

Abstract
It has been suggested that soluble urokinase receptor (suPAR) is a causative circulating factor for and a biomarker of focal and segmental glomerulosclerosis (FSGS). Here we undertook validation of these assumptions in both mouse and human models. Injection of recombinant suPAR in wild-type mice did not induce proteinuria within 24 hours. Moreover, a disease phenotype was not seen in an inducible transgenic mouse model that maintained elevated suPAR concentrations for 6 weeks. Plasma and urine suPAR concentrations were evaluated as clinical biomarkers in 241 patients with glomerular disease from the prospective, longitudinal multi-center observational NEPTUNE cohort. The serum suPAR concentration at baseline inversely correlated with estimated glomerular filtration rate (eGFR) and the urine suPAR/creatinine ratio positively correlated with the urine protein/creatinine ratio. After adjusting for eGFR and urine protein, neither the serum nor urine suPAR level was an independent predictor of FSGS histopathology. A multivariable mixed-effects model of longitudinal data evaluated the association between the change in serum suPAR concentration from baseline with eGFR. After adjusting for baseline suPAR concentration, age, gender, proteinuria and time, the change in suPAR from baseline was associated with eGFR, but this association was not different for patients with FSGS as compared to other diagnoses. Thus, these results do not support a pathological role for suPAR in FSGS.
Introduction
suPAR, the soluble form of the urokinase-type plasminogen activator receptor (uPAR encoded by PLAUR), garnered immense standing and scrutiny by renal scientists since it was suggested that it was linked causally and by association to induction of focal and segmental glomerulosclerosis (FSGS) in humans and in animal models 1, 2. uPAR is a glycophosphatidylinositol (GPI) anchored three domain (commonly referred to as D1, D2, D3) protein that is found in a variety of cell types including monocytes, lymphocytes, macrophages, endothelial and tumor cells and in various tissues throughout the body 1, 3. Although the primary function of this receptor remains unclear, it has been proposed to be involved in cell migration, proliferation, survival and has been extensively studied in cancers 3, 4. suPAR is the 60 kDa soluble form of the receptor that is formed as a result of removal of the GPI-anchor from the full length protein and thus contains only the D1, D2, D3 domains of uPAR 1. This peptide undergoes additional proteolytic cleavage to produce peptides containing domains D1 and D2–D3. suPAR is abundantly present in plasma, urine, blood, serum and cerebrospinal fluid and immune system activation has been shown to further increase suPAR levels in bodily fluids 1.
Since its discovery in 1991, suPAR has been extensively studied in many human disease states. An increased suPAR concentration has been documented in diseases such as paroxysmal nocturnal hemoglobinuria, human immune-deficiency type 1, malaria, bacteremia and sepsis 1. Elevated suPAR was also reported in multiple types of cancers including lung, breast, colon, and ovarian 5. Such observations have intensified research on suPAR, examining its value as a biomarker predictive of risk associated with cancer, cardiovascular disease, diabetes, and for mortality in the general population 6.
FSGS histopathology is often associated with nephrotic syndrome and progressive loss of renal function, culminating in end stage kidney disease 7. It has long been hypothesized that a large fraction of patients with FSGS have a causal circulating factor because more than 30% of patients receiving a renal allograft have recurrent disease 8. Identifying this circulating causal factor would be a critical step in improving FSGS treatment and prevention of post-transplant recurrence of FSGS. Recently, there has been growing interest in establishing a relationship between suPAR and FSGS. Enthusiasm for this relationship was initiated by a study performed by Xu et al. 9 who first demonstrated a strong uPAR expression in glomerular endothelial, mesangial, and epithelial cells. In a 2008 study, Wei et al. 10 proposed a role for uPAR signaling in regulation of podocyte morphology and glomerular permeability. More recently, the same group identified suPAR as a possible causal factor for FSGS 2. In three distinct animal model experiments, Wei et al. reported that introduction of ectopic recombinant suPAR to Plaur −/− mice induced proteinuria, podocyte effacement, and histopathology consistent with FSGS 2.
Due to potentially large clinical implications of identifying suPAR as a causal factor for FSGS, additional research was warranted to help clarify the role of suPAR in human disease. The work of Wei et al. stimulated worldwide research where investigators at multiple institutions invested resources in analyzing the association between suPAR and FSGS 2, 11–15. A few studies have shown that increased suPAR concentration is higher in groups with FSGS when compared to either a control population or patients with other types of glomerular disease 2, 12. Recent papers have challenged this relationship and have fueled controversy by showing that suPAR concentration does not distinguish patients with FSGS from other glomerular histopathology 11, 13–15. There is growing evidence suggesting that glomerular filtration rate (GFR) is the strongest determinant of suPAR concentration 11, 14, 15. However, the majority of the published studies are based on conclusions derived using cross-sectional data, and single center or single racial groups. Additionally, these studies have provided conflicting results regarding the relationship between serum and urine suPAR concentration 16. Finally, until very recently no work had re-examined Wei et al.’s proof of principle experiments performed in mice 17. For these reasons, we re-examined whether suPAR is sufficient to induce FSGS in mice. We also analyzed suPAR concentrations in plasma and urine samples from a large, prospective, longitudinal cohort of glomerular disease subjects obtained by the Nephrotic Syndrome Study Network (NEPTUNE) to test the hypothesis that suPAR concentration can be used to predict glomerular histopathology 18.
Results
Administration of recombinant suPAR protein into wild-type mice does not induce proteinuria
An enzyme-linked immunosorbent assay (ELISA) was used to assess suPAR concentration in normal mouse serum. Recombinant Fc-mouse suPAR was diluted into pooled mouse serum from mice deleted of uPAR (PLAUR−/−) to define a standard curve for circulating mouse uPAR in mouse serum (fig. 1A). Using the relationship obtained, the concentration of suPAR in normal wild-type mouse serum was 3.3 ng/ml +/− 0.82 ng/ml (n=25).
Figure 1. Injection of mice with recombinant suPAR protein does not induce proteinuria.

A) A mouse suPAR ELISA standard curve was generated by adding indicated amounts of purified recombinant Fc-suPAR protein to serum from Plaur −/− mice diluted 1:4. B) Serum suPAR concentration concentration at indicated time points following injection of purified recombinant suPAR protein was estimated using ELISA. Experimental mice (n=5) injected with recombinant protein (solid line) were compared to control mice (n=4) injected with PBS (dashed line). C) Urine albumin and creatinine ratios were calculated at 12 h and 24 h post-Fc-suPAR injection; control (white columns), experimental (gray columns) group. All values are expressed as mean ± standard error of the mean. ** p <0.01, * p <0.05, comparing experimental to control groups at each time point.
