

Abstract
Several members of protocadherins have been found involved in human carcinogenesis, but little is known about PCDH20 in HCC. Here in this study, using quantitative real-time RT-PCR assay, we demonstrated the downregulation of PCDH20 expression in 6 of 7 HCC cell lines tested. Similarly, PCDH20 expression in primary HCC tissues was also significantly downregulated in comparison with that in either disease-free normal liver tissues or the adjacent nontumour liver tissues (P < 0.001, respectively). Among HCC tumour tissues studied, about 48% (51/107) of them showed reduced PCDH20 mRNA level. Further statistic analysis revealed that the reduced PCDH20 mRNA level in tumour tissues was much more common in younger patients group (aged <50 years) than that in older group (≥50 years) (60% vs 33%, P = 0.0303). Loss of heterozygosity (LOH) and promoter hypermethylation analysis revealed that deletion and/or aberrant epigenetic modulation of PCDH20 gene account for its downregulation, at least in a fraction of tumour specimens. Moreover, ectopic expression of PCDH20 in HCC cells significantly suppressed cell proliferation, clonogenicity, migration and tumour formation. Notably, we proved for the first time that, via activating GSK-3β, PCDH20 could inhibit Wnt/β-catenin signalling pathway. Furthermore, our data suggest that PCDH20 may conduct its Wnt/β-catenin signalling antagonizing function through suppressing Akt and Erk activities and promoting GSK-3β signalling activities. However, the detailed mechanism remained undiscovered. In conclusion, our data here strongly suggested that PCDH20 may act as a candidate tumour suppressor in HCC.
Keywords: hepatocellular carcinoma, methylation, PCDH20, tumour-suppressor gene, Wnt/β-catenin signalling pathway
Introduction
Hepatocellular carcinoma (HCC) is a primary malignancy of the liver, and its incidence is rising in China and around the world [1]. Although the main etiologies of HCC are now well defined, the molecular mechanisms involved in tumour initiation and progression have yet to be fully characterized [2]. It is known that chronic HBV and HCV infection causes persistent liver inflammation, regeneration and cirrhosis, increasing cellular turnover, and as a consequence, inducing malignant transformation [3]. During the initiation and progression of hepatocarcinogenesis, multiple genetic and epigenetic events accumulated, leading to deregulated expression of various genes [4–6].
The cadherin superfamily is known as a large family of calcium-dependent adhesion molecules which plays an important role in the adhesion and communication between adjacent cells [7]. Cadherins have been identified by the presence of extracellular cadherin repeats of about 110 amino acid residues and can be classified into at least three subfamilies based on shared properties and sequence similarity: the classical cadherins, desmosomal cadherins and protocadherins (PCDHs) [8]. PCDHs constitute the largest subgroup (about 80 members) of the cadherin superfamily [7,9,10]. They are characterized by seven extracellular domains, a single transmembrane region and distinct cytoplasmic portions [9,11]. In addition to the cell-to-cell adhesion activity, PCDHs are thought to have other important activities as well, although the major functions of each have not been well elucidated [8,12]. Several members of the PCDH family such as PCDH8, PCDH10, PCDH24 and PCDH17 have been suggested to be involved in the carcinogenesis of breast cancer, nasopharyngeal carcinoma, colon cancer and oesophageal squamous cell carcinoma, through their overexpression or inactivation. However, the association between PCDH proteins and the pathogenesis of many other cancers remains unclear [13–16].
PCDH20 is a novel protocadherin which is expressed predominantly in nervous system [17,18]. Like other members of PCDHs, PCDH20 has also attracted attention for their implications in the neurological disorders and tumourigenesis, but little is known about their functions and intracellular signal transduction. It has been reported that PCDH20 was frequently silenced by promoter methylation in non-small cell lung cancers [19]. Restoration of PCDH20 expression in NSCLC cells reduced cell numbers in colony formation and anchorage-independent assays [19]. These results suggest that epigenetic silencing of PCDH20 may be a factor in the carcinogenesis of NSCLC. We have previously performed a 2-Mb array-based CGH analysis for copy number aberrations in 25 paired HCC tissues and their corresponding nontumour tissues [20]. Based on these aCGH data, we identified the LOH status of PCDH20 (13q21.2) in three HCC tissues. The deletion of PCDH20 suggests the possibility of PCDH20 acting as a candidate tumour-suppressor gene in HCC development.
This study was designed to examine the biological function and molecular mechanism of PCDH20 in HCC using complementary in vitro and in vivo approaches. We show herein that expression of PCDH20 mRNA is downregulated in HCC cell lines and HCC tumour tissues. Ectopic expression of PCDH20 in HCC cell lines led to inactivation of Wnt signalling pathway and significant repression of cell growth, migration and colony formation. Our data disclose an important PCDH20-mediated inactivation of Wnt signalling that suppresses hepatocarcinogenesis and tumour progression.
Materials and Methods
Clinical samples
Hepatocellular carcinoma tumour tissues and paired nontumour tissues were collected from 107 patients with HCC at Henan Province Oncology Hospital (Zhengzhou, China) from 2008 to 2012. The patient's age ranged from 34 to 76 years (51.25 ± 9.91 years old). After resection, tumour tissues and the surrounding nontumour tissues were routinely fixed for clinical histological evaluation and the remained tissues were snap-frozen in liquid nitrogen and stored for future use. All histological slides were diagnosed by two pathologists independently.
Our investigation was conducted according to the principles expressed in the Declaration of Helsinki. This study protocol was approved by the Ethics Committee of Peking University Health Science Center. Written informed consents were obtained from each participant involved in our study.
