


Abstract
RNA interference (RNAi) is a simple and powerful tool widely used for studying gene function in a number of species. Recently, inducible regulation of RNAi in mammalian cells using either tetracycline- or ecdysone-responsive systems has been developed to prevent potential lethality or non-physiological responses associated with persistent suppression of genes that are essential for cell survival or cell cycle progression. Here we show that the inducible regulation of RNAi also can be achieved by using a Cre–LoxP approach. We demonstrate that the insertion of a loxP-flanked neomycin cassette into RNA polymerase III promoter, which controls a vector-based RNAi unit, impairs the promoter activity. However, the expression of RNAi construct can be completely restored upon the removal of the neo cassette using a tamoxifen inducible Cre construct. We show that this system works with high efficiency in suppression of two endogenous genes, Fgfr2 and Survivin, in mouse embryonic stem (ES) cells, as evidenced by the decrease of levels of gene expression, reduced cell proliferation and colony formation. This system provides a potentially important yet simple approach to establish mutant mouse strains for functional study at defined stages upon turning on the inducible switches controlled by the Cre–LoxP system.
INTRODUCTION
Recent advances in RNAi have made it possible to degrade specific mRNAs of any endogenous genes in mammals (1–3). The most common procedure consists of the introduction of a 21 nt double-stranded RNA (dsRNA) into the cells using a specific transfectant. The sequence of this dsRNA or small hairpin RNA (shRNA) has to be perfectly homologous and unique to a region of the targeted mRNA. After 1–3 days, expression of the target gene is dramatically reduced and this allows investigations on the function of the gene. Although powerful on the cell culture level, this procedure is limited by the short period of action of the dsRNA. This limitation leads to the development of plasmid- and viral-based vectors, which can be stably integrated into host genome and produce shRNAs to suppress target genes (4–9).
Logically, several recent studies developed inducible regulation of RNAi in mammalian cells using either tetracycline- or ecdysone-responsive systems (10,11). These approaches prevent potential lethality or non-physiological responses associated with persistent suppression of genes that are essential for cell survival or cell cycle progression. As the initial applications, a number of endogenous genes, including DNA methyltransferase 1, p53 and p21, have been successfully knocked down in cultured cells (10,12).
Another inducible system is Cre-LoxP system, which has been most widely used in gene targeting field (13,14). Numerous transgenic mouse strains expressing Cre at specific tissues/organs have been developed to achieve spatial and temporal control of gene expression (15). Apparently, if the RNAi vectors can be modified to allow inducibility by the Cre–LoxP system, it will eventually allow the utilization of the large collection of Cre transgenic mice and significantly enhance our ability to use RNAi system to decipher gene function in vivo. Here, we demonstrate that the combination of RNAi and Cre–LoxP can be used in ES cells for inducible suppression of both an exogenous gene, GFP, and endogenous genes, Fgfr2 and Survivin.
MATERIALS AND METHODS
Vector design and engineering
The original mouse U6 promoter (8) was remodeled to allow insertion of the neomycine cassette. First, we removed the SalI site of the polylinker, deleted 45 bp from −128 to −83 of the U6 promoter and replaced it with a SalI site by site-directed mutagenesis (Stratagene, La Jolla, CA). After restriction by SalI, a 1938 bp XhoI and SalI fragment containing the ploxPneo (16) was inserted. We then mutated the ApaI site of the neomycine cassette using the previous Stratagene kit (the insertion of the first oligonucleotide in the polylinker required one ApaI-restriction step). This final version of the construct can be used to perform ligation of both oligonucleotides as previously described (8) for eGFP and subsequent RNAi experiments. For FGFR2, the oligonucleotides used are 1A, 5′-GGCCTCAGCGTTCCTGAGCGA-3′; 1B, 5′-AGCTTCGCTCAGGAACGCTGAGGCC-3′; 2A, 5′-AGCTTCGCTCAGGAACGCTGAGGCCCTTTTTG-3′; 2B, 5′-AATTCAAAAAGGGCCTCAGCGTTCCTGAGCGA-3′. For Survivin, the oligonucleotides used are 1A, 5′-GGCCTCCTAGCAGGATCTTAA-3′; 1B, 5′-AGCTTTAAGATCCTGCTAGGAGGCC-3′; 2A, 5′-AGCTTTAAGATCCTGCTAGGAGGCCC TTTTTG-3′; 2B, 5′-AATTCAAAAAGGGCCTCCTAGCAGGATCTTAA-3′.
