

Abstract
Although recent studies have shown that adenosine-to-inosine (A-to-I) RNA editing occurs in microRNAs, its effects on tumor growth and metastasis are not well understood. We present evidence of CREB-mediated low expression of ADAR1 in metastatic melanoma cell lines and tumor specimens. Re-expression of ADAR1 resulted in the suppression of melanoma growth and metastasis in vivo. Consequently, we identified 3 miRs undergoing A-to-I editing in the low-metastatic melanoma but not in highly metastatic cell lines. One of these miRs, miR-455-5p has two A-to-I RNA editing sites. The biological function of edited miR-455-5p is different from the unedited form as it recognizes different set of genes. Indeed, w.t. miR-455-5p promotes melanoma metastasis via inhibition of the tumor suppressor gene CPEB1. Moreover, w.t. miR-455 enhances melanoma growth and metastasis in vivo while the edited form inhibits these features. These results demonstrate a previously unrecognized role of RNA editing in melanoma progression.
Introduction
Melanoma is the most aggressive type of skin cancer. In 2014, an estimated 76,100 new cases of melanoma were diagnosed in the United States, and 9,710 will result in death 1. One important step for progression to metastatic disease is the transition from radial growth phase (RGP) to the vertical growth phase (VGP) 2. Previous reports from our laboratory have shown that CREB regulates many important functions during this transition3, 4, including acting as a survival factor and increasing cell invasion by regulating MMP2, IL8, BCL2, MCAM/MUC18, and the tumor suppressor CYR615-8. Furthermore, CREB regulates other important transcription factors involved in melanoma progression such as, MITF and AP2α 9, 10. Herein, we identified an important and previously unknown target for CREB, the RNA-editing enzyme adenosine deaminase acting on RNA 1 (ADAR1).
RNA editing, as well as other post-transcriptional modifications, increases protein diversity from a limited set of genes and can create proteins with several different functions from the same pre-mRNA. In cancer cells, this process can promote tumor growth and progression11, 12. During RNA editing, a site-specific alteration in the RNA sequence occurs and is then copied to the DNA sequence, excluding capping, polyadenylation, or splicing 13. The most common form of RNA editing is adenosine-to- inosine (A-to-I) editing, which is mediated by the action of ADAR enzymes14-16. The translational machinery then reads the inosine as guanosine, leading to altered amino acid sequences, a shift in the codon reading frame, or, if the editing occurs in a non- coding region, a disruption in the stability of the RNA transcript 17-20. More recently, ADAR-mediated RNA editing has been shown to occur in regulatory RNAs such as miRNAs, which can interfere with miRNA biogenesis, alter their stability within the cell, or alter their target binding21-23.
In this study, we found that CREB negatively regulates ADAR1 and that ADAR1 inhibits melanoma tumor growth and metastasis. Moreover, we identified two ADAR1-mediated A-to-I RNA editing sites in miR-455-5p as well as binding targets for the wild-type and the A-to-I edited version of miR-455-5p. Indeed, wild-type but not the edited form specifically targets the tumor suppressor gene CPEB1. In addition, w.t. miR-455-5p enhances melanoma growth and metastasis in vivo, while the edited form inhibited these features. These results fill a key missing link in the mechanistic understanding of the role of RNA editing in the acquisition of the melanoma metastatic phenotype.
Results
ADAR1 expression decreases with melanoma progression
To further evaluate the role of ADAR1 in melanoma progression, we determined the ADAR1 protein expression levels in established highly metastatic human melanoma cell lines (TXM18, MeWo, WM2664, C8161, and A375SML1), a melanoma cell line with low metastatic potential (SB2), and normal melanocytes via Western blot analysis. ADAR1 expression was significantly lower in all the highly metastatic melanoma cell lines than in the SB2 cells and normal melanocytes (Fig. 1A, top panel). Collectively, these data demonstrate that ADAR1 expression is reduced in metastatic melanoma cell lines and in clinical metastatic melanoma specimens 24 during melanoma progression.
Figure 1. ADAR1 expression and function is lost in metastatic melanoma.
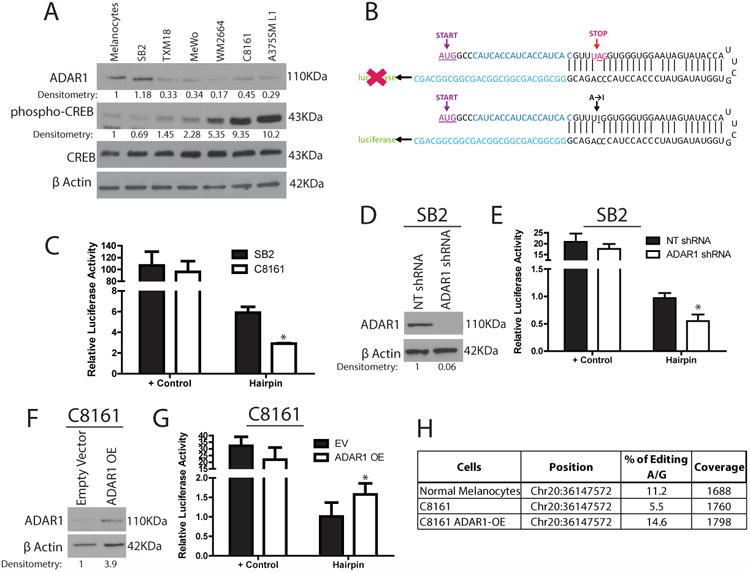
(A) Western blot analysis of a panel of melanoma cell lines shows decreased ADAR1 expression in highly metastatic cell lines. An inverse correlation is observed between ADAR1 expression and phospho-CREB expression, while total CREB expression is similar in all cell lines (data are representative of 3 biologically independent experiments). (B) Schematic representation of ADAR1 A-to-I RNA editing hairpin loop luciferase reporter. The stop codon leads to no luciferase expression (top panel), while A-to-I editing within the stop codon leads to increased expression, (bottom panel). (C) Luciferase activity from the hairpin luciferase construct decreases by more than 2 fold in C8161 cells compared to SB2 cells (right side of figure), *p<0.05. Positive control shows similar levels between both cell lines (left side of figure). Positive controls represent the constantly active form of the hairpin in which the stop codon was mutated to the edited form. (D) ADAR1 shRNA transduction leads to a ∼95% reduction in ADAR1 expression in SB2 cells, as shown by western blot analysis (data are representative of 3, biologically independent experiments). (E) Hairpin luciferase activity is decreased upon silencing of ADAR1 in SB2 cells, *p<0.05. (F) Western blot analysis of overexpression of ADAR1 in C8161 cells indicates a 4 fold increase in ADAR1 expression compared to empty vector control (data are representative of 3 biologically independent experiments). (G) Hairpin luciferase activity is increased after overexpression of ADAR1 in C8161 cells, *p<0.05. (H) Results of BLCAP editing by ADAR1. BLCAP is a known substrate for ADAR1 editing activity24, 33, 34. Metastatic melanoma C8161 cells have significantly lower editing rate (%) compared to normal melanocytes. Overexpressing ADAR1 in C8161 rescued the editing percentage by ∼3 fold. Each experiment in C, E, and G are the result of n=3 samples per group biologically independent; error bars represent s.d.. Statistical significance was determined by two-tailed Student t-test. Uncropped images of the blots are shown in Supplementary Figure 8.
RNA editing activity of ADAR decreases with melanoma progression
We next sought to determine whether the RNA editing activity of ADAR1 was affected. In this construct, the stop codon (UAG) prevents luciferase expression, while, an A-to-I editing event within the stop codon leads to luciferase expression (Fig. 1B). Using these luciferase constructs, we found that the ability of highly metastatic C8161 cells to perform A-to-I editing was lower (by 50%) than that of low metastatic potential SB2 cells (Fig. 1C).