Cell lines and cell culture
HEK 293T cell line and human liver cancer cell lines (Huh-7, SNU-449, SNU-182, SNU-387, PLC/PRF/5 and Hep3B) were obtained from ATCC and stored in our laboratory. HEK 293T, Hep3B and Huh-7 cells were maintained in Dulbecco's modified Eagle Medium supplemented with 10% foetal bovine serum (Gibco, Carlsbad, CA, USA). SNU-449, SNU-182, SNU-387, SMMC-7721 and PLC/PRF/5 cells were maintained in RPMI-1640 medium supplemented with 10% foetal bovine serum (Gibco). All cell lines were maintained in a humidified incubator containing 5% CO2 at 37 °C.
Loss of Heterozygosity (LOH) analysis
The SNP assay was used to detect the potential LOH in HCC tissues as described previously [20]. In brief, 10 SNP sites (rs3829388, rs3812872, rs35945287, rs3812873, rs9539157, rs9570571, rs79312776, rs3812874, rs3812875 and rs3812876) within exon 1 and exon 2 of PCDH20 were selected to analyse the allelic imbalance. The two exons were amplified by four independent PCRs, and the PCR products were directly sequenced to determine the allelotype at each locus. The primers used for PCR were shown in Table S1. Through comparing the electrophograms of the heterozygote SNP sites between tumour and adjacent nontumour tissues, LOH was defined by quantitatively calculating the LOH index.
Quantitative real-time reverse transcription-PCR
Quantitative real-time reverse transcription-PCR (qRT-PCR) was performed as described previously [21]. In brief, 3 μg of total RNA was reversely transcribed using Reverse Transcription kit (Fermentas, Vilnius, Lithuania) and assayed for gene expression by SYBR Green technology using a Light Cycler 480II Real-Time PCR System (Roche, Indianapolis, IN, USA). The primers used for qRT-PCR were shown in Table S1.
Methylation assay
The quantificational methylated DNA analysis was performed by combined DNA methylation-sensitive and methylation-dependent restriction endonuclease digestion, followed by subsequent quantitative PCR assay as described previously [21]. The primers used for quantitative PCR were shown in Table S1. For determination of a specimen's CGI hypermethylation status, the cut-off value was set at 10%, similar to our previous reports [21].
Plasmid construction
Plasmids expressing C-terminally His-tagged PCDH20 (pIRES2-EGFP-PCDH20) were constructed by cloning the full-length coding sequence of PCDH20 into the eukaryotic expression vector pIRES2-EGFP (Clontech, Palo Alto, CA, USA). The sequence of the plasmid recombinant was verified by DNA sequencing and Western blot assay.
Western blot assay
Proteins were extracted from cells and tissues using lysis buffer (Applygen, Beijing, China). The protein lysates were loaded into 10% or 12% sodium dodecyl sulphate-polyacrylamide gels, and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking with 5% fat-free dried milk in phosphate-buffered saline (PBS), membranes were incubated with primary antibodies overnight at 4 °C and then with secondary antibodies for 2 h in room temperature. Band signals were visualized by Odyssey Imager (LI-COR Biosciences, Lincoln, NE, USA). The antibodies used in Western blot analysis are listed in Table S2.
Luciferase reporter assay
Cells were transiently transfected with TOP FLASH plasmids bearing an optimal TCF-binding site and phRL-TK Renilla control plasmid (Promega, Madison, WI, USA). To determine Wnt activity in response to PCDH20, TOP-transfected cells were co-transfected with a constitutively active β-catenin mutant (CA-β-catenin) [22], together with or without PCDH20. Reporter activity was assayed using the Dual Luciferase Assay System (Promega). Results were normalized to total protein amounts and Renilla values of each sample. The reporter assay results represent the average of three independent transfection experiments.
Cell proliferation and cell cycle analysis
Cell proliferation was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. Cells (2000 per well) were allowed to grow in 96-well plates. MTT solution was added into each well at a final concentration of 5 μg/mL and then remained in culture for four hours before measurement. Cell proliferation was measured every 24 h by reading the absorbance at 570 nm using a microplate reader (Perkin Elmer, Framingham, MA, USA).
For cell cycle analysis, 1 × 106 cells were harvested and fixed with 70% ethanol at 4 °C overnight. The fixed cells were rinsed twice with PBS and resuspended in 500 μL PBS containing 50 μg/mL RNaseA (Sigma, St. Louis, MO, USA), then incubated at 37 °C for 30 min. The DNA content was stained in PI solution (50 μg/mL) and was then measured by flow cytometry (FACS Caliber; Becton Dickinson, Mountain View, CA, USA), and the percentage of cells in G0/G1, S and G2/M phase were calculated by the ModFit program (Becton Dickinson), respectively.
Soft agar assay
Soft agar assay was performed using a soft agar kit (GenMed Scientifics, Inc, Arlington, MA, USA) in 6-well plates. Cells (4000 per well) were suspended in top agar and incubated for 2–3 weeks in a humidified incubator at 37 °C with 5% CO2 until the visible colony was formed. Colonies were counted under a microscope. The experiment was performed in triplicate.
Tumourigenicity assay in nude mice
2 × 107 Cells were suspended in 100 μL PBS and then injected subcutaneously into either side of the posterior flank of the same male BALB/c nude mouse at age of 5–6 week. Five mice were used. Tumour growth was monitored for 5 weeks. Tumour volume (V) was observed by measuring the length (L) and width (W) with calipers and calculated with the formula (L × W2) × 0.5. All animal experiments were performed according to the ethical guidelines of the Ethics Committees of Peking University Health Science Center for animal experiment.
Scratch-wound-healing assay
Cells were seeded into a 12-well plate to form a confluent monolayer on the following day. The cell layers were carefully wounded using a sterile 20-μL tip to create a straight ‘scratch’. After PBS washing to discard the fallen cells by scratching, the plate was added with medium and incubated until scratch disappeared. Images of the wound monolayer were acquired on a phase contrast microscope linked to a charge-coupled device camera, and the wound area was measured using CT-AS software (Nikon, Tokyo, Japan) to count the number of pixels after the photographs had been converted to TIFF images.