Cell culture and treatment
eGFP-expressing transformed mammary tumor cells have been produced following transfection of W069 cells (17) with pCMS-eGFP (Clonetech, Palo Alto, CA) using lipofectamine 2000 following instructions of the manufacturer (Invitrogen, Carlsbad, CA). The eGFP-expressing cells were sorted twice at 7 and 30 days after transfection using an FACS Calibur (Becton-Dickson, San Jose, CA). Transient and stable transfections were performed on BOSC23 and W069 cells with lipofectamine 2000. Briefly, cells are seeded 1 day prior to transfection at 80% confluence in six-well plates and transfected with 100 ng of pCMS-eGFP and 3 μg of the different pBS vectors. For stable transfections and selection of Cre-ERT2 clones, we used a ratio of 1/10 for Cre-ERT2 expressing vector on hygromycine resistance vector. ES cells are transfected by electroporation with 20 μg of vector for two 10 cm plates. Selections with hygromycine (200 μg/ml) and/or G418 (300 μg/ml) are performed 36 h following transfections. Resistant colonies were picked to a 96-well plate after 8 days of selection and several clones were analyzed (PCR on genomic DNA, RT–PCR on total RNA). Treatments with 4HT (Sigma, St Louis, MO) (1 μM) were performed one day after transfection. Growth curves are obtained by counting replicated six-wells of ES cells. Treatment by 4HT or vehicle (ethanol) was started one day after plating. Cells were counted every 24 h for monitoring proliferation. For colony formation assay, ES cells were plated into a six-well plate at a density of 300 cells/well and treatment by 4HT or vehicle (ethanol) was started 24 h later. Counting was performed on six different wells for each condition 6 days after plating. Staining was performed using Accustain (Giemsa Stain, modified from Sigma) following instructions provided by the manufacturer.
Fluorescence experiments
Fluorescent levels of eGFP-transfected cells have been monitored every day and images have been analyzed and captured using a green fluorescent microscope (Olympus IX81, IX2-UCB) and the IPLab software 3.6.3 on a Power Macintosh G4.
RNA preparation and reverse transcription (RT) experiments
Total RNAs are prepared using the Rneasy Mini kit from Qiagen, 0.5–1 μg of RNA are used to perform RT using the kit provided by Roche Applied Science (Indianapolis, IN) following the manufacturer instructions.
PCR experiments
PCR experiments are performed on a Perkin-Elmer 9700 machine using 2.5 U of Taq polymerase from GeneChoice, 2.5 mM MgCl2, 0.2 mM dNTP mix, 100 ng of each oligonucleotide. During these experiments, 1 μL of cDNA or 100 ng of genomic DNA are used.
Primers for Fgfr2 (RT) (Tm = 60°C): 5′-AAGGTTTACAGCGATGCCCA-3′; 5′-ACCACCATGCAGGCGATTAA-3′.
Primers for Neo (Tm = 68°C): 5′-CGCACAGACTTGTGGGAGAA-3′; 5′-GCTGCTAAAGCGCATGCTCC-3′.
Primers for Recomb (Tm = 60°C): 5′-CGCACAGACTTGTGGGAGAA-3′; 5′-CACAATTACTTTACAGTTAG-3′.
RESULTS
To develop an inducible approach for RNAi, we modified the polymerase III U6 promoter (18) to make a ‘spatio–temporal’ switch that is suitable for the Cre–LoxP system (19–21). The U6 promoter contains three major regulatory elements, which include a TATA box between −31 and −26, a proximal sequence element (PSE) between −68 and −49 and a distal sequence element (DSE) between −247 and −225 (Figure 1A). It has been shown that all three elements are required for the cooperative binding of proteins, and the DSE is critical for the stability of the DNA–protein complex and the activation of both proximal elements (18). We assumed that the distances between these three elements are critical while the actual sequences of nucleotides are not. We therefore deleted 45 bp of sequences between the DSE and PSE, and replacedthem with a neomycin cassette flanked by two LoxP sites (ploxPneo) (16) (Figure 1B). The insertion of the ploxPneo (total length: 1938 bp) dramatically increases distance between DSE and PSE and is predicted to severely disrupt the U6 promoter activity. After deleting the ploxPneo with Cre, the critical distance is restored due to the retention of a loxP and the flanking restriction sites (Figure 1C). We predicted that the promoter activity should be restored. If these predictions are correct, the combination of the Cre–loxP system and the RNAi technology could be used to regulate gene expression both in vitro and in vivo.