Next, ADAR1 was stably silenced in the low-metastatic potential SB2 cells using a lentiviral ADAR1-targeted shRNA construct. Western blot analysis revealed that ADAR1 protein expression was knocked down by more than 95% compared with the non- targeting (NT) shRNA (Fig. 1D). Additionally, ADAR1 was overexpressed (ADAR1 OE) in highly metastatic C8161 cells (which expresses low levels of ADAR1). ADAR1 expression was nearly 4 times higher in the ADAR1 OE cells than in the empty vector control (Fig. 1F). Using the hairpin luciferase construct to assess the RNA editing ability of these cells, we confirmed that loss of ADAR1 in SB2 cells or overexpression of ADAR1 in C8161 cells either decreased or increased the A-to-I RNA editing activity, respectively (Fig. 1E, G). We also analyzed the editing rate of BLCAP mRNA (a known substrate for ADAR1 editing25) and confirmed that the C8161 cells had less than 50% ability to perform A-to-I editing when compared to normal melanocytes. Re-expression of ADAR1 in C8161 (ADAR1 OE) rescued their ability to perform A-to-I editing (Fig. 1H). These results confirmed that the RNA editing activity of ADAR1 is functional in melanoma cells and that the loss of ADAR1 in metastatic melanoma leads to a decrease in A-to-I RNA editing.
CREB acts as a negative regulator of ADAR1 expression in melanoma
Next, we sought to identify the mechanism(s) by which ADAR1 expression is regulated in melanoma. Gene expression profiling data indicated a 2- to 3-fold increase in ADAR1 expression after CREB silencing in metastatic melanoma cells. To validate these findings, a panel of melanoma cell lines was analyzed via Western blotting for both ADAR1 and phosphorylated CREB expression. The data revealed an inverse correlation between activated CREB and ADAR1 expression. In the normal melanocytes and low metastatic potential SB2 cells, ADAR1 expression was high, but phospho-CREB was low, whereas in the highly metastatic cell lines, phospho-CREB was high and ADAR1 expression was low (Fig. 1A). Next, we stably silenced CREB using shRNA in the highly metastatic C8161 and MeWo melanoma cell lines. CREB was knocked down by more than 95% in both cell lines compared with the NT control (Fig. 2A). Silencing of CREB in these cells resulted in a 3- to 5-fold upregulation of ADAR1, thus corroborating the cDNA expression data (Figure 2A). To confirm that these findings were specific to CREB and not an off-target effect of the shRNA, CREB expression was rescued in both silenced cell lines. Rescue of CREB indeed restored CREB expression and resulted with decreased ADAR1 expression in both cell lines (Fig. 2B). Silencing of CREB in C8161 or overexpressing CREB in SB2 cells did not affect the expression of ADAR2 in these cells (Supplemental Fig. 1).
Figure 2. CREB negatively regulates ADAR1 expression.
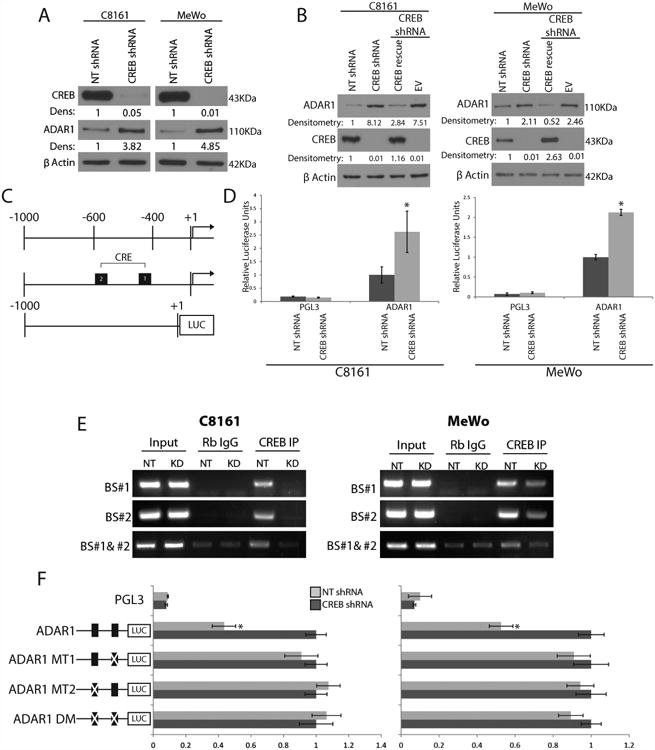
(A) Western blot analysis of shRNA CREB-transduced C8161 and MeWo cells show a 3-5 fold increase in ADAR1 expression as compared to NT shRNA controls (data are representative 3, biologically independent experiments). (B) Rescue of CREB expression in the CREB- silenced cells results in downregulation of ADAR1 expression in both C8161 and MeWo cells (data are representative of 3 biologically independent experiments). (C) Schematic representation of the ADAR1 promoter region fused to the luciferase reporter gene and its predicted CRE binding sites. (D) ADAR1 promoter driven luciferase expression increased by 2 fold after CREB silencing as compared to NT control, *p<0.01 (n=3 biologically independent samples per group, statistical significance was determined by two-tailed Student t-test; error bars represent s.d.). (E) CHIP analyses showed no binding of CREB to the ADAR1 promoter at either CRE binding site after CREB silencing in either C8161 or MeWo cell lines. IgG antibodies were used as negative controls. Input DNA was used as a loading control (data are representative of 3, biologically independent experiments). Statistical significance was determined by two- tailed Student t-test. (F) Schematic representation of the ADAR1 promoter point mutations is depicted left of the panel. Luciferase activity driven by the ADAR1 promoter increased in the NT shRNA group when mutations were made at either or both of the CRE binding sites, *p<0.01 (n=3 biologically independent samples per group, statistical significance was determined by two-tailed Student t-test; error bars represent s.d.). Uncropped images of blots and gels are shown in Supplementary Figure 8.
The promoter region of ADAR1 contains two CREB-binding sites within 600 base pairs (bp; -403 and -549) from the translation initiation site A (Fig. 2C). CREB silencing increased ADAR1 luciferase promoter activity by 2 to 3 fold in both cell lines, indicating regulation at the transcriptional level (Fig. 2D). Binding of CREB at positions -403 and -549 was analyzed by CHIP. Decreased binding of CREB was detected at both binding sites following CREB silencing in both cell lines (Fig. 2E). Single-point mutations were made at the two CREB binding sites individually or in combination in the ADAR1 promoter luciferase construct. Mutations in either of the CREB binding sites individually or together led to an increase in luciferase expression in the NT shRNA group to levels similar to those in the CREB shRNA (Figure 2F). Taken together, these data reveal a new role for CREB: negative regulation of ADAR1 and RNA editing.
Suppression of ADAR1 contributes to tumorigenicity and metastatic potential of melanoma
To elucidate the role of ADAR1 in melanoma tumorigenicity, we used the ADAR1- silenced SB2 cells and the ADAR1 OE C8161 cells (Fig. 1D and F) and analyzed their abilities to produce subcutaneous tumors and experimental lung metastasis in vivo. When ADAR1-silenced SB2 cells were injected subcutaneously into nude mice, a significant increase in tumor growth was observed compared with NT shRNA SB2 cells (Fig. 3A). In addition, when ADAR1 OE C8161 cells were injected subcutaneously into nude mice, tumor growth was significantly inhibited compared with control mice (Fig. 3C). Additionally, when ADAR1 KD SB2 cells were injected intravenously (IV), an increase in the median number of experimental lung metastases was observed compared with the NT shRNA control (ADAR KD, 37, NT shRNA, 2; p<0.01) (Fig. 3B). Overexpression of ADAR1 in C8161 cells injected IV led to a significant decrease in the median number of lung metastases compared with the control mice that received the empty vector p<0.01 (Fig. 3D).
Figure 3. ADAR1 expression leads to decreased melanoma tumor growth and metastasis.