In an initial experiment we attempted to replicate the previously published observation that intravenous administration of commercially available recombinant Fc-mouse suPAR induces proteinuria acutely in mice. In accordance with the observations of Wei, we used 20 μg of this recombinant protein or vehicle (PBS). In distinction to Wei, we employed wild-type mice rather than PLAUR−/− mice, reasoning that a wild-type—rather than null—mouse model was more physiologically relevant. Mice injected with Fc-chimeric uPAR had a 12-fold increase relative to baseline in serum suPAR concentration at 4 h post-injection; a nearly 6-fold increase in serum suPAR concentration persisted at 24 h (fig. 1B). Analysis of urine albumin/creatinine ratio demonstrated no increase in urine protein excretion at 12 h or 24 h in Fc-suPAR injected mice relative to control (fig. 1C).
Chronic expression of suPAR from liver in transgenic mice does not induce proteinuria
Despite the results above, we explored whether increased proteinuria could be induced by continuously expressing soluble uPAR in mice. A new transgenic mouse model was prepared that allowed inducible expression of suPAR (D1D2D3) from liver as depicted in figure 2A. “suPAR-floxed-STOP” mice were created bearing a transgene cassette that included a CMV promoter, that carried a floxed cassette bearing stop codons in all three reading frames, and that contained a 3′-cDNA cassette encoding the uPAR signal peptide and domains 1 through 3 of uPAR without the uPAR carboxyl-terminal GPI-linkage motif. Liver-specific expression of suPAR was induced by delivery of a vector encoding Cre recombinase to the liver. This vector was built on a hepatocyte-specific adenoviral virus backbone and contained a hepatocyte-specific promoter driving Cre recombinase (AAV8.TBG.PI.Cre.rBG). Preliminary experiments using a reporter mouse detailed in Supplemental Figure 1 confirmed liver-specific Cre-mediated recombination by 10 days following intravenous viral injection.
Figure 2. Chronic suPAR expression does not induce proteinuria in transgenic mice.

Transgenic expression of recombinant suPAR from liver was induced following intravenous injection of AAV8.TBG.PI.Cre virus as described in text. A) Schematic representation of protocol for conditional induction of recombinant suPAR. B) Western blot analysis using polyclonal uPAR antibody demonstrates induced expression of suPAR in liver of experimental (1–4) versus control (5–8) mice. Mouse #5 and mouse #8 were injected with AAV8.LacZ virus while mouse #6 and #7 received PBS. C) Mean serum suPAR concentration over a period of 44 days in the AAV8.TBG.PI.Cre-injected (white columns; n=8) and combined PBS and AAV8.LacZ control-injected (gray columns; n=9) mice assayed using mouse suPAR ELISA. D) Urine albumin/creatinine ratios of AAV8.TBG.PI.Cre-injected (gray columns) and control (white columns) mice for the indicated time points. All values are expressed as mean ± standard error of the mean. * p <0.05, comparing experimental to control groups at each time point.
A 44 day experiment was performed using this model in which eight suPAR-flox-STOP mice received AAV8.TBG.PI.Cre.rBG and nine suPAR-flox-STOP mice received a control adenoviral vector (AAV8.TBG.LacZ.bGH) or PBS. Induction of expression of recombinant suPAR in suPAR-flox-STOP mice following AAV8.TBG.PI.Cre injection was confirmed by ELISA of serum suPAR concentration and by immunoblot analysis of liver lysates (figures 2B–C). Following viral transduction of Cre recombinase, serum suPAR concentration rose approximately 2-fold by day 13 (relative to day 0) to approximately 11 ng/ml and rose nearly 3-fold to a mean concentration of 18 ng/ml at day 44 when the experiment was terminated. At 44 days, induction of suPAR was identified in liver lysates obtained from experimental animals but not control virus injected animals. Despite a prolonged increase in serum suPAR concentration, increased proteinuria was not detected in the experimental group relative to control (figure 2D). These results support a conclusion that prolonged exposure to increased serum suPAR concentration does not induce proteinuria in wild-type mice.
Cohort Characteristics
For this study, all 241 participants, who had been enrolled in the prospective, multi-center NEPTUNE observational study of glomerular disease from August 2010 to July 2013 with a diagnosis of FSGS, minimal change disease (MCD), membranous nephropathy (MN) or IgAN, were examined. All patients had proteinuria ≥500mg/day prior to their diagnostic kidney biopsy. Baseline characteristics of these participants are summarized in Table 1. Participants with FSGS and MCD were younger than patients with MN and IgAN (median ages 36 and 14 vs. 54 and 42 years, respectively). The median eGFR (mL/min/1.73m2) was lower in participants with FSGS (60) and IgAN (57) relative to those with MCD (104) and MN (82), overall p-value <0.001. Participants with MN had the greatest median urine protein to creatinine (UPCR) (3.8 mg/mg) relative to FSGS (2.1 mg/mg), overall p-value <0.001. Median plasma suPAR level for the cohort was 2954 pg/mL.
Table 1. Baseline Participant Characteristics (n=241).
All characteristics were recorded at the participant’s baseline study visit, conducted from September, 2010 to June, 2013. Continuous variables are presented as median (IQR). Categorical variables are presented as percentages.