Transwell assay
Cell migration was performed using transwell chambers (24-well) with 8.0-μm-pore membranes purchased from Corning (New York, NY, USA). The bottom chambers were filled with RPMI-1640 containing 10% FBS. The upper chambers were filled with 2 × 104 cells with FBS-free RPMI-1640 and incubated at 37 °C. After incubation for 48 h, nonmigratory cells in the top of the transwell were removed with a cotton swab. The cells migrated to the bottom side of the membrane were fixed by methanol, stained with 5% crystal violet and counted under a light microscope (Olympus, Lake Success, NY, USA). Cells from at least six random fields were counted, and mean ± standard deviation values were calculated for the graphic presentations.
Statistical analysis
Student's t-test and Wilcoxon rank test were used for statistical analysis with SAS software (version 9.1, SAS Institute, Cary, NC, USA). P < 0.05 was considered statistically significant.
Results
Identification of PCDH20 deletion in HCC
Using array-based comparative genomic hybridization (aCGH), we have previously examined the copy number abnormalities in 25 HCC tissues. Loss of 13q21.2 where PCDH20 located was found in three tumour tissues (Table S3). Concordantly, the LOH status of PCDH20 in these three tumour tissues was confirmed by allelotyping using SNP assay. In addition, direct sequencing of PCDH20 exons in these three tumour tissues and seven HCC cell lines identified no mutation. As LOH is the most common genetic alteration for tumour-suppressor gene in human cancers, the loss of PCDH20 in HCC suggests that PCDH20 may be a candidate TSG involved in hepatocarcinogenesis.
Reduced expression of PCDH20 in HCC
To investigate whether PCDH20 was aberrantly expressed in HCC, we first examined PCDH20 mRNA level in an array of HCC cell lines using real-time PCR. Compared to the tumour-free liver tissues, reduced PCDH20 mRNA level was found in six (SNU-182, SNU-387, Huh-7, PLC/PRF/5, SMMC-7721 and SNU-449) of seven HCC cell lines (Fig.1a). More importantly, we found that PCDH20 expression was significantly downregulated in 48% (51/107) of human HCC tumour tissues when compared to the adjacent nontumour tissues (P < 0.001) (Fig.1b). Similar trend was also observed at the protein level by Western blot assay (Fig.1c). The clinicopathologic significance evaluation of PCDH20 downregulation in patients with HCC revealed that downregulation of PCDH20 was much more common among patients younger than 50 years old, in comparison with the elderly patient groups (≥50 years) (60.0% vs 33.3%, P = 0.0303) (Table 1).
Fig 1.
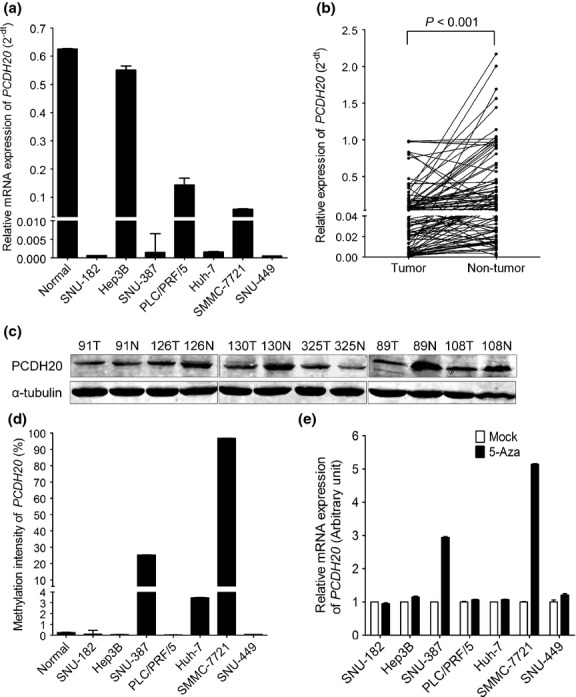
Expression and methylation analysis of PCDH20 in HCC cell lines and tumour tissues. (a) Relative mRNA level of PCDH20 in normal tissues and seven HCC cell lines (mean ± SD; n = 3). (b) Expression of PCDH20 in 107 pairs of HCC and adjacent nontumour tissues as detected by real-time RT-PCR. The line in the grouped column scatter represented mean values of PCDH20 mRNA. (c) Expression level of PCDH20 in six pairs of HCC and adjacent nontumour tissues as detected by Western blot assay: α-tubulin was used as loading control. (d) The promoter methylation status of PCDH20 in HCC cell lines detected by quantificational methylation assay. (e) The expression of PCDH20 in HCC cells after treated by 5 μm of 5-Aza for 4–7 days (mean ± SD; n = 3). The PCDH20 expression level in mock cells was set as 1 (arbitrary unit).
Table 1.
Correlation between PCDH20 mRNA level and clinicopathologic characteristics
| Characteristics | PCDH20 downregulation in tumour tissues | P value | |
|---|---|---|---|
| Yes (n = 51) | No (n = 56) | ||
| Gender | |||
| Male | 37 | 35 | 0.2707 |
| Female | 14 | 21 | |
| Age | |||
| <50 | 27 | 18 | 0.0303 |
| ≥50 | 24 | 38 | |
| Portal vein invasion | |||
| Present | 12 | 12 | 0.6205 |
| Absent | 39 | 43 | |
| N/A | 0 | 1 | |
| Tumour size | |||
| ≥5 cm | 35 | 34 | 0.4897 |
| <5 cm | 15 | 20 | |
| N/A | 1 | 2 | |
| Tumour encapsulation | |||
| Complete | 41 | 35 | 0.0803 |
| Incomplete | 7 | 18 | |
| N/A | 3 | 3 | |
| Intrahepatic dissemination | |||
| Present | 23 | 19 | 0.2605 |
| Absent | 27 | 37 | |
| N/A | 1 | 0 | |
| Abdominal dropsy | |||
| Present | 10 | 6 | 0.1997 |
| Absent | 41 | 50 | |
| Fibrosis stage | |||
| I–III | 35 | 40 | 0.1072 |
| IV–VI | 16 | 16 | |
N/A, not available. The number of patients was not identical in all features because of clinical data absence. Values in bold indicate significant P-value of <0.05.