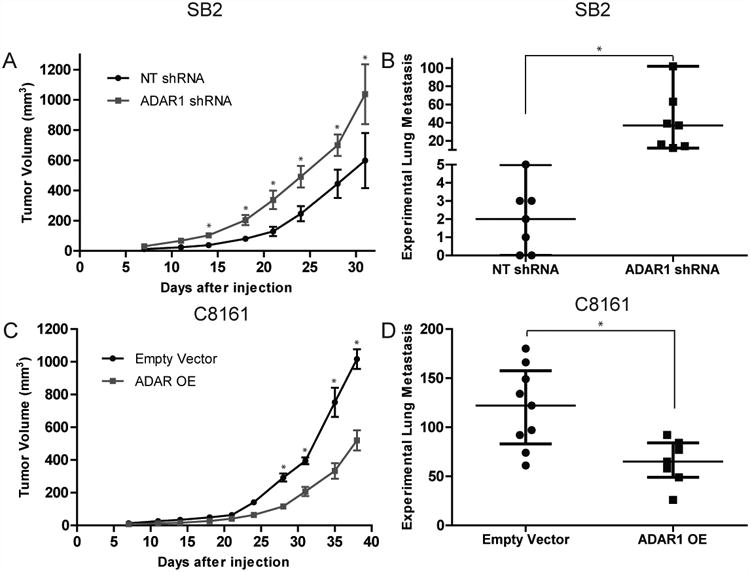
(A) Effect of ADAR1 silencing in SB2 cells on subcutaneous tumor growth. ADAR1 silencing led to a significant increase in tumor growth, *p<0.05 (n=10 mice in each group. Two-tailed Student t-test; error bars represent s.d.). (B) Effect of ADAR1 silencing on metastatic potential of melanoma cells. An increase in experimental lung metastasis was observed in mice injected with ADAR1-silenced SB2 cells as compared to NT shRNA injected control mice, *p<0.01 (n=7 mice in each group. Statistical significance was determined by two-tailed Student t-test; error bars represent s.d.). (C) Effect of ADAR1 overexpression in C8161 cells on subcutaneous tumor growth. ADAR1 overexpression led to a significant decrease in tumor growth, *p<0.05 (n=10 mice in each group. Two-tailed Student t-test; error bars represent s.d.). (D) Effect of ADAR1 overexpression on metastatic potential of melanoma cells. A significant decrease in experimental lung metastasis was observed in mice injected with ADAR1 overexpressing C8161 cells as compared to empty vector injected control mice, *p<0.01 (Empty vector, n=9 mice; ADAR1 OE, n=7 mice). Statistical significance by two-tailed Student t-test; error bars represent s.d..
ADAR1 expression inhibits spontaneous metastasis
We next determined the role of ADAR1 in spontaneous melanoma metastasis. To that end, another murine model was used whereby melanoma cells were injected into the skin ridge of the external ear, and then local lymph node metastasis were monitored. SB2 and C8161 melanoma cells were first transduced with the luciferase gene, and then ADAR1 was silenced or overexpressed. C8161 Luc/empty vector or Luc/ADAR1 OE and SB2 Luc/NT shRNA or Luc/ADAR1 shRNA transduced melanoma cells were injected into the skin ridge of the external ear. Overexpression of ADAR1 in C8161 melanoma cells significantly decreased tumor growth (Fig 4A). Silencing ADAR1 in SB2 cells resulted in increased tumor growth (Fig. 4B). The tumors were then completely resected along with the ear, and metastases to the regional lymph nodes were monitored. In C8161 melanoma cells, 6/8 parental and 7/8 Luc/Empty Vector tumors generated regional lymph node metastasis. ADAR1 OE in C8161 melanoma cells decreased the incidence of lymph node metastasis (2/8 mice) (Fig. 4C, Supplemental Fig. 2). Of the mice injected with low-metastatic potential SB2 cells (both the SB2 parental and Luc/NT shRNA transduced groups), none (0/8) developed lymph node metastasis. However, in the Luc/ADAR1 silenced group, 2/8 mice developed lymph node metastasis (Fig. 4C, Supplemental Fig. 2). Collectively, these results demonstrated that loss of ADAR1 directly contributed to an increase in melanoma growth and metastasis.
Figure 4. Spontaneous metastasis of SB2 and C8161 melanoma Cells.
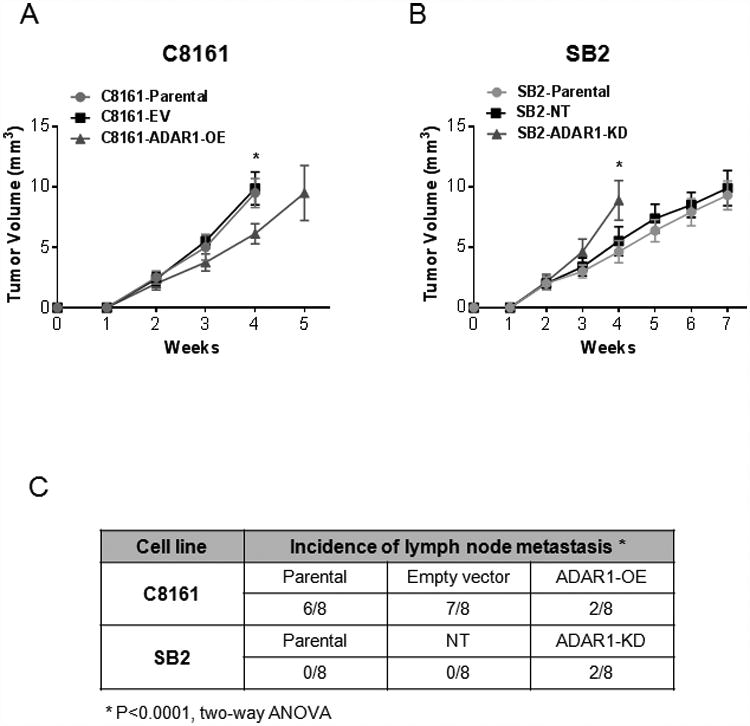
(A) Tumor growth in the skin ridge of the external ear is reduced after overexpression of ADAR1 in C8161 melanoma cells compared to the empty vector control (n=8 mice in each group; statistical significance was determined by two-way ANOVA test, *p<0.0001; error bars represent s.d.). (B) SB2 cells grow at a faster rate in the skin ridge of the external ear when ADAR1 is silenced. Measurements of tumor growth from groups C8161-EV and SB2-ADAR1-KD were stopped on week 4 due to tumor size (n=8 mice in each group. Statistical significance was determined by two-way ANOVA test, *p<0.0001; error bars represent s.d.). (C) Overexpression of ADAR1 in C8161 melanoma cells reduced the number of lymph node metastases while silencing ADAR1 allowed for two SB2 tumors to locally metastasize to regional lymph nodes. (n=8 mice in each group; two-way ANOVA test; *p<0.0001; error bars represent s.d.).
miRNA-455-5p is edited by ADAR1
We next sought to determine how loss of ADAR1 in metastatic melanoma affects A-to-I RNA editing in miRNAs and whether the altered editing affects their function and contributes to melanoma metastasis. First, to identify potentially edited miRNAs, two separate miRNA expression arrays were analyzed: data from the array of SB2 cells after ADAR1 silencing and from C8161 cells after CREB silencing (Fig. 2A). We identified changes in the expression of 131 miRNAs following ADAR1 silencing (Supplementary Table 3 at Nemlich et al.24) and changes in 239 miRNAs after CREB silencing (Supplemental Fig 3A). Only 12 were common in both arrays (Supplemental Table 1 marked in yellow, Supplemental Fig. 3A). Next, we sequenced the pri-miRNA of these 12 identified miRNAs using high-throughput sequencing. Among these 12 miRNAs, A-to-I RNA editing was identified in miR-378-3p, miR-324-5p and miR-455-5p (Supplemental Fig. 3B highlighted in yellow). We will concentrate on miR-455-5p, as two ADAR1-mediated A-to-I RNA editing sites were identified (Fig. 5A). The A-to-I RNA editing sites in miR-378-3p and miR-324-5p are shown in Supplemental Fig 4. SB2 cells transduced with ADAR1 shRNA had a decrease in the guanine peak at both sites compared with the NT shRNA control, indicating a decrease in A-to-I editing (Fig. 5B, black peak under arrows). ADAR1 OE C8161 cells showed an increase in the guanine peak at both sites compared with empty vector control, indicating an increase in A-to-I editing (Fig. 5C). After confirming that the changes in RNA editing were ADAR1 dependent, we determined whether the changes in RNA editing were also CREB dependent. To that end, CREB-silenced C8161 and CREB-silenced cells transduced with a CREB rescue vector were sequenced. Indeed, upon silencing of CREB, we observed an increase in A-to-I RNA editing at both sites, and upon rescue of CREB expression, a decrease in editing was found at these sites back to levels similar to those in the NT shRNA control (Fig. 5D). Taken together A-to-I RNA editing occurs in the non-metastatic SB2 cells but not in the C8161 metastatic cells. No editing was found at the DNA level.