| All (n=241) | FSGS (n=95) | MCD (n=62) | MN (n=52) | IgAN (n=32) | |
|---|---|---|---|---|---|
| Age, years | 37 (17, 55) | 36 (17, 52) | 14 (6, 25) | 54 (41, 61) | 42 (32, 54) |
| Female | 90 (37%) | 31 (33%) | 26 (42%) | 20 (38%) | 13 (41%) |
| Black Race | 69 (29%) | 36 (38%) | 21 (34%) | 11 (21%) | 1 (3%) |
| Serum Albumin (g/dL) | 3.1 (2.5, 3.9) | 3.4 (2.4, 4.1) | 3.2 (2.2, 4) | 2.8 (2.3, 3.1) | 3.5 (3.2, 3.8) |
| Total Cholesterol (mg/dL) | 242 (194, 307) | 236 (180, 291) | 227 (192, 311) | 272 (243, 348) | 220 (195, 256) |
| eGFR (mL/min/1.73m2) | 82 (50, 105) | 60 (42, 93) | 104 (86, 126) | 82 (66, 100) | 57 (39, 95) |
| Urine Protein/Creatinine Ratio (mg/mg) | 2.3 (0.8, 4.3) | 2.1 (1.1, 3.9) | 0.5 (0.1, 2.7) | 3.8 (2.5, 7.1) | 2.6 (1.2, 3.3) |
| Plasma suPAR (pg/mL) | 2954 (2360, 3601) | 3178 (2681, 3763) | 2464 (2101, 2973) | 3061 (2668, 3952) | 2974 (2290, 3728) |
| Urine suPAR | 2324 (1742, 3457) | 2135 (1671, 2913) | 2327 (1749, 3457) | 2861 (2013, 4599) | 2128 (1584, 3068) |
| Follow-up Time (months) | 17 (10, 25) | 15 (11, 23) | 17 (8, 24) | 16 (8, 26) | 25 (19, 30) |
| >50% loss of eGFR | 26 (12%) | 14 (16%) | 1 (2%) | 4 (9%) | 7 (23%) |
| ESRD* | 14 (6%) | 6 (7%) | 0 (0%) | 2 (4%) | 6 (19%) |
| Ever Reached Complete or Partial Remission** | 183 (77%) | 65 (70%) | 47 (92%) | 34 (65%) | 27 (84%) |
ESRD is defined as initiation of dialysis, receipt of kidney transplant or eGFR<15 mL/min/1.73m2
Complete Remission is defined as UPCR<0.3mg/mg. Partial Remission is defined as UPCR decline by 50% from baseline and ≤3.5mg/mg
Correlations of plasma and urine suPAR concentration with eGFR and proteinuria
We examined correlates of baseline plasma and urine suPAR concentration as depicted in Figure 3. Plasma suPAR concentration was negatively correlated with eGFR (ρ = −0.58, P <0.01, figure 3A). Plasma suPAR concentration and urine suPAR/creatinine ratio were correlated with each other (ρ = 0.36, P <0.001, figure 3B). Nevertheless, urine suPAR/creatinine did not correlate with eGFR (ρ = 0.11, P = 0.14). Both plasma suPAR concentration and urine suPAR/creatinine positively correlated with urine protein/creatinine ratio (UPCR) (ρ = 0.38, P <0.001; ρ = 0.30, P <0.001, respectively) (figures 3C & D).
Figure 3. Correlates of Baseline Plasma and Urine suPAR.

A) Plasma suPAR (pg/mL) concentration was negatively correlated with eGFR (ρ = −0.58, p-value <0.001). B) Urine suPAR/creatinine ratio and plasma suPAR concentrations were positively correlated (ρ = 0.36, p-value <0.01). C) Plasma suPAR (pg/mL) was positively correlated with UPCR (ρ = 0.38, p-value 0.006). D) Urine suPAR/creatinine ratio (pg/mg) was positively correlated with urine protein/creatinine ratio (mg/mg) (ρ = 0.30, p-value <0.01).
suPAR concentration and histopathology cohort
Figure 4 depicts baseline plasma suPAR concentration and urine suPAR/creatinine by histopathology cohort. Participants with FSGS did not have greater suPAR concentration or urine suPAR/creatinine as described previously by others 2, 12, 16, 19–21. Median plasma suPAR concentration was lower in MCD as compared to FSGS, MCD and IgA (p-value <0.01), but in a multivariable linear regression, plasma suPAR concentration was not associated with histopathology cohort after adjustment for eGFR. Similarly, unadjusted median urine suPAR/creatinine ratios were greater in participants with MN as compared to FSGS and IgAN (p-value 0.01 and 0.02, respectively), but in multivariable linear regression, urine suPAR/creatinine was not associated with cohort after adjustment for UPCR.
Figure 4. Baseline Plasma and Urine suPAR concentrations by pathologic diagnosis.

A) Baseline plasma suPAR concentration from 183 NEPTUNE participants is plotted by diagnosis. Although MCD participants had a statistically significantly lower median plasma suPAR concentration as compared to all other groups (p-value <0.01), this difference did not persist after adjustment for differences in eGFR. B) Baseline urine suPAR/creatinine ratio from a 24 hour urine specimen in 173 NEPTUNE participants is plotted by diagnosis. Although MN participants had a statistically significantly greater median urine suPAR/creatinine ratio as compared to FSGS and IgAN (p-value 0.01 and 0.02, respectively), this difference did not persist after adjustment for differences in baseline urine protein.
suPAR concentration and clinical endpoints
All suPAR measurements were performed on biological samples collected during the first 12 months of follow-up. Additional follow-up for clinical endpoints was available. We included 1260 study visits that were obtained over a total median follow-up time of 17 months. Overall, 26 (12%) participants lost >50% of baseline eGFR and 14 (6%) progressed to end stage renal disease (ESRD), whereas 183 (77%) experienced either a complete or partial remission during follow-up (Table 1). Neither baseline plasma or urine suPAR were independent predictors of occurrence of ESRD, 50% loss of eGFR or complete and partial remission, after adjusting for baseline GFR and UPCR.
Change in suPAR and renal function
The results of the mixed effects model fit to assess the association of eGFR with change in plasma suPAR concentrations from baseline are presented in Table 2, stratified by adult and pediatric participants. Both greater baseline plasma suPAR concentrations and greater increase of plasma suPAR concentration from baseline were independently associated with lower eGFR levels. However, the association of change in plasma suPAR concentration with eGFR was not different by histopathology cohort as noted by the non-significant interaction terms in the model. The predicted eGFR values by change in suPAR for each histopathology cohort are graphed in Figure 5. The slope of the line for FSGS was not statistically different from the other histopathology cohorts in both adult (MCD p-value 0.61, MN p-value 0.78, IgAN p-value 0.30) and pediatric patients (MCD, p-value 0.64). The parallel lines indicate that although a greater increase in suPAR concentration was associated with lower eGFR, that association is not different for participants with FSGS as compared to other cohorts as might be expected of a pathogenic factor.
Table 2. Results of Longitudinal Mixed Effects Model of eGFR.