Promoter methylation of PCDH20 in HCC
Hypermethylation of the CpG-rich region between −290 and +156 around exon 1 of PCDH20 has been reported accounting for the expressional suppression of PCDH20 in NSCLC [19]. Therefore, we performed quantificational DNA methylation analysis at the same region of PCDH20 using DNA specimens from HCC cells and tumour tissues. As shown in Figs1d, 2, HCC cell lines (SMMC-7721 and SNU-387) were tested positive of hypermethylation, when methylation intensity value ≥10% was used as the threshold value to define the presence of hypermethylation, as previously reported [21]. The methylation status that silencing the expression of PCDH20 in SMMC-7721 and SNU-387 was confirmed, as treatment with DNA methyl-transferase inhibitor 5-Aza markedly enhanced PCDH20 expression in these two cell lines. In contrast, treatment with 5-Aza could not increase the PCDH20 expression in SNU-182, Hep3B, PLC/PRF/5, Huh-7 and SNU-449 cell lines which have no detectable hypermethylation in their PCDH20 CpG-rich region (Fig1e). These data suggested that hypermethylation of PCDH20 promoter may be a mechanism leading to the silencing of PCDH20 in some liver cancers. However, when primary HCC tissues were analysed, only 7 in 66 cases (10.6%) showed PCDH20 CpG-rich region hypermethylation.
Fig 2.
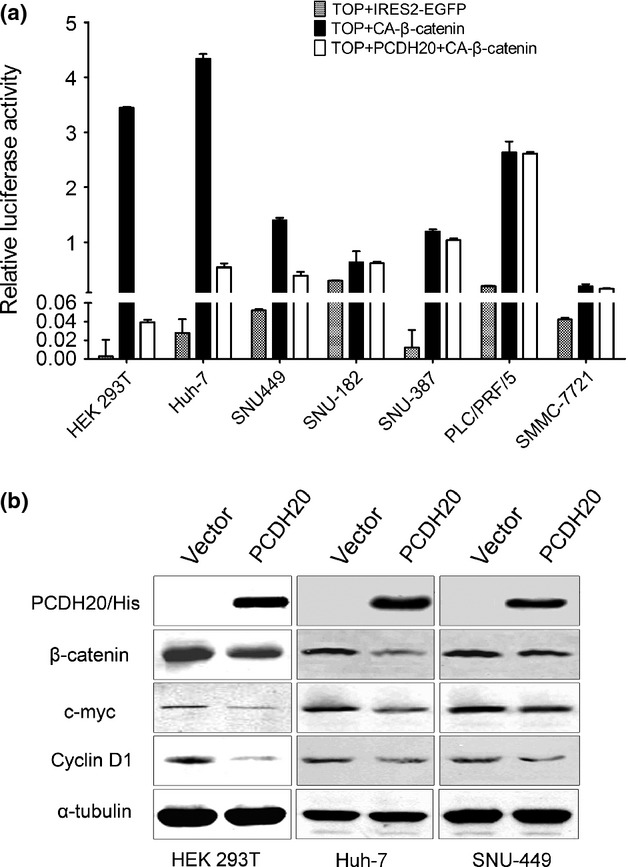
Ectopic expression of PCDH20 inhibits Wnt/β-catenin signalling pathway in HCC cells (a) A TCF luciferase reporter assay was performed using TOP FLASH (the wild-type TCF reporter) in HEK 293T cells and six HCC cell lines (Huh-7, SNU-449, SMMC-7721, SNU-387, PLC/PRF/5 and SNU-182). Experiments were performed in triplicate, and the results are presented as the mean ± SD. (b) Western blot analysis was used to analyse Wnt/β-catenin signalling targeted protein levels in HEK 293T, SNU-449 and Huh-7 cells that were transfected with PCDH20 or control plasmids. α-tubulin was used as loading control.
PCDH20 inhibits Wnt/β-catenin signalling pathway in HCC cells
It has been reported that some members of PCDHs superfamily such as PCDH24, PCDHGA and PCDHGC3 could regulate canonical Wnt signalling [23–25]. To explore the possible involvement of PCDH20 in regulating Wnt/β-catenin signalling pathway, TOP FLASH reporter assay was conducted firstly in HEK 293T and then in PCDH20 downregulated HCC cell lines SNU-182, SNU-387, Huh-7, PLC/PRF/5, SMMC-7721 and SNU-449. As shown in Figure2a, ectopic expression of PCDH20 could inhibit the transcription activity of β-catenin/TCF in HEK 293T, Huh-7 and SNU-449 cells, but not in SNU-182, SNU-387, PLC/PRF/5 and SMMC-7721 cells. We then measured the expression changes of typical Wnt signalling transcriptional target genes in HEK 293T, Huh-7 and SNU-449 cells. Consistent with the results of TOP FLASH reporter assay, Western blotting assay showed that ectopic PCDH20 expression markedly reduced β-catenin protein level, along with the repressed expression of c-myc and cyclin D1, as compared with vector-only transfected control (Fig.2b).
PCDH20 inhibits Wnt signalling pathway through antagonizing ERK and Akt-mediated activation of GSK-3β
It is known that phosphorylation of β-catenin by GSK-3β initiates its degradation by a multiprotein ‘destruction complex’ [22]; therefore, we examined whether PCDH20 reduced β-catenin protein level via activation of GSK-3β. As shown in Figure3a, the level of phospho-GSK-3β (Ser9) (inactivation form of GSK-3β) in PCDH20 overexpressing cells was much less than that detected in controls. Accordantly, pretreatment of cells with GSK-3β inhibitor lithium chloride (LiCl) could reverse the PCDH20-mediated β-catenin downregulation, accompanied with the increase of c-myc and cyclin D1 levels. These results suggest that PCDH20 may inhibit β-catenin through activation of GSK-3β.