Figure 1.
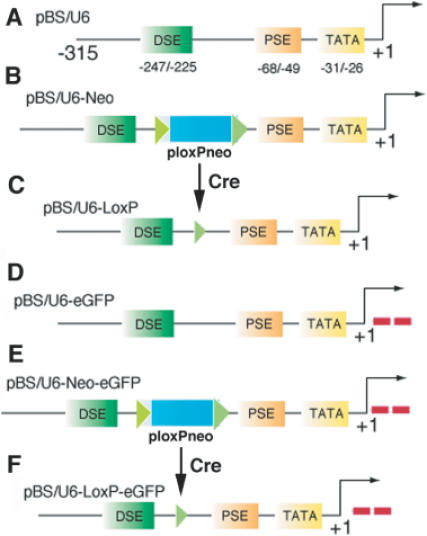
Vectors using for interference study. (A) Structure of the mouse U6 (mU6) promoter and of its three major regulatory elements (TATA box, TATA; Proximal Site Enhancer, PSE; Distal Site Enhancer, DSE). +1 corresponds to the transcription start site (polylinker in the empty vector). (B) The pBS/U6-Neo vector carried a ploxPneo cassette inserted between the PSE and the DSE inside the mU6 promoter. Expression of the neomycine cassette in the cells enables selection of positive stable clones with G418. (C) The pBS/U6–LoxP is the result of the recombination event by one Cre recombinase enzyme on the pBS/U6-Neo. (D–F) Same constructs as shown in (A–C), respectively, but with two double oligonucleotides specifically against eGFP inserted for shRNA.
To provide a quick assay on the efficiency of our plasmids, we first targeted the eGFP mRNA in transient transfections. Oligonucleotides against eGFP were introduced in the above three constructs to generate the pBS/U6-eGFP (Figure 1D), pBS/U6-Neo-eGFP (Figure 1E) and pBS/U6-LoxP-eGFP (Figure 1F) constructs. BOSC23 cells were used in the assay because of their high transfection efficiency. We showed that co-transfection of an eGFP-expressing vector (pCMS-eGFP) with pBS/U6 (shown in Figure 1A) produced high levels of eGFP expression (Figure 2A). The expression of eGFP was repressed by co-transfection of the pBS/U6-eGFP (shown in Figure 1D) (Figure 2B), indicating that the U6 promoter was active in BOSC23 cells. Next, we co-transfected pCMS-eGFP with pBS/U6-Neo-eGFP (shown in Figure 1E) and found that this transfection yielded eGFP levels comparable to those produced by the co-transfection of pCMS-eGFP and pBS/U6 vector (Figure 2C). This observation indicated that the presence of the ploxPneo between DES and PES disrupted the activity of the U6 promoter. Then, we co-transfected pCMS-eGFP with pBS/U6-LoxP-eGFP vector (shown in Figure 1F) and found that this vector could sufficiently suppress eGFP expression (Figure 2D). This observation indicated that the presence of a loxP site did not have an obvious effect on U6 promoter activity.
Figure 2.
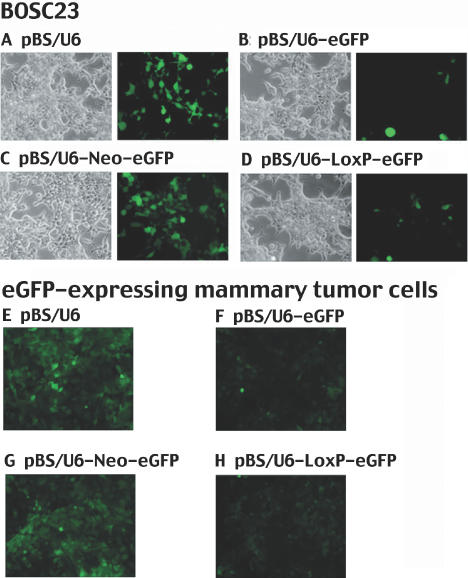
Suppression of eGFP in BOSC23 and W069 cells. (A–D) Transient transfections of BOSC23 cells with pCMS-GFP and pBS/U6 (A), pBS/U6-eGFP (B), pBS/U6-Neo-eGFP (C) and pBS/U6-LoxP-eGFP (D). Left, bright field control; right, green fluorescence. (E–H) W069 cells that are stably express eGFP are transiently transfected with constructs indicated. Pictures are taken three days after transfections using lipofectamine 2000.