Data from 178 participants with complete data and 493 visits. Results stratified by adult and pediatric patients. Model was adjusted for Urine Protein/Creatinine ratio, Age, Sex and Follow-up time.
| Adult | Effect | Estimate | CI | p-value |
|---|---|---|---|---|
| Baseline suPAR (per 100 pg/mL) | −1.3 | (−1.6, −0.9) | <.0001 | |
| Change in suPAR from baseline (per 100 pg/mL) | −0.7 | (−1.1, −0.2) | 0.0044 | |
| Cohort | ||||
| FSGS | ref | -- | -- | |
| IGA | −3.4 | (−15.2, 8.4) | 0.5697 | |
| MN | 24.8 | (14.5, 35.2) | <.0001 | |
| MCD | 26 | (12.9, 39.1) | 0.0001 | |
| Interaction of Change in suPAR by Cohort | ||||
| Change in suPAR * FSGS | ref | -- | -- | |
| Change in suPAR * IGA | 0.3 | (−0.3, 0.9) | 0.3515 | |
| Change in suPAR * MN | 0.1 | (−0.5, 0.6) | 0.779 | |
| Change in suPAR * MCD | 0.2 | (−0.8, 1.1) | 0.7436 |
| Child | Effect | Estimate | CI | p-value |
|---|---|---|---|---|
| Baseline suPAR (per 100 pg/mL) | −0.6 | (−1.8, 0.6) | 0.3231 | |
| Change in suPAR from baseline (per 100 pg/mL) | −0.6 | (−2.1, 0.8) | 0.3854 | |
| Cohort | ||||
| FSGS | ref | -- | -- | |
| MCD | 25.4 | (12.4, 38.4) | 0.0002 | |
| Interaction of Change in suPAR by Cohort | ||||
| Change in suPAR * FSGS | ref | -- | -- | |
| Change in suPAR * MCD | 0.3 | (−1.6, 2.1) | 0.7911 |
Figure 5. Predicted eGFR values by Change in suPAR from baseline.
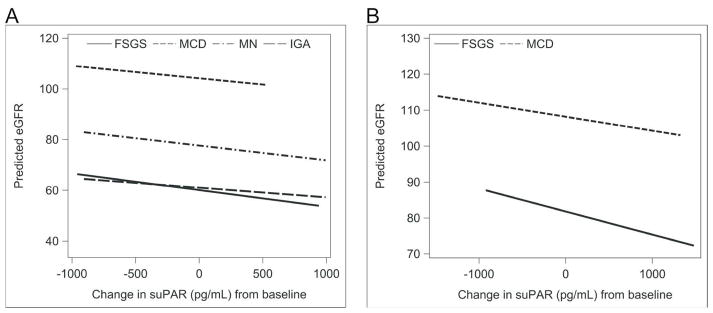
Median values for each covariate (age, follow-up time, baseline urine protein and baseline suPAR) were used in the linear mixed effects model to generate the predicted line for each histologic cohort. The graph was restricted to overlapping regions of observed values of change in suPAR. Difference in slope as compared to FSGS was not significant in either adult (MCD p-value 0.74, MN p-value 0.78, IgAN p-value 0.35) or pediatric participants (MCD, p-value 0.79).
Discussion
Identification of a circulating factor explaining pathogenesis of FSGS has been anticipated for decades. For this reason, it is not surprising that the purported discovery of soluble uPAR as this pathogenic factor met with great enthusiasm from both renal clinical and scientific communities when it was published in 20112. Predictably, a discovery of this potential impact engendered scrutiny and immediate attempts at replicating and extending the initial observations. During the subsequent few years, several reports examining for a correlation between serum and/or urine suPAR concentration and FSGS histopathology in humans either challenged or added support to the original observations made by Wei et al. 2. These conflicting initial reports established a need to probe the described causative relationship between suPAR and FSGS and its predictive value as a FSGS biomarker. Using a large prospective, longitudinal observational cohort of glomerular disease patients, we did not find a correlation between serum or urine suPAR concentration and FSGS histopathology. In addition, we could not replicate results obtained in one of Wei’s published proof-of-principle experiments, nor did we find evidence that prolonged two- to three-fold increased concentration of circulating suPAR engenders proteinuria in a new inducible transgenic mouse model.
Wei et al. provided evidence that increased concentration of recombinant suPAR can induce proteinuria, foot process effacement, and FSGS histopathology using both short-term and more prolonged exposure of PLAUR null mice to recombinant suPAR (2). We designed experiments to replicate and extend these results, which are fundamental to supporting the conclusion that suPAR is a causative factor for FSGS. In short-term experiments, Wei injected a commercially available mouse recombinant Fc-fusion protein that coupled a human IgG1 Fc-domain to mouse uPAR lacking a GPI-linkage motif. While this recombinant protein was targeted to the glomerulus, it was not determined whether this IgG1 Fc domain containing protein engendered complement fixation-dependent glomerular injury. Nevertheless, as a first experiment, we chose to repeat these experiments using wild-type mice rather than null mice because it was uncertain that experiments performed on a PLAUR null background had relevance to human disease where PLAUR was present. Despite obtaining at least a 12-fold increase in serum suPAR concentration above baseline, we were unable to detect proteinuria after 12 h or 24 h in our experiments. Our negative results are identical to those recently reported by Cathelin et al. 17. Why our results and those obtained by Cathelin are distinct from those obtained by Wei is unclear; possibly, the discrepancy is due to the fact that the later experiment was conducted on a PLAUR null mouse background.
To extend experiments suggesting that prolonged elevation of serum suPAR concentration leads to proteinuria and histopathology consistent with FSGS, we designed an inducible transgenic mouse model that expresses mouse suPAR from liver. We chose to employ a plasmid that encodes suPAR D1-D2-D3 to explore whether prolonged exposure of wild-type mice to increased suPAR concentration results in proteinuria. We anticipated that this construct would undergo proteolytic cleavage typical of that observed in human to distinct D1 and D2–D3 fragments. No proteinuria was obtained by this approach despite a 3-fold increase in serum suPAR concentration sustained to 44 days, an increase in suPAR concentration much greater than that seen in glomerular disease patients, even those with advanced loss of glomerular filtration rate.