Fig 3.
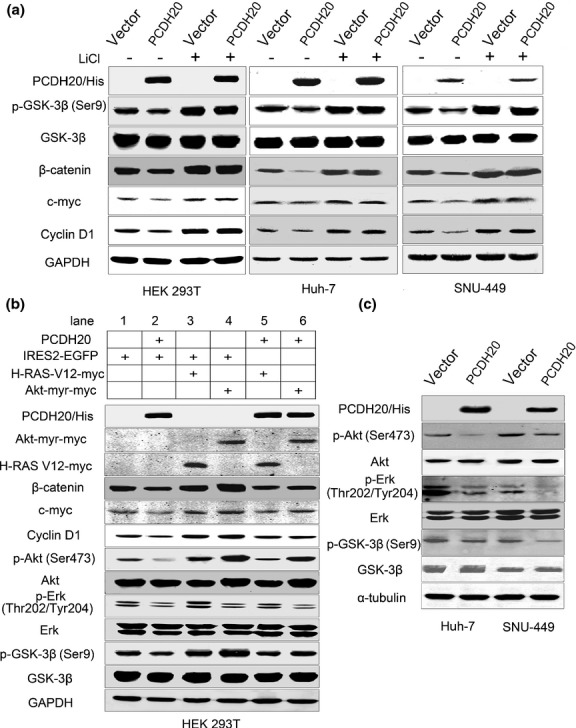
Ectopic expression of PCDH20 inhibits Wnt signalling pathway through antagonizing Erk and Akt-mediated activation of GSK-3β. (a) Western blot was used to analyse GSK-3β and Wnt/β-catenin signalling target protein levels in HEK 293T, Huh-7 and SNU-449 cells with LiCl treatment. (b) Western blot analysis of GSK-3β and Wnt/β-catenin signalling target protein levels in HEK 293T cells co-transfected PCDH20 with or without H-Ras-V12 and Akt-myr plasmids. Western blot analysis pathway (‘+’ indicates treatment for 24 h before harvested). HEK 293T, Huh-7 and SNU-449 cells were transfected with total 4 μg plasmid in a 35-mm dish, and cells were harvested 48 h after transfection. (b) Western blot analysis was used to prove the PCDH20 antagonized Wnt/β-catenin signalling pathway by inhibiting Erk, Akt pathway in HEK 293T cells. GAPDH was used as loading control. (c) Western blot analysis of Erk, Akt and GSK-3β in Huh-7 and SNU-449 cells transfected with PCDH20 plasmids.
We then investigated which signal pathway is involved in PCDH20-mediated activation of GSK-3β. It is well known that Erk and Akt can phosphorylate GSK-3β at the Ser9 residue and inactivate its kinase activation [26,27]. To elucidate whether Erk and Akt are responsible for PCDH20-mediated activation of GSK-3β, we detected the level change of activated Akt and Erk first in HEK 293T cells transfected with PCDH20-expressing plasmid. We found that ectopic expression of PCDH20 could down-regulate p-Akt (Ser473) and p-Erk (Thr202/Tyr204) when compared with vector-transfected control (Fig.3a,b). Moreover, the involvement of Akt and Erk in PCDH20-induced GSK-3β activation and consequently Wnt signalling pathway inactivation was supported by the following experiments: constitutively, active form of H-Ras (H-Ras-V12) could activate both Akt and Erk, while co-transfecting PCDH20 with H-Ras-V12 could partially block Akt and Erk, and subsequent Wnt signalling pathway activation induced by H-Ras-V12 in HEK 293T cells (Fig.3b lane 3 vs lane 5). Similarly, co-transfected Akt-myr (a constitutively active Akt mutant) with PCDH20 could also antagonize the active Akt-induced GSK-3β inactivation and Wnt/β-catenin signalling pathway activation (Fig.3b, lane 4 vs lane 6). Furthermore, the ectopic PCDH20 expression caused decrease of p-Akt (Ser473) and p-Erk (Thr202/Tyr204) in Huh-7 and SNU-449 HCC cell lines confirmed the phenomena seen in HEK 293T cell (Fig.3c). These findings demonstrate that the effect of PCDH20 on GSK-3β and Wnt/β-catenin signalling pathway is associated with Akt and Erk activities.
PCDH20 suppresses HCC cell growth in vitro and in vivo
To further investigate the potential tumour-suppressor role of PCDH20, SNU-449 cell was chosen for the generation of stable transfected cell lines with ectopic PCDH20 expression. After G418 selection, we established SNU-449 stable clones with high PCDH20 expression which was confirmed by qRT-PCR and Western blot assay (Fig.4a). When compared to cells transfected with empty vector, PCDH20-overexpressed SNU-449 cells showed a significant decrease of cell growth measured by MTT (P < 0.05, Fig.4b). Consistently, flow cytometry analysis showed that PCDH20 overexpression increased cells in G0/G1 phase (55.5% vs 70.0%) and decreased the cells in S phase (27.6% vs 15.2%) (Fig.4c). In addition, soft agar clonogenic assay showed that ectopic expression of PCDH20 reduced SUN-449 cells clonogenic growth, with the colony formation rates dropped from mock cells’ 23.4 ± 4.5% to ectopic PCDH20-expressing cells’ 5.6 ± 1.59% (P < 0.05) (Fig.4d). Furthermore, nude mice were used to examine whether PCDH20 affected HCC cell growth in vivo. As shown in Figure4e,f, the tumour size of PCDH20-overexpressed SNU-449 cells in nude mice decreased to approximately one-tenth of the controls (440.4 ± 196.6 mm3 vs 36.6 ± 33.1 mm3, P = 0.036). Taken together, these findings demonstrate that PCDH20 could induce HCC cell cycle arrest at G1/S transit and inhibit HCC cell proliferation, clonogenic growth and tumourigenesis.