Next, we checked the efficiency of our system on the GFP reporter after it was stably integrated into the genomic DNA. We transfected a cell line, WO69, which was derived from a mouse mammary tumor (17), with pCMS-eGFP, and enriched GFP expressing cells by sorting them twice at 7 and 30 days after transfection. We then transfected the eGFP-expressing cells with the pBS/U6 (Figure 2E), pBS/U6-eGFP (Figure 2F), pBS/U6-Neo-eGFP (Figure 2G) or pBS/U6-LoxP-eGFP (Figure 2H) vectors. We obtained similar results as shown in transient system, i.e. the presence of the neo blocked the U6 promoter activity and the removal of the neo restored the promoter function. It is important to notice that we did not modify the distance between each element of the promoter. Even in an inserted LoxP site, we deleted on purpose the corresponding number of base pairs that we add following the insertion. As a result, we did not observe any differences between the effects of the unmodified U6 and inserted LoxP-U6 promoters. These data indicated that it is possible to use the combination of RNAi and Cre–LoxP system to knock-down the expression of genes that are stably integrated in the genome in an inducible manner.
Encouraged by these results, we tested the efficiency of our system on an endogenous gene, Fgfr2, in mouse ES cells. We chose to study this gene for several reasons. First, FGFR2 is expressed in ES cells and we surmised that down-regulation of this protein could have an effect on the growth of those cells as demonstrated in other cell types (22–25). Second, we have previously created a mouse strain carrying Fgfr2 mutation through gene targeting (23,26). The comparison between inducible Fgfr2 ‘knock-down’ and Fgfr2 ‘knock-out’ mice would allow us to assess efficiency of the RNAi system. Furthermore, mutant mice bearing inducible suppression of Fgfr2 would be very useful after crossing them with different Cre expressing mice to assess functions of this gene in multiple biological processes during mammalian development.
To this end, we constructed two plasmids carrying oligonucleotides specific to different regions of Fgfr2 (pBS/U6–Neo-Fgfr2). After testing the corresponding pBS/U6–LoxP-Fgfr2 constructs (obtained through recombination in Cre-positive bacteria) in transient transfections in mouse embryonic fibroblasts, we found that one of the constructs appeared to work better than the others (data not shown). We then transfected it into an ES clone (referred to D4) that we generated by stably expressing the recombinase Cre-ERT2 (27,28). Seven days after transfection of the pBS/U6-Neo-Fgfr2 into D4 ES cells, we picked 24 G418-resistant colonies and amplified two of them (clones 1 and 75) for further analysis. As both ES clones yielded similar results, only the data obtained from clone 1 are presented below.
First, we performed RT-PCR of Fgfr2 following treatment with vehicle or 4OH-tamoxifen (4HT) (1 μM) at varying time-points from 24 to 96 h. A clear reduction of Fgfr2 is evident 24 h after 4HT treatment, with a dramatic effect after 96 h compared to vehicle treatment (Figure 3A). To determine if the decreased expression of Fgfr2 was a direct consequence of Cre-mediated deletion of the pLoxPneo, we extracted the genomic DNA from the ES cells and amplified by PCR a specific region of pBS/U6-Neo-eGFP construct overlapping both distal fragment of the U6 promoter and the 5′ region of the neomycine cassette. As shown in figure 3B, 24 h after the 4HT treatment, Cre action, as evidenced by the decreased intensities of PCR products, was already perceptible and continued throughout the treatment until 72 h, while the intensities of PCR products from vehicle treated cells remained unchanged. This effect is observed even if the efficiency of the recombination event does not reach 100%. Indeed, this ‘mosaic’ effect is commonly observed in mouse studies using the Cre–LoxP system. However, this did not interfere with our following study.
Figure 3.
