range of EOT values for each KpnAI site are shown on the right. Standard deviations are shown as error bars.
We then developed an additional series of plasmids (pE), which contain fragments of E.coli genomic DNA ligated into pUC9 (Figure 1B, pE2 to pE45). EOT values for the pE series plasmids are shown in Figure 2B. EOT values of less than 0.1 indicated the presence of at least one recognition site. Our results showed that half of the pE series plasmids (13/26) contain a KpnAI site.
Modification tests were done to confirm the presence of a recognition site in each positive plasmid. When the EOT was less than 10−1, modified plasmids were isolated from ampicillin resistant colonies and were transferred again into M5a1 and M5a1R. In every case, EOT values were close to 1.0 (values varied from 0.8 to 1.3). These results confirm that surviving plasmids were completely modified by the KpnAI methyltransferase.
When the restriction data of the first few pE series plasmids were entered into the RM search program in addition to the pL series sequence data, only one sequence, GAA(NNNNNN)TGCC, a typical type I bipartite sequence, was found. Further analysis showed that this sequence exists in all positive pE series plasmids and is absent from all negative plasmids. Thus we concluded that this sequence is the best candidate for the KpnAI recognition sequence.
For confirmation, a 19 bp oligonucleotide containing this KpnAI sequence (Figure 3A) was synthesized and cloned into the EcoRV site of pMECA. This oligonucleotide contains an MluI site, which does not exist in the vector, to easily identify the insert. An EcoRV ligation site was created at both ends of the oligonucleotide for cloning purposes. A restriction test using this plasmid, pKpnAI, resulted in an EOT of 2 × 10−2 confirming the presence of the KpnAI recognition site. pKpnAI DNA was purified from the surviving colonies and a modification test showed that plasmids were completely modified by M5a1 as expected (EOT = 1.0).
Figure 3.

Oligonucleotides used to confirm the recognition sequence (A) and to determine the methylation sites for KpnAI (B). Oligonucleotides B-1 and B-2 were used to determine the methylated adenine in the prime strand and B-3 was used for the complementary strand. The KpnAI sequence is shown in bold. Plasmids containing these oligonucleotides are designated, from top to bottom: pKpnAI, pKpnAIH1, pKpnAIH2 and pKpnAIH3.
Computer analysis found that all degenerate forms of this sequence exist in the negative plasmid DNA. Therefore, we conclude that KpnAI recognizes only the candidate sequence listed above.
The relationship between the number of KpnAI recognition sites and the EOT is also shown in Figure 2. Plasmid DNA with additional sites was more strongly restricted as shown by a further reduction in EOT. These results support the original observation that the presence of additional restriction sites contributes to a further reduction of EOT values (6,11).
Location of KpnAI methylation sites
The top strand of the KpnAI recognition sequence, as written in Figure 3A, has two potential target adenines (A) in the trinucleotide component. The complementary strand (lower) has only one target A in the tetranucleotide component. To determine which adenines are methylated by the KpnAI methyltransferase, the following approach was used. HindIII recognizes the sequence AAGCTT and cleaves DNA between the two adenines. HindIII methyltransferase modifies m6AAGCTT; and HindIII endonuclease cannot cut this methylated sequence. However, when the second adenine is modified, HindIII endonuclease can cleave the sequence, Am6AGCTT (21).
With this knowledge, we designed three oligonucleotides containing overlapping KpnAI and HindIII recognition sequences. Two plasmids, pKpnAIH1 and pKpnAIH2 (Figure 3B-1 and B-2) were used to determine the methylated adenine in the top strand and pKpnAIH3 (Figure 3B-3) was used for the lower strand. These oligonucleotides were cloned into plasmid pMECA and transformed into Klebsiella strain M5a1.