Fig 4.
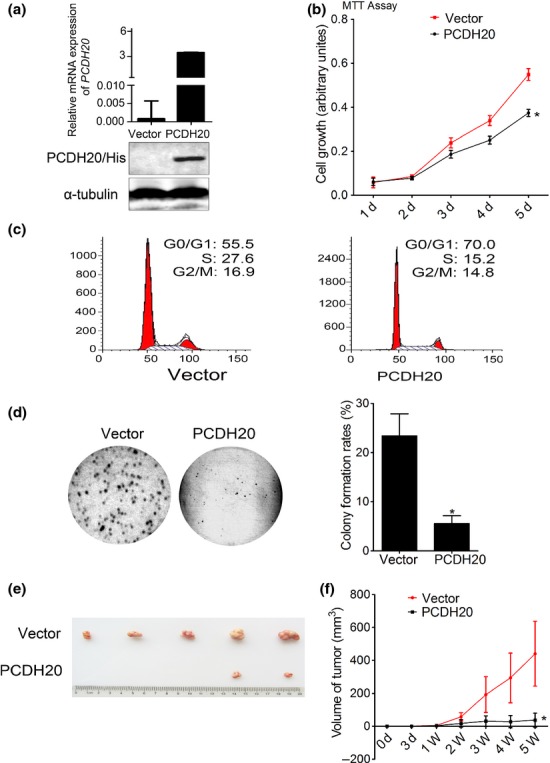
The effect of PCDH20 on HCC cell growth in vitro and in vivo. (a) After selected by G418 for three weeks, the expression level of PCDH20 in stably transfected cell clones was measured by quantitative RT-PCR and Western blot assay, respectively. (b) The growth rate of SNU-449 cells with stable expression of PCDH20 and vector control was measured using MTT assay according to the number of days (d) in culture. Values are presented as the mean ± SD of three independent experiments. *P < 0.05. (c) Cell cycle of SNU-449 cells with ectopic PCDH20 expression was analysed by flow cytometry. (d) Colony formation was measured by soft agar assay. Representative images of colony formed in soft agar and the histograms of relative percentage of colony numbers of the indicated clones were shown. (e, f) Effect of PCDH20 on tumourigenicity in nude mice xenograft model. Tumour volume was measured and plotted as mean ± SD. The curve of tumour growth was shown (n = 5/group).
PCDH20 suppresses HCC cell migration in vitro
Cadherins, including protocadherins coordinate cell polarity, enable cells to stick to each other to make cells clingy [28]. Therefore, it is reasonable to investigate whether PCDH20 exerted influence on HCC cell migration. Results of transwell chamber assay and wound-healing assay showed that ectopic expression of PCDH20 in SUN-449 cells remarkably suppressed its migration ability (Fig.5a,b). These results suggested that PCDH20 may inhibit metastasis in HCC cancer by impeding cell migration.
Fig 5.
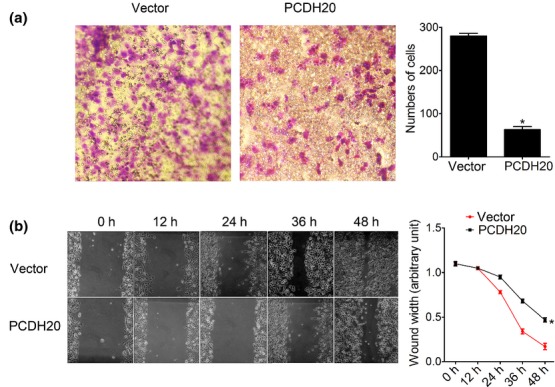
Effects of PCDH20 on HCC cell migration. (a) Transwell migration was carried out in 24-well modified Boyden chambers. Columns, means cells passed the membrane; bars, SD. P < 0.05 vs mock-transfected control at 48 h (Student's t-test). (b) The cell motility was determined by wound-healing assay. The wound width at 0 h, 12 h, 24 h, 36 h and 48 h was measured, and 0 h was arbitrarily assigned as 1.0. *P < 0.05.
Discussion
At present, although the involvements of PCDHs in the pathogenesis of tumour are relatively well established, the endeavours to understand the molecular functions of PCDH in tumour are still in its infancy and more detailed mechanism exploring is required at cellular and molecular levels [17]. This study provides direct evidence for the tumour-suppressing role of PCDH20 in HCC. Our results suggest that PCDH20 induces Erks and Akt-mediated activation of GSK-3β, which in turn inhibits Wnt/β-catenin signalling and leads to inhibition of HCC growth and migration.
PCDH20, also known as PCDH13, is located at 13q21.2 and belongs to the δ subgroup of nonclustered protocadherin gene family [17]. Till now, only one paper has discussed the function of PCDH20 in carcinogenesis [19]. PCDH20 was frequently silenced by promoter methylation in NSCLC cancers, suggested that it might function as a candidate tumour suppressor [19]. However, the expression pattern and functions of PCDH20 in the progression of HCC remain elusive. In this study, we detected PCDH20 mRNA level in 107 paired human HCC tumour tissues and eight human HCC cancer cell lines. The results showed that PCDH20 had a higher probability of being downregulated in HCC tissues (51 of 107 tissues, 48%). Downregulation of PCDH20 was also observed in 6 of 7 HCC cell lines compared with the normal liver tissues. Our data clearly demonstrate that expression of PCDH20 is frequently decreased in human HCC. Moreover, further statistical analysis revealed that reduced expression of PCDH20 was significantly associated with younger age at diagnosis of patients with HCC. Whether it implicates an early onset of carcinogenesis should be an interesting question.