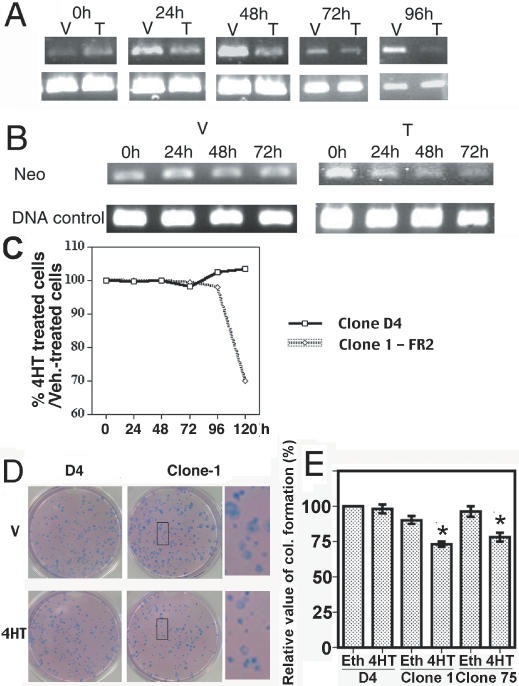
Suppression of Fgfr2 in ES cells. (A) Kinetics of Fgfr2 levels revealed by RT–PCR after vehicle (V) or 4HT (T) treatment in Cre-ERT2 expressing ES cells (Clone-1) that bear the pBS/U6-Neo-Fgfr2 construct. Gapdh is used as control. (B) Kinetics of removal of the neomycine cassette (Neo) evaluated by PCR on genomic DNA isolated from ES cells after treatment by vehicle or 4HT (DNA control = Gapdh). (C) Growth curves of Cre-ERT2 expressing ES clones that bear (Clone-1) or do not bear (parental D4) the pBS/U6-Neo-FGFR2 construct expressed by percentage ratio of the number cells after 4OH-Tamoxifen treatment (1 μM) on the number of cells treated with ethanol (vehicle). (D and E) Colony formation of Clone D4 (parental), Clone-1 and Clone-75 ES (U6-Neo-FR2) cells. (D) The cells are seeded one day before beginning of treatment by vehicle (ethanol) or 4HT. Pictures are taken 6 days later after staining by Accustain. (E) Relative values of colony formation in D4 and Clone-1 ES cells. Number of colonies obtained from vehicle-treated are set as 100%, and those of 4HT-treated are calculated as following: nb. of colonies from 4HT treatment/nb. of colonies from vehicle treatment ×100/100. Six independent counts have been performed. Astral indicates significant difference as assayed by Student's t-test (p < 0.01).
Previous studies conducted in various types of cells indicated that FGF/FGFR2 signals play an essential role in cell proliferation (22–25). As our approach decreased Fgfr2 transcripts by over 90% (Figure 3A), we were interested to know whether the acute suppression of Fgfr2 could affect ES cell proliferation. We performed a growth curve of clone 1 ES cells in the presence or absence of 4HT. As a control, we used the parental D4 clone expressing only the Cre-ERT2 to ensure that an eventual effect could not be attributed to action of the recombinase. Our data revealed no obvious differences between 4HT treated, untreated and vehicle treated cells in both clone 1 and D4 cells between 24 and 96 h (Figure 3C). However, a significant effect of 4HT on the proliferation of clone 1 cells was observed at 120 h (30% of decrease) (Figure 3C). Those effects are also observed with the clone 75 (data not shown). The delayed response of cell proliferation compared with decreased Fgfr2 transcripts may suggest that the depletion of Fgfr2 protein to the critical levels that impairs cell proliferation requires time. Alternatively, the delay may happen somewhere in the Fgf/Fgfr2 signaling transduction pathway instead of at the receptor level.
In addition, we have also assessed the impact of Fgfr2 suppression upon the treatment of 4HT on colony formation in clone 1 cells in comparison with the parental D4 cells. Figure 3D and E showed a significant decrease of the number and the size of colonies in 4HT-treated clones 1 and 75, but not in vehicle-treated clone 1 cells. These effects were consistent with the experiments on cell growth. In contrast, such differences were not observed in D4 cells (Figure 3D and E).