Methylated plasmid DNA was obtained from strain M5a1 and digested with HindIII. Because one HindIII site is already present in the multi-cloning site of pMECA, the plasmid was linearized (Figure 4, lane 2). If the KpnAI methylase modifies the first adenine, creating the sequence m6AAGCTT, the site will be resistant to cleavage by the HindIII endonuclease and the plasmid will be cut only once. Thus, no additional cleavage of the plasmid will be seen when compared to plasmid pMECA. When KpnAI and HindIII methylation sites do not overlap, HindIII will cut the plasmid into two fragments. Figure 4 (lane 4) shows KpnAI methylated pKpnAIH1 cut into two fragments by HindIII, 2.6 kb and 209 bp. On the other hand, methylated pKpnAIH2 was only linearized (Figure 4, lane 6). Similarly, plasmid pKpnAIH3 was also linearized and no further cleavage was observed (Figure 4, lane 8). These results show that the second adenine in the top strand 5′-GAm6A(6N)TGCC -3′ and the single adenine in the lower strand 5′-GGCm6A(6N)TTC-3′ are modified by the KpnAI methyltransferase.
Figure 4.
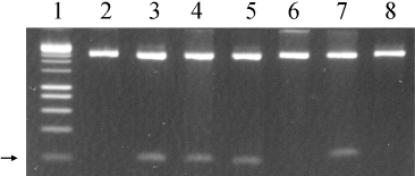
Determination of the methylation sites of KpnAI. Plasmid DNA was digested with HindIII. Lane 1, 1 kb marker; lane 2, pMECA control; lane 3, pKpnAIH1 unmodified; lane 4, pKpnAIH1 modified; lane 5, pKpnAIH2 unmodified; lane 6, pKpnAIH2 modified; lane 7, pKpnAIH3 unmodified; lane 8, pKpnAIH3 modified. The arrow shows the position of the 209 bp fragments.
Recognition sequences for Salmonella R-M systems
E.coli–Salmonella hybrids, each containing the genes for one Salmonella type I R-M system in E.coli, were used to determine DNA recognition sequences for StySENI, StySEAI and StySGI enzymes. These strains were constructed by transferring Salmonellas serB-hsd regions in to E.coli with P1 transduction (16).
The results of the R-M tests were simplified to show either the presence (+) or absence (−) of the recognition site (Table 2). All test plasmids with EOT values less than 0.1 were designated positive (+) and when EOT was greater than 0.1 the plasmid was negative (−). The vector pMECA, does not contain recognition sequences for any of the R-M systems tested. Analysis of positive and negative plasmid DNA using R-M search identified the recognition sequences for the StySEAI, StySENI and StySGI as ACA(6N)TYCA, CGA(6N)TACC and TAAC(7N)RTCG, respectively (Table 2).
Table 2. Plasmid restriction test for Salmonella R-M systems.
| Plasmid | R-M system | ||
|---|---|---|---|
| StySEAI | StySENI | StySGI | |
| pMECA | − | − | − |
| pL1 | − | − | − |
| pL2 | + | + | + |
| pL3 | + | − | − |
| pL4 | + | + | − |
| pL6 | + | − | − |
| pE2 | + | − | − |
| pE3 | + | − | − |
| pE4 | − | + | − |
| pE5 | + | + | − |
| pE6 | + | − | − |
| pE8 | + | + | − |
| pE9 | − | − | − |
| pE10 | − | + | − |
| pE11 | + | − | − |
| pE12 | + | + | − |
| pE14 | − | − | − |
| pE15 | + | + | − |
| pE16 | + | − | − |
| pE17 | − | + | − |
| pE18 | + | − | − |
| pE19 | + | + | + |
| pE22 | + | − | − |
| pE23 | − | − | − |
| pE24 | − | − | + |
| pE26 | − | + | − |
| pE28 | − | − | − |
| pE29 | − | − | − |
| pE31 | − | − | − |
| pE32 | + | + | − |
| pE33 | − | + | − |
| pE38 | − | + | − |
| pE41 | − | + | − |
| pE44 | − | − | − |
| pE45 | + | + | − |
| Recognition sequence | ACA(6N)TYCA | CGA(6N)TACC | TAAC(7N)RTCG |
| Type | I (new) | I (StySBLI isoschizomer) | I (new) |
Plus (+) or minus (−) indicates the presence or absence of a recognition sequence in each plasmid.