Increased DNA methylation in the promoter region of genes is well established as a common epigenetic mechanism for the silencing of tumour-suppressor genes in cancer cells [29]. Aberrant hypermethylation status (about 54%) of PCDH20 promoter was reported in primary NSCLC tissues [19]. In this study, we examined whether transcriptional silencing of PCDH20 was due to the promoter hypermethylation. We found that two (SNU-387 and SMMC-7721) of the five PCDH20 downregulated HCC cell lines showed hypermethylation in PCDH20 promoter. The downregulation of PCDH20 in these two cell lines could be reversed by 5-Aza treatment, indicating that promoter methylation is one of the mechanisms for PCDH20's inactivation in HCC. In addition, we also found that PCDH20 was downregulated in four HCC cell lines (SNU-182, Huh-7, SNU-449 and PLC/PRL/5) without detectable PCDH20 promoter hypermethylation. When we examined the promoter hypermethylation in 66 primary HCC tissues, only seven tumour specimens (10.6%) were PCDH20 CpG-enriched region hypermethylated. As the frequency of the silencing of PCDH20 mRNA expression in primary HCC tissues and cell lines was higher than that expected from the methylation and gene loss, other mechanisms including transcription regulation and/or post-translational modification may contribute to the downregulation of PCDH20 in HCC.
Wnt signalling plays a key role in the embryonic development of multicellular animals, and aberrant alterations of Wnt signalling pathway are the most common events associated with human carcinogenesis [30]. It has been reported that some protocadherins such as PCDHGA, PCDH24 and PCDHGC3 can affect the expression of some downstream targets of Wnt signalling pathway and growth properties [23–25]. In this study, we are the first to demonstrate that PCDH20 suppresses the canonical Wnt/β-catenin signalling in HEK 293T and two HCC cell lines Huh-7 and SNU-449 via suppressing the kinase activities of Akt and Erks, consequently unable their abilities to inactive GSK-3β. As expected, the main target of Wnt/β-catenin signalling, c-myc, cyclin D1 and β-catenin was also downregulated in PCDH20-overexpressed cells. The downregulation of cyclin D1 and c-myc induced by PCDH20 provided a possible role of PCDH20 to cause HCC cell G1-phase arrest and decrease tumourigenicity of HCC cells. It has been reported that PCDHs could act as a regulator via interaction with a variety of intracellular binding partners [8], and further studies should be conducted to determine which intracellular molecule(s) mediating the inactivation of Akt and Erks activities induced by PCDH20.
In consistent with above-suggested mechanisms, re-expression of this gene in HCC cell lines in vitro and in vivo further confirmed the tumour-suppressive function of PCDH20 in HCC. We demonstrated that restoration of PCDH20 expression could decrease cell proliferation, clonogenicity in soft agar and tumour growth in nude mice. In addition, flow cytometry analysis showed a PCDH re-expression inducing G1 arrest in HCC cancer cell. All these observations strongly suggest that PCDH20 acts as a candidate tumour-suppressor gene in HCC. The same observations that ectopic expression of PCDH20 inhibited colony formation are also identified in NSCLC cells by anchorage-independent assays [19]. In addition, we observed that re-expression of PCDH20 induced inhibition of cell migration and invasion. Different from classic cadherins, it is known that PCDHs are not just function as a regulator of cell–cell adhesive proteins and their heterophilic interactions with other molecules may be more important for their various physiologic functions [31–34]. Therefore, PCDH20 may have multiple tumour-suppressive functions potentially contributing to cell–cell adhesion, signal transduction and growth control, although the exact function of PCDH20 has not been reported.
In summary, we demonstrated in this study that PCDH20 can inhibit cell proliferation and cell migration, through antagonizing Wnt/β-catenin signalling pathway in HCC. Our data strongly support the notion that PCDH20 is a candidate tumour suppressor in HCC.
Acknowledgments
This work was supported by the National S & T Major Project for Infectious Diseases (Grant no. 2012ZX10004-904), the National Natural Science Foundation of China (Grant No. 81101539), the Leading Academic Discipline Project of Beijing, and the 111 Project (B07001).
Glossary
- CGI
CpG island
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- LOH
loss of heterozygosity
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PCDHs
protocadherins
- qRT-PCR
quantitative real-time reverse transcription-PCR
- TSG
tumour-suppressor gene
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article:
Table S1: Primers used in PCR assays.
Table S2: Antibodies used in western blot assays.
Table S3: PCDH20 heterozygous-deletion in 3 HCC cases.