Figure 5. miR-455-5p is edited by ADAR1 at two A-to-I RNA editing sites.
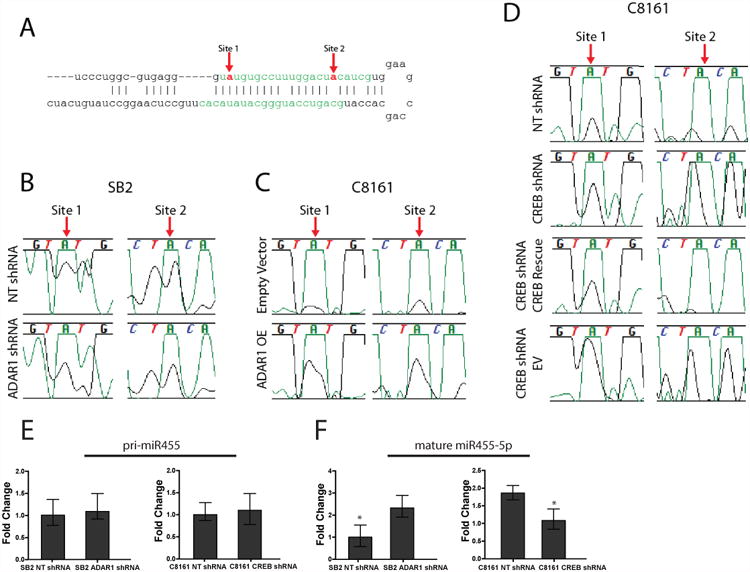
(A) miR-455-5p with mature miRNA sequence highlighted in green. The red arrows indicate the RNA editing sites. (B) RNA sequencing data indicates a decrease in A-to-I editing in SB2 cells after ADAR1 silencing. (C) An increase in RNA editing is observed in C8161 cells when ADAR1 is overexpressed. (D) A-to-I editing is increased upon CREB silencing and is returned to NT levels upon rescue of CREB expression in C8161 cells. In B-D green peak represents Adenosine and black peak represents Guanosine. (E) The pri-miR-455 expression is not changed after ADAR1 silencing in SB2 cells (left panel) or CREB silencing in C8161 cells (right panel). (F) Silencing ADAR1 in SB2 cells results in increased mature miR-455-5p expression (left panel) while silencing CREB in C8161 cells causes a decrease in mature miR-455-5p expression (right panel), *p<0.01. Data in E and F are the mean of n=3 biological independent samples. Statistical significance for E and F was determined by two-tailed Student's t-test; error bars represent s.d..
To determine whether RNA editing in miR-455-5p affects the microRNA biogenesis, pri- miRNA and mature levels were measured in SB2 ADAR1 KD and C8161 CREB KD cells using qRT-PCR. When pri-miRNA-455 levels were analyzed, no significant differences were observed in either SB2 or C8161 cells after ADAR1 KD or CREB KD, respectively (Fig. 5E). However, when the levels of mature miR-455-5p were analyzed, an increase in the mature form was seen in the SB2 ADAR1 KD, and a decrease was observed in the C8161 CREB KD cells compared with the NT controls, suggesting that ADAR1-mediated RNA editing may affect miR-455 biogenesis or could act in an RNA editing–independent role to affect its biogenesis (Fig. 5F). Therefore we next evaluated binding of Drosha and Dicer to miR-455. Indeed, the amount of miR-455 bound to Drosha and Dicer inversely correlated with ADAR1 expression (Supplemental Fig. 5). Silencing ADAR1 in SB2 melanoma cells induces the binding of Dicer to pre-miR-455, whereas overexpressing ADAR1 in C8161 reduces the amount of bound pre-miR-455 to Dicer (Supplemental Fig. 5A, B respectively). Pri-miR-455 expression after pulldown of Drosha demonstrated similar results (Supplemental Fig. 5C, D). We concluded that ADAR1 reduces the ability of pri-miR-455 to bind to Drosha and be processed to mature miR-455-5p.
The function of miRNA-455 is altered after A-to-I RNA editing
To compare the functions of wild-type and edited miR-455 (EDmiR-455), we created a set of lentiviral expression vectors overexpressing either wild-type miR-455 (WTmiR-455), miR-455 edited at site 1 only (ED1miR-455), miR-455 edited at site 2 only (ED2miR-455), or miR-455 edited at both sites 1 and 2 (DEDmiR-455) as well as an antagomir to silence miR-455 (anti-miR-455). These vectors were then transduced into ADAR1 KD SB2 cells, and qRT-PCR was performed to confirm that the expression of the experimentally manipulated miR-455 caused changes in miR-455-5p expression (Supplemental Fig. 6A). RNA was isolated and submitted for cDNA gene expression profiling to identify potential miR-455-5p targets. Targetscan was then used to identify the genes in the wild-type overexpressing group that had potential miR-455-5p binding sites in their 3′UTR. Both data sets were crossed. One of the genes that was downregulated in the wild-type group, with two miR-455-5p binding sites in its 3′UTR, was the tumor suppressor CPEB1. The main function of CPEB1 is to activate dormant mRNAs by polyadenylating them26, 27. qRT-PCR and Western blotting were used to assess CPEB1 expression in the C8161 ADAR1 OE and SB2 ADAR1 KD cells (Fig. 6A, B). When ADAR1 was overexpressed in C8161 cells (leading to increased EDmiR-455 and less WTmiR-455), CPEB1 expression was increased by 2-fold at the mRNA level and 1.5-fold at the protein level, whereas when ADAR1 was silenced in SB2 cells (leading to increased WTmiR-455), CPEB1 expression was decreased by about 70% at the mRNA level and 80% at the protein level, confirming that ADAR1 affects CPEB1 expression (Fig. 6A, B).
Figure 6. miR-455-5p regulates CPEB1 expression.