To confirm the viability of our system, we decided to perform the same experiments with another gene: Survivin, which is a member of the inhibitor of apoptosis gene family. As indicated by its name, Survivin inhibits apoptosis in vivo and in vitro but is also implicated in cell division regulation (29,30). We then designed an RNAi construct for Survivin using the same approach for Fgfr2 and transfected it into the parental ES clone D4. After neomycin selection of more than 75 clones, clones 1 and 35 have been chosen to perform the experiments according to their capacity to delete the neo cassette following treatment by 1 μM of 4HT (Figure 4A). Our time course RT–PCR revealed a sharp decrease of Survivin mRNA levels after 24 h, a continuous ‘knock-down’ effect even 120 h in the presence of 4HT (Figure 4B). This observation correlates with decreased growth (almost 40% after 96 h) after several days of 4HT treatment compared to vehicle (Figure 4C). Finally, we also performed a colony formation experiment with both clones 1 and 35 and yielded reduced sizes and numbers of colonies (Figure 4D and E). The observed decreased growth is particularly correlated with the dramatic effect on colony sizes (Figure 4D). These results confirmed that combination of the RNAi and the Cre-LoxP system works efficiently in ES cells bearing the shRNA conditional constructs.
Figure 4.
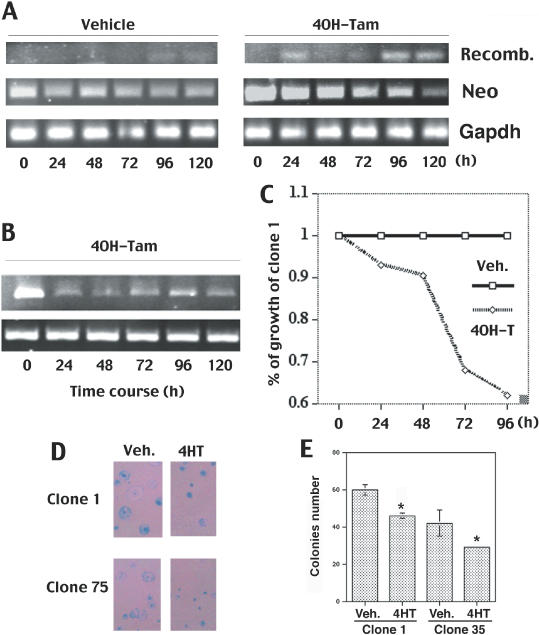
Suppression of Survivin in ES cells. (A) Kinetics of removal of the neomycine cassette and recombination event evaluated by PCR on genomic DNA isolated from ES cells after treatment by vehicle or 4HT. (B) Kinetics of Survivin levels revealed by RT–PCR after 4HT (T) treatment in Cre-ERT2 expressing ES cells (Clone-1) that bear the pBS/U6-Neo-Survivin construct. Gapdh is used as control. (C) Growth curves of Cre-ERT2-expressing ES clone 1 that bear the pBS/U6-Neo-Survivin construct after 4OH-Tamoxifen or ethanol (vehicle) treatment expressed by ratio versus ethanol-treated cells. (D and E) Colony formation of D4, Clone-1 and Clone-35 ES cells. (D) The cells are seeded one day before beginning of treatment by vehicle (ethanol) or 4HT. Pictures are taken 6 days later after staining by Accustain. (E) Real values of colony formation in Clone-1 and Clone-35 ES cells. Astral indicates significant difference as assayed by Student's t-test (p < 0.01).
DISCUSSION
In summary, we have demonstrated the feasibility of a novel technique that allows the inducible production of shRNA using the U6 promoter and the Cre–LoxP system upon 4HT treatment. At the cellular level, this system is applicable for establishing stable cell lines to decipher functions of genes, which are essential for cell growth and viability. Indeed, even with an uncompleted efficiency of recombination, we have observed significant effects after down-regulation of Fgfr2 and Survivin. Similarly, this system should be very useful in vivo for functional analysis of genes in which the targeted disruption is not compatible for embryonic or early postnatal development. The techniques from ES cells to mouse germline transmission have been well established (14,31); we believe that the generation of mice via blastocyst injection of modified ES cells, although time consuming, should not be a major concern. To this end, successful germline transmission of ES cells carrying non-inducible RNAi constructs have been achieved recently (32). It has also been demonstrated that embryos generated by tetraploid aggregation from ES cells carrying an RNAi construct against Rasa1 recapitulated the null phenotype (33). Our method aims at generation of mice carrying inducible RNAi constructs, which would allow the utilization of large collection of Cre-transgenic mice to study the consequence of decreased levels of gene expression in vivo in animal models in a spatio–temporal dependant manner.