References
- 1.Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127(5 Suppl 1):S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 2.But DY, Lai CL, Yuen MF. Natural history of hepatitis-related hepatocellular carcinoma. World J Gastroenterol. 2008;14(11):1652–1656. doi: 10.3748/wjg.14.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 4.Jones PL, Veenstra GJ, Wade PA, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19(2):187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 5.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21(1):103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 6.Coleman WB, Tsongalis GJ. Multiple mechanisms account for genomic instability and molecular mutation in neoplastic transformation. Clin Chem. 1995;41(5):644–657. [PubMed] [Google Scholar]
- 7.Morishita H, Yagi T. Protocadherin family: diversity, structure, and function. Curr Opin Cell Biol. 2007;19(5):584–592. doi: 10.1016/j.ceb.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Kim SY, Yasuda S, Tanaka H, Yamagata K, Kim H. Non-clustered protocadherin. Cell Adh Migr. 2011;5(2):97–105. doi: 10.4161/cam.5.2.14374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blevins CJ, Emond MR, Biswas S, Jontes JD. Differential expression, alternative splicing, and adhesive properties of the zebrafish delta1-protocadherins. Neuroscience. 2011;199:523–534. doi: 10.1016/j.neuroscience.2011.09.061. [DOI] [PubMed] [Google Scholar]
- 10.Yagi T, Takeichi M. Cadherin superfamily genes: functions, genomic organization, and neurologic diversity. Genes Dev. 2000;14(10):1169–1180. [PubMed] [Google Scholar]
- 11.Angst BD, Marcozzi C, Magee AI. The cadherin superfamily: diversity in form and function. J Cell Sci. 2001;114(Pt 4):629–641. doi: 10.1242/jcs.114.4.629. [DOI] [PubMed] [Google Scholar]
- 12.Frixen UH, Behrens J, Sachs M, et al. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 1991;113(1):173–185. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu JS, Koujak S, Nagase S, et al. PCDH8, the human homolog of PAPC, is a candidate tumor suppressor of breast cancer. Oncogene. 2008;27(34):4657–4665. doi: 10.1038/onc.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ying J, Li H, Seng TJ, et al. Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene. 2006;25(7):1070–1080. doi: 10.1038/sj.onc.1209154. [DOI] [PubMed] [Google Scholar]
- 15.Okazaki N, Takahashi N, Kojima S, Masuho Y, Koga H. Protocadherin LKC, a new candidate for a tumor suppressor of colon and liver cancers, its association with contact inhibition of cell proliferation. Carcinogenesis. 2002;23(7):1139–1148. doi: 10.1093/carcin/23.7.1139. [DOI] [PubMed] [Google Scholar]
- 16.Haruki S, Imoto I, Kozaki K, et al. Frequent silencing of protocadherin 17, a candidate tumour suppressor for esophageal squamous cell carcinoma. Carcinogenesis. 2010;31(6):1027–1036. doi: 10.1093/carcin/bgq053. [DOI] [PubMed] [Google Scholar]
- 17.Frank M, Kemler R. Protocadherins. Curr Opin Cell Biol. 2002;14(5):557–562. doi: 10.1016/s0955-0674(02)00365-4. [DOI] [PubMed] [Google Scholar]
- 18.Lee W, Cheng TW, Gong Q. Olfactory sensory neuron-specific and sexually dimorphic expression of protocadherin 20. J Comp Neurol. 2008;507(1):1076–1086. doi: 10.1002/cne.21569. [DOI] [PubMed] [Google Scholar]
- 19.Imoto I, Izumi H, Yokoi S, et al. Frequent silencing of the candidate tumor suppressor PCDH20 by epigenetic mechanism in non-small-cell lung cancers. Cancer Res. 2006;66(9):4617–4626. doi: 10.1158/0008-5472.CAN-05-4437. [DOI] [PubMed] [Google Scholar]
- 20.Jiang S, Yang Z, Li W, et al. Re-evaluation of the carcinogenic significance of hepatitis B virus integration in hepatocarcinogenesis. PLoS One. 2012;7(9):e40363. doi: 10.1371/journal.pone.0040363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Cheng J, Xu C, et al. Quantitative methylation analysis reveals gender and age differences in p16INK4a hypermethylation in hepatitis B virus-related hepatocellular carcinoma. Liver Int. 2012;32(3):420–428. doi: 10.1111/j.1478-3231.2011.02696.x. [DOI] [PubMed] [Google Scholar]
- 22.Morin PJ. Beta-catenin signaling and cancer. BioEssays. 1999;21(12):1021–1030. doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 23.Ose R, Yanagawa T, Ikeda S, Ohara O, Koga H. PCDH24-induced contact inhibition involves downregulation of beta-catenin signaling. Mol Oncol. 2009;3(1):54–66. doi: 10.1016/j.molonc.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Junghans D, Haas IG, Kemler R. Mammalian cadherins and protocadherins: about cell death, synapses and processing. Curr Opin Cell Biol. 2005;17(5):446–452. doi: 10.1016/j.ceb.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Dallosso AR, Oster B, Greenhough A, et al. Long-range epigenetic silencing of chromosome 5q31 protocadherins is involved in early and late stages of colorectal tumorigenesis through modulation of oncogenic pathways. Oncogene. 2012;31(40):4409–4419. doi: 10.1038/onc.2011.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baba HA, Stypmann J, Grabellus F, et al. Dynamic regulation of MEK/Erks and Akt/GSK-3beta in human end-stage heart failure after left ventricular mechanical support: myocardial mechanotransduction-sensitivity as a possible molecular mechanism. Cardiovasc Res. 2003;59(2):390–399. doi: 10.1016/s0008-6363(03)00393-6. [DOI] [PubMed] [Google Scholar]
- 27.Ding Q, Xia W, Liu JC, et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell. 2005;19(2):159–170. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 28.Unterseher F, Hefele JA, Giehl K, De Robertis EM, Wedlich D, Schambony A. Paraxial protocadherin coordinates cell polarity during convergent extension via Rho A and JNK. EMBO J. 2004;23(16):3259–3269. doi: 10.1038/sj.emboj.7600332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA. 1994;91(21):9700–9704. doi: 10.1073/pnas.91.21.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14(15):1837–1851. [PubMed] [Google Scholar]
- 31.Stockinger A, Eger A, Wolf J, Beug H, Foisner R. E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity. J Cell Biol. 2001;154(6):1185–1196. doi: 10.1083/jcb.200104036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yasuda S, Tanaka H, Sugiura H, et al. Activity-induced protocadherin arcadlin regulates dendritic spine number by triggering N-cadherin endocytosis via TAO2beta and p38 MAP kinases. Neuron. 2007;56(3):456–471. doi: 10.1016/j.neuron.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci. 1999;112(Pt 8):1237–1245. doi: 10.1242/jcs.112.8.1237. [DOI] [PubMed] [Google Scholar]
- 34.Yang X, Chen MW, Terry S, et al. A human- and male-specific protocadherin that acts through the wnt signaling pathway to induce neuroendocrine transdifferentiation of prostate cancer cells. Cancer Res. 2005;65(12):5263–5271. doi: 10.1158/0008-5472.CAN-05-0162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Primers used in PCR assays.
Table S2: Antibodies used in western blot assays.
Table S3: PCDH20 heterozygous-deletion in 3 HCC cases.