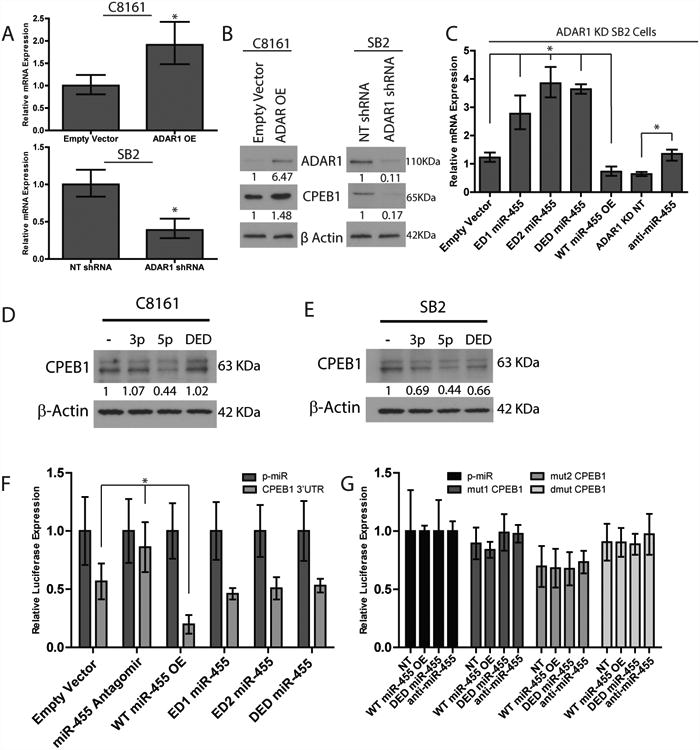
(A) rtPCR analysis of the mRNA levels of CPEB1. Overexpression of ADAR1 in C8161 cells resulted with increased CPEB1 expression (top panel). Silencing ADAR1 in SB2 cells resulted with decreased CPEB1 expression (bottom panel), *p<0.05 (n=3 RNA samples per group, biologically independent experiments). Statistical significance by two-tailed Student t- test; error bars represent s.d.. (B) Western blot analysis of CPE1B expression. A 1.5 fold increase in CPEB1 protein expression was observed in C8161 ADAR1 OE cells, while a reduction of about 80% was seen in SB2 ADAR1 KD cells (data are representative of 3, biologically independent experiments). (C) rtPCR analysis of CPEB1 expression after miR-455 manipulation. Upon overexpression of WT miR-455 a significant decrease in CPEB1 was observed, while expression of any of the three edited forms of miR-455 led to a significant increase of CPEB1 expression, *p<0.05. Silencing miR-455 resulted with increased CPEB1 expression, *p<0.05. n=3 RNA samples per group, biologically independent. Statistical significance by two-tailed student t-test; error bars represent s.d.. (D) Western blot analysis demonstrating a direct and specific binding of miR-455-5p but not miR-455-3p or miR-455-5p DED to the 3′UTR of CPEB1 in C8161 metastatic melanoma cells. CPEB1 is downregulated by ∼60% by miR-455-5p and not by the edited form or miR-455-3p (data are representative of 3 biologically independent experiments). (E) In SB2 cells, a significant higher downregulation of CPEB1 was observed after transfection with miR-455-5p WT compared to miR-455-5p DED or miR-455-3p (data are representative of 3 biologically independent experiments). (F) Luciferase expression increases upon expression of anti- miR-455-5p, and decreases when wild-type miR-455-5p is overexpressed, *p<0.01. No change was observed when the edited miR-455-5p was overexpressed, as compared to control (n=3 biologically independent samples). (G) Luciferase activity of the mutational analysis of the two miR-455-5p binding sites in the 3′UTR of CPEB1. No significant differences were observed between any of the experimental groups (n=3 biologically independent samples). Statistical significance by two-tailed student t-test, error bars represent s.d. for F and G. Uncropped images of blots in B, D, and E are shown in Supplementary Figure 8.
Next, we determined whether WTmiR-455 is responsible for the change in CPEB1 expression. Indeed, when WTmiR-455 was overexpressed, CPEB1 expression was decreased by 1.7-fold, whereas when any of the edited constructs were overexpressed, CPEB1 expression was increased by at least 2-fold when compared to the empty vector control (Fig. 6C). Additionally, when miR-455 was silenced, we found a ∼2 fold increase in CPEB1 expression compared to the NT control (Fig. 6C, right 2 columns).
To show that CPEB1 regulation is specific to miR-455-5p and not to miR-455-3p, we performed a transient transfection with miR-455-5p WT, miR-455-5p DED and miR-455- 3p to both C8161 and SB2 parental cells. Results clearly show that in C8161, CPEB1 is downregulated by ∼60% when miR-455-5p WT was transfected into the cells, and not by the edited form or miR-455-3p (Fig. 6D). In SB2 we observed a significant higher downregulation of CPEB1 by miR-455-5p when compared to miR-455-3p and DED (Fig. 6E). Taken together these data demonstrate that miR-455-5p is biologically active and is involved in CPEB1 gene regulation.
We next cloned the 3′UTR of CPEB1 into the pmiR luciferase reporter construct, transfected it into manipulated miR-455 ADAR1 KD SB2 cells, and measured luciferase activity 48 hours later. Luciferase expression was significantly decreased when miR-455 WT was overexpressed and significantly increased when anti-miR-455 was expressed compared with the empty vector control. In addition, when any of the three edited miR-455s were overexpressed, levels were similar to those in the empty vector control, indicating that these edited miR-455s were unable to bind to the 3′UTR and suppress CPEB1 (Fig. 6F). To confirm these findings, the two miR-455-5p binding sites within the CPEB1 3′UTR (mut1CPEB1, mut2CPEB1, and dmutCPEB1) were mutated to assess the binding of miR-455-5p and luciferase expression. Luciferase expression in WTmiR-455 cells was similar to that in the empty vector control in all three mutated versions of CPEB1 (Fig. 6G). Taken together, these data confirm that wild-type miR-455-5p can suppress CPEB1 expression, but the edited version cannot. Thus, the effect of the unedited miR-455-5p is mediated by its sequence-dependent ability to target CPEB1. To further confirm the role of CPEB1 as a tumor suppressor, we next rescued the expression of CPEB1 in SB2 ADAR1 KD cells. Rescue of CPEB1 in these cells reduced their ability to invade through matrigel coated filters, without affecting their proliferation rate (Supplemental Fig. 6B, C). Further analysis to determine potential binding sites for the edited miR-455-5p in the 3′ UTR of the targets was performed. Three genes that had potential binding sites, RHO-C, MDM4, and integrin α2 were identified, all of which have known tumor promoting functions (Supplemental Fig. 7), and suggest that a different set of genes are regulated by the edited vs. the wild-type form of miR-455-5p.
miR-455 contributes to melanoma tumor growth and metastasis
To elucidate the role of miR-455 in melanoma growth in vivo, SB2 and C8161 cells (empty vector, WTmiR-455, DEDmiR-455, and anti-miR-455) were injected subcutaneously into nude mice. A significant increase in tumor growth was observed in mice injected with WTmiR-455 SB2 cells but not in mice injected with C8161 cells. (Fig. 7A, C). In contrast, injection of cells with either the DEDmiR-455 or anti-miR-455 resulted in a significant decrease in tumor growth in mice injected with SB2 cells and in mice injected with C8161 cells compared with control cells (Fig. 7A, C). In addition, in mice injected with SB2 cells and in mice injected with C8161 cells, WTmiR-455 overexpression led to increased incidence of experimental lung metastasis compared with EV controls. Overexpression of either DEDmiR-455 or anti-miR-455 led to a significant decrease in lung metastasis in mice injected with either cell type (Fig. 7B, D). Taken together, these data show that overexpression of WTmiR-455 results in increased tumor growth and incidence of lung metastasis, whereas overexpression of the edited form of miR-455 results in diminished tumor growth and lung metastasis.
Figure 7. miR-455 overexpression leads to increased melanoma tumor growth and metastasis.