To confirm these sequences, the same strategy that was used for KpnAI was adopted and oligonucleotides containing each of the candidate sequences were designed (Figure 5) and cloned into pMECA. The plasmids were designated pSEAI-S1, pSENI-S1 and pSGI-S1 (Figure 5). As described below, these plasmids were also used for the determination of the methylated adenine in each recognition sequence. Plasmid R-M tests using these plasmids resulted in a reduced EOT for the restriction test (1.0 × 10−2, 1.8 × 10−2 and 4.0 × 10−2 for pSEAI-S1, pSENI-S1 and pSGI-S1, respectively) and no reduction of EOT for the modification (M) test (EOT = 0.9–1.2). These results verify the DNA recognition sequences predicted for each Salmonella R-M system (Table 2).
Figure 5.

Design of oligonucleotides used for confirmation of recognition sequences and determination of methylation sites in Salmonella R-M systems. Oligonucleotides SEA-1, SEN-1 and SG-1 were used to determine the methylated adenine in the trinucleotide portion of each recognition sequence (marked in bold). SEA-2, SEN-2 and SG-2 represent the oligonucleotide used for the tetranucleotide component. Each oligonucleotide was inserted into the EcoRV site (or SspI for SG-1) in pMECA and the plasmids were designated from the top, pSEA-S1, pSEA-S2, pSEN-S1, pSEA-S2, pSG-S1 and pSG-T1. The type II restriction enzyme site is shown with double arrows below each oligonucleotide sequence. Each methylated adenine is shown in large font.
Methylation sites for StySEAI, StySENI and StySGI
Using the methylation sensitivity method described above for KpnAI, the methylation sites for three Salmonella systems were also determined. Figure 5 also shows the composition of the additional oligonucleotides used to determine the methylation sites for StySEAI, StySENI and StySGI. The plasmids containing the additional oligonucleotide were designated pSEA-S2, pSEN-S2 and pSG-T1. The activities of the all three restriction enzymes used here: ScaI (AGTACT) and SpeI (ACTAGT) and Tth111I (GACNNNGTC) can be blocked by the methylation of the first adenine of each recognition sequence [(22), Stickel and Roberts, unpublished observation cited in (3)].
The plasmids containing each sequence were modified in the corresponding restriction and modification proficient bacterial strains. These modified plasmids were then digested with the appropriate restriction enzyme (Figure 5) in vitro. All the modified plasmids were protected from restriction whereas the original unmodified plasmids were digested. These results confirmed the target adenine for each methylase, which is shown in large font (Figure 5). These results are also consistent with the former observation that if more than one adenine exists in the recognition sequence, the adenine closer to the central (random nucleotide) region is the methylation target (23).
DISCUSSION
A total of four type I DNA recognition sequences were elucidated using plasmid R-M tests and the RM search computer program. All the sequences are typical type I sequences with bipartite structures. With the exception of StySENI (isoschizomer of StySBLI) (3), we describe three novel type I sequences (prototypes).