Recent investigations pointed out the fact that RNA interference could target other non-specific genes (34,35) while others indicated that the repression by RNAi is highly specific (36,37). Because the non-specific effect appears to be primarily sequence-dependent, careful screening of RNAi sequences is recommended to avoid potential problems. Furthermore, majority of the studies used siRNA transiently transfected at high levels, the observed non-specific response could be due to the transient transfection of large amounts of siRNA. In this case, we predict that a stable integrated vector strategy combined with an inducible system could attenuate this problem. Nonetheless, like all other newly developed technologies, RNAi system is not perfect at its current stand and needs to be consistently improved in such a way that high specific gene suppression at any given stages in vivo can be achieved.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Y. Shi for providing the vector pBS/U6, P. P. Chambon for ERT2 plasmids, and S. Lee and members of Deng laboratory for critically reading and commenting on the manuscript.
REFERENCES
- 1.Cerutti H. (2003) RNA interference: traveling in the cell and gaining functions? Trends Genet., 19, 39–46. [DOI] [PubMed] [Google Scholar]
- 2.Dykxhoorn D.M., Novina,C.D. and Sharp,P.A. (2003) Killing the messenger: short RNAs that silence gene expression. Nature Rev. Mol. Cell Biol., 4, 457–467. [DOI] [PubMed] [Google Scholar]
- 3.Famulok M. and Verma,S. (2002) In vivo-applied functional RNAs as tools in proteomics and genomics research. Trends Biotechnol., 20, 462–466. [DOI] [PubMed] [Google Scholar]
- 4.Paddison P.J., Caudy,A.A. and Hannon,G.J. (2002) Stable suppression of gene expression by RNAi in mammalian cells. Proc. Natl Acad. Sci. USA, 99, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- 6.Paul C.P., Good,P.D., Winer,I. and Engelke,D.R. (2002) Effective expression of small interfering RNA in human cells. Nature Biotechnol., 20, 505–508. [DOI] [PubMed] [Google Scholar]
- 7.Miyagishi M. and Taira,K. (2002) U6 promoter-driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nature Biotechnol., 20, 497–500. [DOI] [PubMed] [Google Scholar]
- 8.Sui G., Soohoo,C., Affar el,B., Gay,F., Forrester,W.C. and Shi,Y. (2002) A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc. Natl Acad. Sci. USA, 99, 5515–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu J.Y., DeRuiter,S.L. and Turner,D.L. (2002) RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc. Natl Acad. Sci. USA, 99, 6047–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta S., Schoer,R.A., Egan,J.E., Hannon,G.J. and Mittal,V. (2004) Inducible, reversible, and stable RNA interference in mammalian cells. Proc. Natl Acad. Sci. USA, 101, 1927–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J., Tekle,E., Oubrahim,H., Mieyal,J.J., Stadtman,E.R. and Chock,P.B. (2003) Stable and controllable RNA interference: investigating the physiological function of glutathionylated actin. Proc. Natl Acad. Sci. USA, 100, 5103–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsukura S., Jones,P.A. and Takai,D. (2003) Establishment of conditional vectors for hairpin siRNA knockdowns. Nucleic Acids Res., 31, e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Y. and Sauer,B. (2000) Conditional gene knockout using cre recombinase. Methods Mol. Biol., 136, 477–485. [DOI] [PubMed] [Google Scholar]
- 14.Capecchi M.R. (1989) Altering the genome by homologous recombination. Science, 244, 1288–1292. [DOI] [PubMed] [Google Scholar]
- 15.Nagy A. and Mar,L. (2001) Creation and use of a Cre recombinase transgenic database. Methods Mol. Biol., 158, 95–106. [DOI] [PubMed] [Google Scholar]
- 16.Yang X., Li,C., Xu,X. and Deng,C. (1998) The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc. Natl Acad. Sci. USA, 95, 3667–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brodie S.G., Xu,X., Qiao,W., Li,W.M., Cao,L. and Deng,C.X. (2001) Multiple genetic changes are associated with mammary tumorigenesis in Brca1 conditional knockout mice. Oncogene, 20, 7514–7523. [DOI] [PubMed] [Google Scholar]