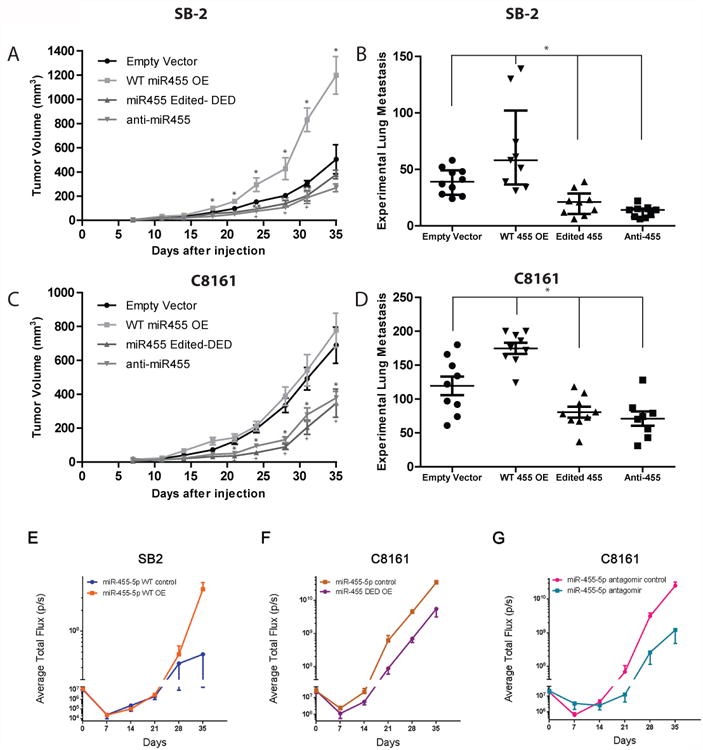
(A) Effect of manipulation of miR-455 on subcutaneous tumor growth in SB2 cells. Overexpression of wild-type miR-455 results in a significant increase in tumor growth as compared to empty vector control, *p<0.001, while silencing miR-455 or overexpressing the double edited form of miR-455 led to a significant decrease in tumor growth, + or **, respectively, p<0.05. Each group n=10 mice, statistical significance by two-tailed Student's t-test, *p<0.001, +p<0.05, and **p<0.05; error bars represent s.d. (B) Effect of miR-455 manipulation in SB2 cells on metastatic potential. Overexpression of wild-type miR-455 led to a significant increase in experimental lung metastasis, while silencing miR-455 or overexpressing the edited form of miR-455 resulted with decreased lung metastases, *p<0.05. Empty vector group n=10 mice, WT 455 OE, Edited 455, and anti-455 n=9 mice per group. Statistical significance by two- tailed Student's t-test; error bars represent s.d.. (C) Overexpression of wild-type miR-455 did not result in a significant increase in the already highly tumorigenic C8161 cells. While silencing miR-455 or overexpressing the edited form of miR-455 led to a significant decrease in tumor growth, * or +, respectively, p<0.01. Each group n=10 mice. Statistical significance by two-tailed Student t-test; error bars represent s.d. (D) Overexpression of wild-type miR-455 led to a significant increase in experimental lung metastasis, while silencing miR-455 or overexpressing the edited form of miR-455 resulted in decreased lung metastasis, *p<0.05. Empty vector, WT miR-455 OE, and miR-455 Edited-DED have n=9 mice in each group; Anti-miR-455 n=8 mice per group. Statistical significance by two-tailed Student t-test; error bars represent s.d. (E-G) SB2 or C8161 luciferase labeled cells were injected i.v and 3 days later the mice were injected i.v with miR-455-5p, double edited or antagomir encapsulated in DOPC particles (E) Delivery of miR-455-5p WT to mice harboring SB2 cells, increased their metastatic potential (p=0.0163). (F) Delivery of double edited miR-455-5p to mice harboring C8161 cells decreased their metastatic potential (p=0.0001) as did the antagomir of miR-455-5p (p=0.0203) shown in (G). Each group (D, F, and G) n=6 mice. Statistical analyses by two-way ANOVA, error bars represent S.E.M.
To further study the specific role of miR-455-5p in melanoma metastasis, luciferase labeled SB2 and C8161 were injected i.v into nude mice. Three days later the mice were systemically treated with nano-liposomes carrying different miR-455-5p sequences. SB2 cells were treated with miR-455-5p WT and C8161 were treated with miR-455-5p WT antagomir and miR-455-5p DED. miR-455-5p WT delivery resulted with an increased ability of SB2 cells to form lung metastasis (Fig. 7E). In contrast systemic delivery of miR-455-5p DED caused a reduction in lung metastases of C8161cells as did the delivery of antagomir to miR-455-5p (Fig. 7F, G). Collectively these results demonstrate that wild-type miR-455-5p has an opposite effect on melanoma experimental metastasis than the edited form, and that the miR-455-5p is biologically active in melanoma cells.
Discussion
We have previously reported that loss of ADAR1 in melanoma contributes to melanoma growth by modulating the processing of several miRs independent of its RNA editing24. Herein, we extended these observations to demonstrate that loss of ADAR1 in metastatic melanoma cells causes hypo A-to-I miRs editing, leading to changes in mRNA selection. Taken together, we propose that ADAR1 contributes to the melanoma metastatic phenotype by RNA editing-dependent and independent mechanisms.
In the current study, we identified RNA editing in 3 miRNAs; miR-324-5p, miR-378-3p, and miR-455-5p. Two ADAR1-mediated RNA-editing sites were identified in miR-455-5p, one at mature position 2 and one at mature position 17. Alon et al. 25 identified the editing site for ADAR2 at position 17 in miR-455-5p in glioblastoma cell lines; our data confirmed their finding and revealed an additional editing site in melanoma cells.
ADAR1-mediated RNA-editing of miRNA has been shown to affect the biogenesis of miRNAs or their binding targets19, 28. When the mature sequence was analyzed, we observed an increase in miR-455-5p WT when ADAR1 was silenced and a decrease in miR-455-5p WT when CREB was silenced. One possible explanation for these changes was detailed in a recent study by Ota et al., who showed that ADAR1 forms a complex with Dicer to regulate miRNA processing and/or their gene-silencing abilities 29. Therefore, loss of ADAR1 expression could lead to abnormal miRNA processing or functioning and changes in total mature levels of miR-455-5p. Another explanation is that the edited form of miR-455-5p is not recognized or cleaved by either Dicer or Drosha for processing, as was previously shown with miR-151 and miR-142, respectively 28, 30. We found that the amount of miR-455 bound to Dicer and Drosha was inversely correlated with ADAR1 expression.
The importance of RNA editing in melanoma growth and metastasis were verified by assessing tumor growth and metastatic capabilities after overexpression or silencing of the wild-type and overexpression of the edited form of miR-455. Indeed, systemic delivery of miR-455-5p WT, its edited form and antagomir in vivo by means of nano particles validated our observations and clearly demonstrated that miR-455-5p is biologically active in melanoma cells.
Using a cDNA microarray and ingenuity pathway analysis, we identified 30 known tumor suppressor genes that were downregulated when wild-type miR-455-5p was overexpressed. Predicted targets for miR-455-5p from TargetScan identified 6 genes as potential targets for miR-455-5p, and 3 of them (CPEB1, JDP2, and VCAN) were on the list of known tumor suppressors (Supplemental Table 2). CPEB1 acts as a tumor suppressor in several cancers, including gastric and thyroid KO cancer31, 32. When ADAR1 was overexpressed, CPEB1 levels also increased, presumably owing to decreased wild-type miR-455-5p. Moreover, CPEB1 is regulated by miR-455-5p and not by miR-455-3p. On the other hand, the edited form of miR-455-5p targets oncogenes such as ITGA2, MDM4 and RhoC.
In summary, herein we provide an evidence of CREB-mediated hypo-expression of ADAR1 in metastatic melanoma. This loss of ADAR1 resulted in differential A-to-I RNA editing in melanoma cell lines, particularly in miR-455-5p. In this model (Fig 8), activation of CREB in metastatic melanoma cells leads to downregulation of ADAR1 expression. Subsequently, there is an accumulation of wild-type miR-455-5p in metastatic cells and more of the edited form in the non-metastatic cells. Wild-type miR-455-5p contributes to melanoma growth and metastasis via downregulation of the tumor suppressor gene CPEB1. At this stage however, the clinical relevance of this editing pathway in human melanoma is yet to be determined.
Figure 8. Model of ADAR1 RNA editing in melanoma progression.
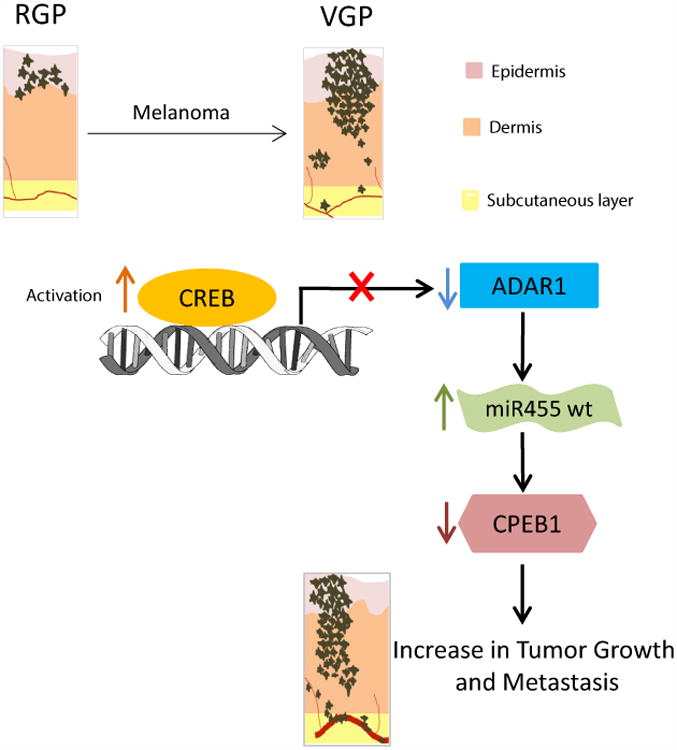
Overall model of the study showing the mechanistic understanding of how a loss of CREB-mediated ADAR1 in metastatic melanoma cells causes a shift towards the accumulation of wild- type miR-455-5p, thus contributing to melanoma growth and metastasis. CREB is activated in the transition from RGP to VGP. Activation of CREB leads to downregulation of ADAR1 in metastatic melanoma cells. Subsequently, miR-455-5p is edited in the non-metastatic melanoma cells. Wild-type miR-455-5p preferentially binds to 3′UTR of CPEB1 and suppresses its expression, thus, contributing to melanoma growth and metastasis.