Previously we reported that KpnAI belongs to the type ID family (14). Subunits HsdR and HsdM of the KpnAI R-M system share extensive sequence identity with the prototype ID system, StySBLI, 95 and 98%, respectively. Less similarity, 44% is shared between the HsdS subunits possibly reflecting the fact that these two R-M systems recognize different DNA sequences. The HsdS amino acid sequence of type I restriction enzymes has variable regions at the N- and C-terminals which recognize the 5′ or 3′ ends of the DNA recognition sequences (5). However, the amino acid sequences of the central and sometimes C-terminal region of the HsdS subunits are highly conserved and unique within the families (23). Comparison of the Hsd subunits of KpnAI and StySBLI using DOTPLOT predicted high similarity between the two systems (14). The recognition sequence of StySBLI was recently identified as CGA(6N)TACC (2). Comparison of the recognition sequences for these two enzymes reveals striking similarities. First, both consist of a trinucleotide component, a 6 bp spacer and a tetranucleotide component. Second, they share five of the seven nucleotides in the recognition sequence, GA in the trinucleotide component (shifted by 1 bp) and T-CC in the tetranucleotide component. Finally, both enzymes methylate adenine in the same position. EcoR9I, also a member of the type ID family, has HsdR and HsdM subunits that complement the corresponding subunits of StySBLI (2). It is interesting to speculate how these type ID systems developed in three closely related species of enteric bacteria: Escherichia, Salmonella and Klebsiella.
Using bacteriophage EOP results, Bullas et al. (16) reported that the restriction-modification patterns are similar between StySENI and StySBLI, and also between StySGI and StySKI. As they predicted, we verified here that StySENI and StySBLI recognize the same DNA sequence, CGA(6N)TACC. Thus, StySENI is the first isoschizomer of StySBLI. The StySGI sequence, TAAC(7N)RTCG, is a degenerate form of the StySKI sequence, TAAC(7N)ATCG. This explains the previous observation that P1 phage propagated on the StySGI-bearing strain is resistant to restriction by StySKI (16). The cloning and sequencing of the specificity gene, hsdS, may reveal the difference in amino acid sequences which caused this delicate change in DNA base recognition.
As shown here, this plasmid transformation method is useful to determine the unknown recognition sequences of previously reported type I enzymes. Plasmid transformation methods are available for many bacterial strains. Theoretically, any active R-M systems could be detected if sufficient DNA sequences are available. To find the recognition sequences in other bacteria, it may be necessary to develop an additional series of plasmid sets with known DNA sequences. Alternatively, one can clone the genes by using genome information into E.coli and then determine the recognition sequences using the series of plasmids described here. The latter method may be also useful in bacterial strains producing several restriction enzymes, since determination of multiple recognition sites is much more complicated.
Acknowledgments
ACKNOWLEDGEMENTS
We thank L. Bullas for the E.coli–Salmonella hybrid strains, Terence Tay and Hiroko Emoto for their contribution to the collection and analysis of the clinical E.coli strains, and S. Kawai and A. Burnett for their contribution to the construction of the lambda plasmid subclones. This work was supported by grant DAMD17-97-2-7016 from the Department of the Army. The content of the information does not necessarily reflect the position or the policy of the Federal Government or of the National Medical Technology Testbed, Inc.
REFERENCES
- 1.Bickle T.A. and Kruger,D.H. (1993) Biology of DNA restriction. Microbiol. Rev., 57, 434–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Titheradge A.J., King,J., Ryu,J. and Murray,N.E. (2001) Families of restriction enzymes: an analysis prompted by molecular and genetic data for type ID restriction and modification systems. Nucleic Acids Res., 29, 4195–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts R.J., Vincze,T., Posfai,J. and Macelis,D. (2003) REBASE: restriction enzymes and methyltransferases. Nucleic Acids Res., 31, 418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagaraja V., Shepherd,J.C., Pripfl,T. and Bickle,T.A. (1985) Two type I restriction enzymes from Salmonella species. Purification and DNA recognition sequences. J. Mol. Biol., 182, 579–587. [DOI] [PubMed] [Google Scholar]