It is possible that Wei obtained proteinuria and segmental sclerosis following chronic expression of suPAR because their experimental approach was distinct from that of Cathelin and that reported here. In addition to employing a mouse genetic background deficient in PLAUR, Wei used a mouse suPAR cDNA fragment that was obtained from a purchased cDNA clone (IMAGE cDNA clone 3158012) containing a retained intron 4 (uPAR-intron 4) that—if not spliced out—would result in a frame shift mutation within the open reading frame corresponding to amino acid residue 133 within the second uPAR domain (D2) and premature termination of translation within uPAR domain 2. This mouse cDNA was first described in 199122 and was felt at the time to explain a mechanism alternative to proteolytic processing by which a circulating form of uPAR is obtained. The mouse splice variant encoding uPAR-intron 4 is rare; its expression has only been described in a subset of mouse gut epithelial cells 22. The expression of the protein associated with this variant has not been reported and based on its predicted structure, some have questioned whether this uPAR variant protein is structurally stable, reducing the probability that it is expressed 23, 24. Additionally, a homologous splice variant of this type has not been identified in human (ENCODE; release GRCh37/hg19)25; while we cannot be certain that this variant is not expressed in human, its absence in the most recent transcriptome dataset release decreases the probability that the mouse uPAR-intron 4 variant is relevant in human.
Wei et al. reported the creation of a control mouse suPAR construct with an E134A mutation intended to abrogate binding of mouse suPAR to beta3 integrin. The creation of this mouse suPAR mutant was predicated on the observation that mutation of GEEG to GAAG in the motif 130IQEGEEGRPKDDR142 in the D2 domain of human uPAR decreased affinity of uPAR for beta3 integrin 26. However, a similar beta3 integrin binding motif has not been described in mouse uPAR. While mouse uPAR might bind to beta3 integrin, we were uncertain what residues were critical in this interaction, and for this reason chose to use either injection with PBS or injection with adenovirus carrying an expression plasmid for LacZ as a control in our transgenic mouse experiments.
Since the initial description of suPAR as a putative causative agent of FSGS 2, several groups have examined this hypothesis in human cohorts 11, 12, 14, 15 with conflicting conclusions. While the results from the FSGS Clinical Trial and Podonet cohorts 12 did indicate elevated concentration of suPAR in FSGS patients relative to controls, the small sample size, discordant protocols, low number of clinical events and limited duration and availability of longitudinal follow-up did not allow for robust evaluation of the relationship with adjustment for relevant confounders. Specifically, more recent studies have shown that serum suPAR concentration is inversely correlated with eGFR 11, 14, 15 and that after accounting for differences in renal function, suPAR is not a predictive marker for FSGS. Aggregated, these recent studies agreed that suPAR was not a valid biomarker for FSGS in human patients. However, each of these reports had some limitations in study design, including small sample size 15, single center 14, and limited 14 or absent 11, 15 longitudinal follow-up.
Our study has several unique strengths based on the design of the NEPTUNE cohort. This multi-center study enrolls patients at the time of first diagnostic kidney biopsy based on uniform inclusion criteria, resulting in a representative, unbiased sample of incipient, proteinuric glomerular disease. Diagnosis of the four disease cohorts is assigned by a central pathology committee and specimen and data collection is done in a uniform and prospective manner. Measurement of plasma and urine suPAR was performed by laboratory personnel blinded to participant information. The resultant large cohort is ethnically and racially diverse with a broad representation of histologic diagnosis and longitudinal data to fully evaluate novel biomarkers for diagnostic and predictive abilities.
Our results agree with the recent growing evidence that suPAR is not a diagnostic biomarker for FSGS and cannot be used to distinguish FSGS from other causes of glomerular disease. In our cohort, plasma and urine suPAR levels were not different in participants with FSGS compared to other diagnostic cohorts after adjusting for differences in the relevant confounders of eGFR and proteinuria. The initial reports of the association 2, 12 did not account for the correlation between serum suPAR and eGFR and urine suPAR and proteinuria. Leveraging the uniform and robust longitudinal data available, we were able to examine the association of change in suPAR over time with disease progression. We hypothesized that if suPAR was a pathogenic factor for FSGS participants, as opposed to other diseases cohorts, for a given increase in suPAR, the eGFR would be lower in FSGS than MCD, MN and IgAN. Using a longitudinal model and adjusting for relevant confounders, there was no difference in this relationship in FSGS patients, relative to MCD, MN and IgAN.
Limitations of our study include the lack of inflammatory makers (e.g. CRP) which have been previously associated with suPAR levels. In this observational cohort, therapy was prescribed during routine clinical care and thus, confounding by indication limits the ability to examine the effect of medication on the association between suPAR and eGFR. However, when we limited our analysis to visits where the participant was not on any immunosuppressive agents, the results were unchanged. Finally, validation of suPAR as a predictor of hard outcomes such as 50% loss of GFR and ESRD was limited by the few number of events. However, future study in the NEPTUNE cohort will be possible as recruitment and follow-up continues.
It has been argued that the presently available commercial uPAR ELISA is insufficient to predict the presence of pathogenic forms of suPAR and that a functional bioassay must be used to identify these forms. However, this commercial ELISA was employed in the original Wei study and in confirmatory human studies by others since 2, 11, 12, 14, 15. It has been suggested that unique, possibly improperly glycosylated forms of suPAR exist in humans to explain pathogenesis of FSGS by suPAR. It has also been suggested that alternatively spliced suPAR forms might exist that cause human FSGS which cannot be identified using the currently available commercial human uPAR ELISA. In support of this contention, there are 7 known human uPAR transcript splice variants expressed with variable abundance from a variety of tissues. A relationship between abnormally elevated expression of some of these variants have been associated with shortened survival in breast cancer and other cancers 27.
Until a time when an alternatively transcribed or atypically post-translationally processed form of suPAR is identified that causes FSGS, it is difficult to ignore the overwhelming confounding effect of GFR on the correlation between suPAR and glomerular histopathology, the uncertainties raised by the experimental proof-of-principle approaches employed by Wei et al. 2, and the inability to reproduce or extend these important proof-of-principle observations here and by others.
METHODS
Construction and validation of suPAR floxed construct
The full-length (FL) mouse uPAR open reading frame (ORF) without GPI anchor was amplified by PCR using Mus Musculus PLAUR cDNA (Sino Biological, Inc. Cat#MG50160-M, Gene Bank Ref.ID: NM-011113.3) as the template. The PCR fragment was cloned into pCMVflox vector 28. The resulting CMVflox-suPAR cDNA construct was validated by restriction analysis and complete nucleotide sequencing. The expression and proper processing of suPAR protein from this construct was confirmed from the supernatant of transfected HEK 293 cells.