Online Methods
Cell lines and culture conditions
All of the human melanoma cell lines used in our studies, were maintained in Eagles minimum essential medium (MEM) supplemented with 2mM glutamine, 1% non- essential amino acids and 10% fetal bovine serum (FBS). The 293FT cells (Invitrogen) used to produce the lentiviral shRNA were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS according to the manufacturer's instructions. All cell lines used in our studies were tested prior to their usage for authentication by DNA fingerprinting using the short tandem repeat (STR) method.
Lentiviral shRNA
CREB-targeting shRNA (target sequence: 5′-GAGAGAGGTCCGTCTAATG-3′), ADAR1- targeting shRNA (target sequence: 5′-CTTCCTGTCAGA-3′), and a non-targetable shRNA (NT shRNA, target sequence 5′-TTCTCCGAACGTGTCACGT-3′) were designed with a hairpin and inserted into the pSIH lentiviral vectors and transfected into 293FT (human embryonic kidney) cells to generate lentiviral particles. The lentivirus system and cell transduction were generated as described previously8. To silence CREB, highly metastatic C8161 and MeWo cell lines plated at 70% confluency in 6-well plates were transduced with the virus. To silence ADAR1, SB2 cells were plated at 60% confluency in 6-well plates and transduced with the virus. After 16 h, the virus- containing medium was removed and replaced with normal growth medium. Transduced cells were sorted using green fluorescent protein.
Nontargetable CREB expression vector
The lentiviral CREB expression vector was developed as described above. Briefly, total RNA was extracted from A375SM cells, and the open reading frame (ORF) of CREB was amplified by PCR from the reverse transcription product with the following two primers: CreB-XbaF: 5′-GCTCTAGAATGACCATGGAATCTGGAGCCGAG-3′ and CreB- Cla-H3R: 5′-CCCAAGCTTatcgaTTAATCTGATTTGTGGCAGTAAAG-3′. To create a nontargetable CREB expression vector we, used the following oligonucleotides: CreB1B-ReF, 5′- GAAGCAGCACGAAAGAGAGAAGTGCGACTGATGAAGAACAGGGAAGCAG-3′; and CreB1B-ReR, 5′- CTGCTTCCCTGTTCTTCATCAGTCGCACTTCTCTCTTTCGTGCTGCTTC-3′. To rescue CREB expression in stably CREB-silenced cells, C8161 and MeWo CREB- shRNA or NT-shRNA cells were plated in 6-well plates and transduced with virus containing either the non-targetable CREB expression vector or an empty vector. After 48 h, the cells were expanded and selected. CREB expression was confirmed via Western blot analysis.
Western blot analysis
To detect the expression of CREB (rabbit mAb #9197L, clone 48H2, cell signaling), phospho-CREB (Ser133 rabbit Ab #9191s, cell signaling), ADAR1 (Rabbit Ab, SAB4200541, SIGMA), ADAR2 (rabbit pAb, GTX114237, genetex)or CPEB1 (Rabbit Ab 13583s, cell signaling), we loaded 20 μg of whole-cell protein lysates on SDS-PAGE and performed Western blotting as described previously8. Blots were incubated with primary antibodies (1:1000 CREB, phospho-CREB, CPEB1; 1:1000, anti-ADAR1). Densitometry was carried out using ImageJ software (NIH). All western blots analyses were performed in 3 independent experiments.
CHIP assay
CHIP assays were performed using ChIP-IT Express kit (Active Motif) according to the manufacturer's protocol. Briefly, cells were fixed with 1% formaldehyde, and the cross- linking reaction was stopped with 0.125 M glycine. The cells were pelleted and resuspended in a hypotonic buffer, and cell nuclei were isolated using a Dounce homogenizer. The chromatin was then sheared into 200 to1000-bp fragments by adding an enzymatic solution for 10 min at 37 °C. Fractions of the chromatin solutions were incubated overnight at 4 °C with either 3 μg of anti-CREB or IgG control antibodies cross-linked to magnetic beads. The immune complexes were then eluted from the magnetic beads, and proteins were reverse cross-linked at 65 °C for 2.5 h. Proteins were digested with 2 μl of proteinase K at 37 °C for 1 h, extracted in elution buffer, and analyzed via PCR. For binding site 1 a 151-bp fragment (primer sequences used were Forward: 5′-CCTTGGTCGTTTGACGAGAT-3′, and Reverse: 5′- GGAAAACAAAGGCACACAAAA-3′), for binding site 2 a 180bp fragment (Forward: 5′- TGCCTTTGTTTTCCTTTTGC-3′, and Reverse: 5′-AACTCCCGGCTTCAAGAGAT-3′) and for both binding sites a 317bp fragment (Forward: 5′- CCTTGGTCGTTTGACGAGAT-3′, and Reverse: 5′-AACTCCCGGCTTCAAGAGAT-3′) of the ADAR1 promoter was amplified by PCR. For miR-455-Dicer or miR-455-Drosha co-immunoprecipitation, melanoma cells were collected with immunoprecipitation buffer [50mM Tris-HCL (pH 7.4), 150mM NaCl, 0.05% IGEPAL, 1mM phenylmethylsulfonyl fluoride (PMSF), proteinase inhibitor cocktail, and RVP. Cells were then sonicated briefly for complete lysis and centrifuged. The supernatant was then immunoprecipitated with the Dicer antibody (1:100 dillution, Ab #14601, Abcam) overnight followed by incubation with magnetic protein G beads for 1h. After three washes with IP buffer, the RNA was collected with 50mM NaHCO3 in 1%SDS. TRIZOL reagent was used to purify the RNA. pri-miR-455 (Applied Biosystems) was used for the Drosha IP. All CHIP analyses were performed in 3 independent experiments.
Pri-miRNA sequencing
Total RNA was extracted from cells and reverse transcription was carried out. PCR was performed to amplify the pri-miRNA sequence from 10 different samples. The PCR product was then sent to Seqwright and subjected to Sanger sequencing. Upon return the sequence chromatograms were analyzed for editing sites. Primers used for pri-miR-455 sequencing were: Forward: 5′-CGAGCTTCCTTCTGCAGGT-3′ and Reverse: 5′- CACCACTGCCATCCCACA-3′.