- 18.Paule M.R. and White,R.J. (2000) Survey and summary: transcription by RNA polymerases I and III. Nucleic Acids Res., 28, 1283–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagy A. (2000) Cre recombinase: the universal reagent for genome tailoring. Genesis, 26, 99–109. [PubMed] [Google Scholar]
- 20.Kwan K.M. (2002) Conditional alleles in mice: practical considerations for tissue-specific knockouts. Genesis, 32, 49–62. [DOI] [PubMed] [Google Scholar]
- 21.Gossen M. and Bujard,H. (2002) Studying gene function in eukaryotes by conditional gene inactivation. Annu. Rev. Genet., 36, 153–173. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y., Li,X., Eswarakumar,V.P., Seger,R. and Lonai,P. (2000) Fibroblast growth factor (FGF) signaling through PI 3-kinase and Akt/PKB is required for embryoid body differentiation. Oncogene, 19, 3750–3756. [DOI] [PubMed] [Google Scholar]
- 23.Li C., Guo,H., Xu,X., Weinberg,W. and Deng,C.X. (2001) Fibroblast growth factor receptor 2 (Fgfr2) plays an important role in eyelid and skin formation and patterning. Dev. Dyn., 222, 471–483. [DOI] [PubMed] [Google Scholar]
- 24.Wilkie A.O., Patey,S.J., Kan,S.H., van den Ouweland,A.M. and Hamel,B.C. (2002) FGFs, their receptors, and human limb malformations: clinical and molecular correlations. Am. J. Med. Genet., 112, 266–278. [DOI] [PubMed] [Google Scholar]
- 25.Yu K., Xu,J., Liu,Z., Sosic,D., Shao,J., Olson,E.N., Towler,D.A. and Ornitz,D.M. (2003) Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development, 130, 3063–3074. [DOI] [PubMed] [Google Scholar]
- 26.Xu X., Weinstein,M., Li,C., Naski,M., Cohen,R.I., Ornitz,D.M., Leder,P. and Deng,C. (1998) Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development, 125, 753–765. [DOI] [PubMed] [Google Scholar]
- 27.Feil R., Brocard,J., Mascrez,B., LeMeur,M., Metzger,D. and Chambon,P. (1996) Ligand-activated site-specific recombination in mice. Proc. Natl Acad. Sci., USA, 93, 10887–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feil R., Wagner,J., Metzger,D. and Chambon,P. (1997) Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem. Biophys. Res. Commun., 237, 752–757. [DOI] [PubMed] [Google Scholar]
- 29.Li F. (2003) Survivin study: what is the next wave? J. Cell Physiol., 197, 8–29. [DOI] [PubMed] [Google Scholar]
- 30.Altieri D.C. (2003) Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene, 22, 8581–8589. [DOI] [PubMed] [Google Scholar]
- 31.Capecchi M.R. (1994) Targeted gene replacement. Sci. Am., 270, 52–59. [DOI] [PubMed] [Google Scholar]
- 32.Carmell M.A., Zhang,L., Conklin,D.S., Hannon,G.J. and Rosenquist,T.A. (2003) Germline transmission of RNAi in mice. Nature Struct. Biol., 10, 91–92. [DOI] [PubMed] [Google Scholar]
- 33.Kunath T., Gish,G., Lickert,H., Jones,N., Pawson,T. and Rossant,J. (2003) Transgenic RNA interference in ES cell-derived embryos recapitulates a genetic null phenotype. Nature Biotechnol., 21, 559–561. [DOI] [PubMed] [Google Scholar]
- 34.Sledz C.A., Holko,M., de Veer,M.J., Silverman,R.H. and Williams,B.R. (2003) Activation of the interferon system by short-interfering RNAs. Nature Cell Biol., 5, 834–839. [DOI] [PubMed] [Google Scholar]
- 35.Scacheri P.C., Rozenblatt-Rosen,O., Caplen,N.J., Wolfsberg,T.G., Umayam,L., Lee,J.C., Hughes,C.M., Shanmugam,K.S., Bhattacharjee,A., Meyerson,M.et al. (2004) Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc. Natl Acad. Sci. USA, 101, 1892–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Semizarov D., Frost,L., Sarthy,A., Kroeger,P., Halbert,D.N. and Fesik,S.W. (2003) Specificity of short interfering RNA determined through gene expression signatures. Proc. Natl Acad. Sci. USA, 100, 6347–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chi J.T., Chang,H.Y., Wang,N.N., Chang,D.S., Dunphy,N. and Brown,P.O. (2003) Genomewide view of gene silencing by small interfering RNAs. Proc. Natl Acad. Sci. USA, 100, 6343–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]