Creation of suPAR transgenic mice
The CMVflox-suPAR construct was linearized using PacI endonuclease, purified and submitted for microinjection. The Transgenic and Chimeric Mouse Facility at the University of Pennsylvania was employed for microinjecting the DNA into FVB oocytes and for oocyte implantation into pseudo-pregnant females. Mice were genotyped prior to three weeks of age by PCR using genomic DNA obtained from tail biopsies. The following primers were used for suPAR transgene identification, supg5.fwd: 5′-aag ctg gct agc gtt taa ac-3′ and supg6.rev: 5′-tag gat agc ggc att gca gg-3. PCR amplification resulted in a product of 473 base pairs representing the transgene. Germline transmission of suPAR transgene was obtained by crossing CMVflox-suPAR-FL transgenic founders (n=5) with wild type FVB mice and thus multiple transgenic lines were established. Mice were raised and all experiments using mice were performed in accordance with protocol 804493 approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
suPAR ELISA
To evaluate suPAR concentration in mouse serum, a kit from R&D systems (DY531) that has been validated by the manufacturer for detection of mouse suPAR in cell supernatant was used. This assay uses polyclonal goat IgG mouse uPAR antibody (R&D systems; Cat# AF534) for suPAR detection. The assays were performed as per the manufacturer’s protocol. suPAR standard protein was serially diluted (concentrations ranging from 337pg/ml to 2500pg/ml) in the provided reagent diluent (0.05% Tween-20 in PBS) to generate standard curves. Standard curve derivation was repeated in serum from PLAUR −/− mice (gift from Dr. Nicholai Sidenius, Unit of Cell Matrix Signaling IFOM, the FIRC Institute for Molecular Oncology, Milan, Italy). Pooled serum from 6 wild type mice was used to estimate the basal concentration of suPAR in mice. All samples were assayed in duplicate. The mean co-efficient of variation in these assays was 5%.
Urine collection and calculations
Urine albumin/creatinine ratio was calculated from clean-catch urine. Mice were placed in containers atop elevated wire mesh for 3–4 hours. Urine free of feces was collected and spun at 8000 x g for 5 min in a centrifuge, and supernatants were frozen at −80°C until assayed. Albumin concentration was determined in duplicate by plate ELISA (Albuwell-M kit, Glycadia/Exocell). Urine creatinine concentration was determined by endpoint assay (TECO diagnostics). Albumin and creatinine plates were read using a multiplate reader (Beckman-Coulter DTX 880).
Adeno Associated Virus 8 (AAV8) in reporter tomato mice
Cre recombination was confirmed in Tomato reporter transgenic mice using PBS, a hepatocyte-specific Adeno Associated Virus 8 (AAV8.TBG.PI.Cre), or a negative control virus (AAV8.TBG.LacZ.bGH) procured from the University of Pennsylvania Vector Core Gene Therapy Program 29. Retro-orbital injections of viral particles (an optimal dose of 4×1011 vector genome copies was determined) were performed in 8 weeks old heterozygous reporter mice. Reporter mice were euthanized 10 days after virus injection and various organs including liver, muscle, kidney and tails were isolated and subjected to PCR analysis. Tissue samples from liver and muscle were analyzed by immunofluorescence and the images were collected on a Zeiss microscope fitted with Metamorph Image Analysis software (Diagnostic Instruments).
Chronic suPAR expression in transgenic mice and proteinuria measurement
22–26 weeks old suPAR transgenic (n=8) and control (n=9) mice were employed. Experimental mice received retro-orbital injections of AAV8.TBG.PI.Cre (4×1011 vector genome copies), whereas the control mice were injected with either PBS (n=6) or AAV8.TBG.LacZ.bGH control virus (n=3). A 200ul bolus of normal saline was given to each mouse following each blood collection. To validate the over-expression of suPAR protein in transgenic mice following induction, liver of transgenic mice was isolated, homogenized and suspended in lysis buffer (1% NP-40, 0.05M HEPES (pH 7.5), 150mM NaCl, 1.5mM MgCl2, 1mM EGTA, 10% glycerol, 0.5% deoxycholate, 0.1% SDS, 100mM NaF, 0.2mM Na3VO4, 10mM sodium pyrophosphate), and lysates were analyzed by immunoblotting using suPAR antibody (R&D systems; Cat# AF534).
Recombinant mouse uPAR Fc-chimera injections in mice
Twenty μg recombinant mouse uPAR Fc-chimera (Sino Biological Inc. Cat#50160-M03H) (n=5) or PBS control (n=4) was injected by retro-orbital vein cannulation into 22 week old FVB wild-type mice (n=9). Results are presented as mean ± SEM. 2-tailed Student t test was used to analyze the difference between the experimental and control groups. A P value less than 0.05 was considered statistically significant.
Cohort Study Population
The NEPTUNE study is a multi-center prospective observational cohort study of patients with nephrotic syndrome established to define mechanistic classifications of disease and identify clinical, histological and genetic disease predictors 18. The study enrolled participants at 21 clinical sites starting in August, 2010 with a target enrollment of 450 patients with MCD, FSGS and MN. Inclusion criteria are ≥500mg/day of proteinuria from either a 24-hour or spot urine collection and clinically indicated renal biopsy. Exclusion criteria include evidence of other renal disease (e.g., lupus, diabetic nephropathy), prior solid organ transplant and life expectancy of <6 months. Participants whose biopsy was not consistent with MCD, FSGS or MN were retained in the study. Approximately 1/3 of this group had IgA nephropathy. The protocol was approved by the institutional review board at each patient recruiting site. Participants provided informed consent. This study included 241 patients who were enrolled in NEPTUNE as of July 2013, had a pathologic diagnosis of MCD, FSGS, MN or IgA nephropathy and had at least one plasma sample or 24 hour urine specimen available.
Data Collection
Detailed information regarding socio-demographics, medical history, and medication exposure were collected by subject interview and chart review, as previously described 18. Blood and urine specimens were collected at baseline and each follow-up visit. Plasma creatinine and urine protein/creatinine ratio were measured at a single, central laboratory. eGFR (mL/min/1.73m2) was calculated using the CKD-Epi formula 30 for participants ≥17 years old and the modified CKiD-Schwartz formula for participants ≤17 years old 31. Disease cohort (MCD, FSGS, MN and IgAN) was assigned after review of pathology material by the NEPTUNE histopathology committee. ESRD was defined as initiation of dialysis, receipt of kidney transplant or eGFR <15 mL/min/1.73m2. Complete remission was defined as UPCR <0.3mg/mg on 24 hour urine. Partial remission is defined as UPCR decline by 50% from baseline and ≤3.5mg/mg and eGFR >= 15 ml/min/1.73 m2.