- 5.Cowan G.M., Gann,A.A. and Murray,N.E. (1989) Conservation of complex DNA recognition domains between families of restriction enzymes. Cell, 56, 103–109. [DOI] [PubMed] [Google Scholar]
- 6.Gann A.A., Campbell,A.J., Collins,J.F., Coulson,A.F. and Murray,N.E. (1987) Reassortment of DNA recognition domains and the evolution of new specificities. Mol. Microbiol., 1, 13–22. [DOI] [PubMed] [Google Scholar]
- 7.Gingeras T.R., Milazzo,J.P. and Roberts,R.J. (1978) A computer assisted method for the determination of restriction enzyme recognition sites. Nucleic Acids Res., 5, 4105–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arber W. (1974) DNA modification and restriction. Prog. Nucleic Acid Res. Mol. Biol., 14, 1–37. [DOI] [PubMed] [Google Scholar]
- 9.Dussoix D. and Arber,W. (1965) Host specificity of DNA produced by Escherichia coli. IV. Host specificity of infectious DNA from bacteriophage lambda. J. Mol. Biol., 11, 238–246. [DOI] [PubMed] [Google Scholar]
- 10.Arber W. and Morse,M.L. (1965) Host specificity of DNA produced by Escherichia coli. VI. Effects on bacterial conjugation. Genetics, 51, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kasarjian J.K.A., Iida,M. and Ryu,J. (2003) New restriction enzymes discovered from Escherichia coli clinical strains using a plasmid transformation method. Nucleic Acids Res., 31, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellrott K.P., Kasarjian J.K.A., Jiang,T. and Ryu,J. (2002) Restriction enzyme recognition sequence search program. Biotechniques, 33, 1322–1326. [DOI] [PubMed] [Google Scholar]
- 13.Streicher S.L., Shanmugam,K.T., Ausubel,F., Morandi,C. and Goldberg,R.B. (1974) Regulation of nitrogen fixation in Klebsiella pneumoniae: evidence for a role of glutamine synthetase as a regulator of nitrogenase synthesis. J. Bacteriol., 120, 815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee N.S., Rutebuka,O., Arakawa,T., Bickle,T.A. and Ryu,J. (1997) KpnAI, a new type I restriction-modification system in Klebsiella pneumoniae. J. Mol. Biol., 271, 342–348. [DOI] [PubMed] [Google Scholar]
- 15.Titheradge A.J., Ternent,D. and Murray,N.E. (1996) A third family of allelic hsd genes in Salmonella enterica: sequence comparisons with related proteins identify conserved regions implicated in restriction of DNA. Mol. Microbiol., 22, 437–447. [PubMed] [Google Scholar]
- 16.Bullas L.R., Colson,C. and Neufeld,B. (1980) Deoxyribonucleic acid restriction and modification systems in Salmonella: chromosomally located systems of different serotypes. J. Bacteriol., 141, 275–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomson J.M. and Parrott,W.A. (1998) pMECA: a cloning plasmid with 44 unique restriction sites that allows selection of recombinants based on colony size. Biotechniques, 24, 922–924, 926,, 928. [DOI] [PubMed] [Google Scholar]
- 18.Kohara Y., Akiyama,K. and Isono,K. (1987) The physical map of the whole E.coli chromosome: application of a new strategy for rapid analysis and sorting of a large genomic library. Cell, 50, 495–508. [DOI] [PubMed] [Google Scholar]
- 19.Blattner F.R., Plunkett,G.,III, Bloch,C.A., Perna,N.T., Burland,V., Riley,M., Collado-Vides,J., Glasner,J.D., Rode,C.K., Mayhew,G.F. et al. (1997) The complete genome sequence of Escherichia coli K-12. Science, 277, 1453–1474. [DOI] [PubMed] [Google Scholar]
- 20.Ryu J. and Hartin,R.J. (1990) Quick transformation in Salmonella typhimurium LT2. Biotechniques, 8, 43–45. [PubMed] [Google Scholar]
- 21.McClelland M., Nelson,M. and Raschke,E. (1994) Effect of site-specific modification on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Res., 22, 3640–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hofer B. (1988) The sensitivity of DNA cleavage by SpeI and ApalI to methylation by M-Ecok. Nucleic Acids Res., 16, 5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murray N.E. (2000) Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol. Mol. Biol. Rev., 64, 412–434. [DOI] [PMC free article] [PubMed] [Google Scholar]