Blood and 24 hour urine samples from all available visits occurring within the first 12 months of follow-up were analyzed for suPAR (pg/mL) concentration. Plasma suPAR were concentration was measured by enzyme-linked immunosorbent assay according to the manufacturer’s instructions (R&D systems; Cat#DUP00). All samples were assayed in duplicate, and pooled human plasma samples were included to assess variability. Plasma suPAR intra- and inter-assay coefficients of variation were 6.6% and 4.2%. Urine suPAR intra- and inter-assay coefficients of variation were 9.5% and 5.5%. Urine creatinine was assayed as described previously 32. Urine suPAR intra- and inter-assay coefficients of variation were 5.9% and 7.1%. Urine suPAR was normalized for creatinine concentration and expressed as urine suPAR/creatinine ratio (pg/mg). Laboratory test results were generated by personnel blinded to the clinical characteristics of the research subjects.
Statistical Analysis
Descriptive statistics, including mean and standard deviation (SD) for normally distributed variables, median and interquartile range (IQR) for skewed variables, and proportions for categorical variables were used to characterize baseline participant characteristics. Spearman’s rank correlation coefficient (ρ) was used to assess the relationship between baseline plasma suPAR concentration and eGFR, UPCR and urine suPAR concentration. Linear regression was used to assess the association of baseline plasma suPAR concentration and cohort, adjusted for demographics, eGFR and UPCR. Logistic regression was used to assess the association baseline plasma and urine suPAR concentrations with clinical outcome.
A mixed-effects model was fit to assess the association of eGFR and change in plasma suPAR concentration from baseline, adjusted for cohort, baseline suPAR, age, sex and UPCR. To test the hypothesis that suPAR is pathogenic in FSGS participants as opposed to all other cohorts, an interaction between cohort and change in suPAR was tested. This model was stratified by pediatric and adult participants due to the different estimating equations for eGFR as well as the different distributions of disease cohort (i.e. nearly all pediatric participants had either MCD or FSGS). The interaction between cohort and change in suPAR was of similar magnitude and significance when the pediatric and adult cohorts were modeled together. Analyses were performed using SAS, v9.2 (SAS Institute Inc.), with two-sided tests of hypotheses and p-value<0.05 as the criterion for statistical significance.
Acknowledgments
Support: Financial support for this work provided by the NIDDK Diabetic Complications Consortium (DiaComp, www.diacomp.org), grant DK076169 to LBH and by support to DN (DK087956) and to JS (DK007378). The Nephrotic Syndrome Study Network Consortium (NEPTUNE), U54-DK-083912, is a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through a collaboration between the Office of Rare Diseases Research (ORDR), NCATS, and the National Institute of Diabetes, Digestive, and Kidney Diseases. Additional funding and/or programmatic support for this project was provided by the University of Michigan, the NephCure Foundation and the Halpin Foundation.
The Nephrotic Syndrome Study Network investigators include: John Sedor, Katherine Dell, Case Western Medical Center, Cleveland, OH; Christine Sethna, Cohen Children’s Medical Center, Manhasset, NY; Gerald Appel, Pietro Canetta, Columbia University, New York, NY; Larry Greenbaum, Titi Ilori, Emory University, Atlanta, GA; Sharon Adler, Harbor UCLA Medical Center, Torrance, CA; Cynthia Nast, Cedars Sinai Health System; Alicia Neu, Michael Choi, Johns Hopkins Medical Institute, Baltimore, MD; Kalyani Perumal, John H. Stroger, Jr. Hospital of Cook County, Chicago, IL; Jeffrey Kopp, Kidney Disease Section, NIDDK, NIH, Bethesda, MD; Frederick Kaskel, Montefiore Medical Center, Bronx, NY; Howard Trachtman, Olga Zhdanova, New York University School of Medicine, New York, NY; Richard Lafayette, Stanford University, Stanford, CA; Fernando Fervenza, Marie Hogan, John Lieske, The Mayo Clinic, Rochester, MN; Crystal Gadegbeku, Duncan Johnstone, Temple University, Philadelphia, PA Daniel Cattran, Michelle Hladunewich, Heather Reich, University Health Network, Toronto, Ontario, Canada; Alessia Fornoni, Gaston Zilleruelo, Laura Barisoni, University of Miami Medical Center, Miami, FL; Matthias Kretzler, Debbie Gipson, Laura Mariani, Matthew Sampson, Peter Song, University of Michigan, Ann Arbor, MI; Patrick Nachman, Keisha Gibson, Susan Hogan, University of North Carolina at Chapel Hill, Chapel Hill, NC; Lawrecne B. Holzman, Kevin Meyer, University of Pennsylvania, Philadelphia, PA; Kevin Lemley, University of Southern California, Children’s Hospital, LA, CA; Kamalanathan Sambandam, Elizabeth Brown, University of Texas Southwest Medical Center, Dallas, TX; Peter Nelson, Sangeeta Hingorani, Katherine Tuttle, University of Washington, Seattle, WA.
Footnotes
Disclosure
The authors have no financial relationship with companies that might have interest in the information contained in this manuscript.
Supplementary information
Supplementary information is available at Kidney International’s website.
References
- 1.Thuno M, Macho B, Eugen-Olsen J. suPAR: the molecular crystal ball. Dis Markers. 2009;27:157–172. doi: 10.3233/DMA-2009-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei C, El Hindi S, Li J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. 2011;17:952–960. doi: 10.1038/nm.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blasi F, Sidenius N. The urokinase receptor: focused cell surface proteolysis, cell adhesion and signaling. FEBS Lett. 2010;584:1923–1930. doi: 10.1016/j.febslet.2009.12.039. [DOI] [PubMed] [Google Scholar]
- 4.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 5.Holst-Hansen C, Hamers MJ, Johannessen BE, et al. Soluble urokinase receptor released from human carcinoma cells: a plasma parameter for xenograft tumour studies. Br J Cancer. 1999;81:203–211. doi: 10.1038/sj.bjc.6690678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eugen-Olsen J, Andersen O, Linneberg A, et al. Circulating soluble urokinase plasminogen activator receptor predicts cancer, cardiovascular disease, diabetes and mortality in the general population. J Intern Med. 2010;268:296–308. doi: 10.1111/j.1365-2796.2010.02252.x. [DOI] [PubMed] [Google Scholar]
- 7.Trimarchi H. Primary focal and segmental glomerulosclerosis and soluble factor urokinase-type plasminogen activator receptor. World J Nephrol. 2013;2:103–110. doi: 10.5527/wjn.v2.i4.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hariharan S, Adams MB, Brennan DC, et al. Recurrent and de novo glomerular disease after renal transplantation: a report from renal allograft disease registry. Transplant Proc. 1999;31:223–224. doi: 10.1016/s0041-1345(98)01511-5. [DOI] [PubMed] [Google Scholar]
- 9.Xu Y, Berrou J, Chen X, et al. Induction of urokinase receptor expression in nephrotoxic nephritis. Exp Nephrol. 2001;9:397–404. doi: 10.1159/000052638. [DOI] [PubMed] [Google Scholar]
- 10.Wei C, Moller CC, Altintas MM, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med. 2008;14:55–63. doi: 10.1038/nm1696. [DOI] [PubMed] [Google Scholar]
- 11.Meijers B, Maas RJ, Sprangers B, et al. The soluble urokinase receptor is not a clinical marker for focal segmental glomerulosclerosis. Kidney Int. 2014;85:636–640. doi: 10.1038/ki.2013.505. [DOI] [PubMed] [Google Scholar]
- 12.Wei C, Trachtman H, Li J, et al. Circulating suPAR in two cohorts of primary FSGS. J Am Soc Nephrol. 2012;23:2051–2059. doi: 10.1681/ASN.2012030302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bock ME, Price HE, Gallon L, et al. Serum soluble urokinase-type plasminogen activator receptor levels and idiopathic FSGS in children: a single-center report. Clin J Am Soc Nephrol. 2013;8:1304–1311. doi: 10.2215/CJN.07680712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinha A, Bajpai J, Saini S, et al. Serum-soluble urokinase receptor levels do not distinguish focal segmental glomerulosclerosis from other causes of nephrotic syndrome in children. Kidney Int. 2014;85:649–658. doi: 10.1038/ki.2013.546. [DOI] [PubMed] [Google Scholar]
- 15.Wada T, Nangaku M, Maruyama S, et al. A multicenter cross-sectional study of circulating soluble urokinase receptor in Japanese patients with glomerular disease. Kidney Int. 2014;85:641–648. doi: 10.1038/ki.2013.544. [DOI] [PubMed] [Google Scholar]
- 16.Franco Palacios CR, Lieske JC, Wadei HM, et al. Urine but not serum soluble urokinase receptor (suPAR) may identify cases of recurrent FSGS in kidney transplant candidates. Transplantation. 2013;96:394–399. doi: 10.1097/TP.0b013e3182977ab1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cathelin D, Placier S, Ploug M, et al. Administration of Recombinant Soluble Urokinase Receptor Per Se Is Not Sufficient to Induce Podocyte Alterations and Proteinuria in Mice. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2013040425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gadegbeku CA, Gipson DS, Holzman LB, et al. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int. 2013;83:749–756. doi: 10.1038/ki.2012.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harita Y, Ishizuka K, Tanego A, et al. Decreased glomerular filtration as the primary factor of elevated circulating suPAR levels in focal segmental glomerulosclerosis. Pediatr Nephrol. 2014 doi: 10.1007/s00467-014-2808-5. [DOI] [PubMed] [Google Scholar]
- 20.Huang J, Liu G, Zhang YM, et al. Plasma soluble urokinase receptor levels are increased but do not distinguish primary from secondary focal segmental glomerulosclerosis. Kidney Int. 2013;84:366–372. doi: 10.1038/ki.2013.55. [DOI] [PubMed] [Google Scholar]
- 21.Segarra A, Jatem E, Quiles MT, et al. Value of soluble urokinase receptor serum levels in the differential diagnosis between idiopathic and secondary focal segmental glomerulosclerosis. Nefrologia: publicacion oficial de la Sociedad Espanola Nefrologia. 2014;34:53–61. doi: 10.3265/Nefrologia.pre2013.Oct.12272. [DOI] [PubMed] [Google Scholar]
- 22.Kristensen P, Eriksen J, Blasi F, et al. Two alternatively spliced mouse urokinase receptor mRNAs with different histological localization in the gastrointestinal tract. J Cell Biol. 1991;115:1763–1771. doi: 10.1083/jcb.115.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Llinas P, Le Du MH, Gardsvoll H, et al. Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 2005;24:1655–1663. doi: 10.1038/sj.emboj.7600635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gårdsvoll H, Gilquin B, Le Du MH, et al. Characterization of the Functional Epitope on the Urokinase Receptor: COMPLETE ALANINE SCANNING MUTAGENESIS SUPPLEMENTED BY CHEMICAL CROSS-LINKING. Journal of Biological Chemistry. 2006;281:19260–19272. doi: 10.1074/jbc.M513583200. [DOI] [PubMed] [Google Scholar]
- 25.An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Degryse B, Resnati M, Czekay RP, et al. Domain 2 of the urokinase receptor contains an integrin-interacting epitope with intrinsic signaling activity: generation of a new integrin inhibitor. The Journal of biological chemistry. 2005;280:24792–24803. doi: 10.1074/jbc.M413954200. [DOI] [PubMed] [Google Scholar]
- 27.Luther T, Kotzsch M, Meye A, et al. Identification of a novel urokinase receptor splice variant and its prognostic relevance in breast cancer. Thromb Haemost. 2003;89:705–717. [PubMed] [Google Scholar]
- 28.Moeller MJ, Soofi A, Sanden S, et al. An efficient system for tissue-specific overexpression of transgenes in podocytes in vivo. American journal of physiology Renal physiology. 2005;289:F481–488. doi: 10.1152/ajprenal.00332.2004. [DOI] [PubMed] [Google Scholar]
- 29.Muzumdar MD, Tasic B, Miyamichi K, et al. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 30.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz GJ, Munoz A, Schneider MF, et al. New equations to estimate GFR in children with CKD. Journal of the American Society of Nephrology: JASN. 2009;20:629–637. doi: 10.1681/ASN.2008030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Interpretation and Techniques. 3. Lea & Febiger; Philadelphia: 1988. Clinical Chemistry. [Google Scholar]