Reporter constructs and luciferase activity analysis
The ADAR1 A-to-I RNA editing hairpin loop luciferase reporter was used as described previously35 The ADAR1 promoter luciferase assay was performed as follows. The ADAR1 promoter region (nucleotides −1000 to +65 from the transcription initiation site) was amplified from C8161 genomic DNA using the following primers, Forward: 5′- GGGGTACCAGCCTCGGTTTCTACACCTGC-3′; Reverse: 5′- CCGCTCGAGGGTTCAATTTCGCTTTCGTTTC-3′. The fragment was digested with KpnI and XhoI and ligated into the pGL3-basic vector (Promega). Analysis of transcription factor binding sites was performed using GENOMATIX software. Site-directed mutagenesis of the CRE sites, replacing CG of the GACGTCA CRE site with AT, was performed using the QuikChange II XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The CPEB1 3′UTR miRNA binding luciferase assay was performed as follows. The 3′UTR region of CPEB1 was was amplified from C8161 genomic DNA using the following primers, Forward: 5′- GGGCCCAAGCTTTCCCAAGGACAAGGGAAAATTG-3′; Reverse: 5′- GGACTAGTCACATGGTCCCACCATTATCCT-3′. The fragment was digested with HindIII and SpeI and ligated into the pmiR vector (Ambion). Site directed mutagenesis was performed using the QuikChange II XL site-directed mutagenesis kit (Stratagene) according to manufacturer's instructions. Transient transfections were performed using Lipofectin or Lipofectamin 2000 (Invitrogen) according to the manufacturer's instructions. In a 24-well plate, a total of 2.5 × 104 cells/well were transfected with 0.8 μg of the empty pGL3 expression vector or with 0.8 μg of the pGL3-ADAR1 or pGL3- ADAR1-promoter-mutant-containing firefly luciferase expression constructs. For each transfection, 2.5 ng of cytomegalovirus-driven Renilla luciferase reporter construct (pRL- CMV, Promega) was included. After 4 h, the transfection medium was replaced with serum-containing growth medium. After 48 h, the cells were harvested and lysed, and the luciferase activity was assayed using a Dual-Luciferase reporter assay system (Promega) according to the manufacturer's instructions. Luciferase luminescence (relative light intensity × 106) was measured with a LUMIstar microplate reader (BMG Labtech). The ratio of firefly luciferase activity to CMV-driven Renilla luciferase activity was used to normalize any differences in transfection efficiency among samples. All constructs were fully sequenced in both directions before use. All luciferase analyses were performed in 3 independent experiments.
BLCAP Sequencing
RNA was extracted from C8161, C8161 ADAR1-OE, and normal melanocytes cells using RNAqueous Phenol-free total RNA Isolation kit (Life Science Technologies). Double stranded cDNA was made using cDNA Synthesis System (Roche) kit using 10 μg of RNA input. PCR reactions were performed to amplify the region on BLCAP gene known to be edited (Exon 2 – on chromosome 20) using the following primers: 5′- CTCCCATTAGGTCGGTTCCT and 5′- AGCAGGTAGAAGCCCATGAA. Amplification parameters were as follows: 1 × [95°C-3′]; 35 × [94°C-0.5′, 55°C-0.5′, 72-1′]; 1 × [72°C- 5′]. Each replicate PCR amplicon (168 nt) was confirm in a 1% agarose gel. Ion Torrent libraries were made using the Ion Plus Fragment Library Kit where each 100ng PCR amplicon was prepared as an individual library by ligating to a unique Ion Xpress Barcode Adaptors following the manufacture's recommendations. The barcoded libraries were amplified for 5 cycles, and re-qualified libraries were pooled and templated using The Ion One Touch 2 and sequenced on The Ion Torrent Personal Genome Machine using a 318 V2 Chip following the manufacture's recommendations. Raw sequencing data were concatenated into one ‘reference’ file and indexed it using BWA. We aligned the sequences to this reference file using BWA36 align and measured the allele frequency of RNA editing sites in each sample.
Animals, tumor growth, and experimental metastasis
Female athymic BALB/c nude mice (National Institutes of Health, NCI, Frederick Cancer Research Institute) were housed in laminar flow cabinets under specific pathogen-free conditions and used at 8 weeks of age. The mice were maintained in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with the current regulations and standards of the U.S. Department of Agriculture, Department of Health and Human Services, National Institutes of Health, and our institutional regulations. The sample size was determined to have a 80% power with 95% confidence. In accordance with the Institutional Animal Care and Use Committee, when the largest dimension of a subcutaneous tumor reached 1.5 cm, the mice were euthanized in a CO2 chamber. Subcutaneous tumors were produced by injecting 5 × 105 C8161 cells or 1 × 106 SB2 cells (single-cell suspensions, >95% viability by a trypan blue exclusion test) in 0.2 ml of Hanks' buffered salt solution into the right flank of each mouse. Tumor growth was recorded twice weekly with a caliper and calculated as a × b2/2 mm3 (a, long diameter; b, short diameter). Mice were euthanized when the tumor volume reached 1.5 cm3 or when ulceration appeared. Each group included 10 mice. To determine metastatic potential, 1 × 106 tumor cells, processed as described above, were injected into the tail veins of mice (0.1 ml/mouse). Thirty days later for C8161 and sixty days later for SB2 cells, the mice were killed and autopsied. The lungs were removed and fixed in Bouin's fixative solution, and the macroscopic surface tumor nodules were counted. Each group included 7-9 mice. Excluded mice were those which died for unknown reason unrelated with tumor burden or lung metastasis. The experiments were not randomized. The investigators were not blinded to the group allocation during the experiments.
Spontaneous metastasis model
To determine the spontaneous metastatic potential of the cell lines, 2×105 cells of C8161 or 5×105 cells of SB2 luciferase expressing clones in 20μl Ca++/Mg++ free HBSS were injected into the skin ridge of the external ear. Mice were ear-tagged and imaged weekly via an IVIS 100 series system (Xenogen Corp., Alameda, CA). When the tumor reached 9-10 mm in diameter, 4-5 weeks later for C8161 and 4-7 weeks later for SB2, the ear was resected, and the ear canal was reconstructed. Mice were followed up to determine the development of regional lymph node metastasis by in vivo imaging and autopsy. Two-way ANOVA test was used for comparison of the results (at week 4 *p<0.0001).
In vivo imaging
For bioluminescence imaging, mice received an i.p. injection of 0.2 ml of 15 mg/ml D- luciferin under 1-2% inhaled isoflurane anesthesia. The bioluminescence signals were monitored using an IVIS 100 series system (Xenogen Corp., Alameda, CA) consisting of a highly sensitive cooled CCD camera. Living Image software (Xenogen Corp., Alameda, CA) was used to grid the imaging data and integrated the total bioluminescence signals in each boxed region. D-luciferin potassium salt (luciferin; Gold Bio Technology, Inc., St.Louis, MO), was dissolved in PBS and then filtered through a 0.22 μm membrane for sterilization. Two kinetic bioluminescent acquisitions were collected between 0 and 20 min after D-luciferin injection to confirm the peak photon emission recorded as the maximum photon efflux per second. Data were analyzed and quantified by using the total photon flux emission (photons/second) in the regions of interest.
In vivo liposomal delivery experiment
To determine whether siRNA mimics delivered by neutral liposomes could reach the experimental lung tumors, siRNA mimics were incorporated into DOPC liposomes as described previously37,38. The liposomes where injected intravenously, twice weekly starting 3 days after melanoma cell injections. The following miRNA mimics were purchased from Life Technologies: Human Mir-455-5p WT: UAUGUGCCUUUGGACUACAUCG, Costumed Mir-455-5p-DED: UGUGCGCCUUUGGACUGCAUCG, Human Mir-455 Inhibitor- antagomir (Cat#4464088, Life technologies), miRNA mimic inhibitor negative control (Cat#4464079, Life technologies), miRNA mimic negative control (Cat#4464060, Life technologies). Statistical significance by two-way ANOVA; S.E.M.
Delivery of miRNA-455 mimics in vitro
The mimics of miRNA-455 were delivered into 2 cell lines (C8161, and SB2 parental) by using Lipofectamine RNAiMAX following the manufacturer's instruction. 25pmol of miR-455 mimics were used for 0.7×106 cells in 6 well plates. Cells were collected 24 hours after transfection for relevant analyses for Western blot and qRT-PCR. The following mimics sequences of miRNA-455 were purchased from Life Technologies: Human Mir-455-5p: UAUGUGCCUUUGGACUACAUCG, Human Mir-455-3p: GCAGUCCAUGGGCAUAUACAC, Costumed Mir-455-5p-DED: UGUGCGCCUUUGGACUGCAUCG and miRNA mimic negative control (Cat#4464060, Life technologies)
Expression profiling studies
Total RNA was isolated from C8161 CREB shRNA or NT shRNA (for miRNA array) or from SB2 cells overexpressing wildtype or edited miR-455-transduced melanoma cells using the mirVana RNA isolation kit (Ambion). RNA quality was assessed using an RNA bioanalyzer chip (Agilent). For cDNA microarray, RNA was submitted to Phalynx Biotech Group for expression profiling and analysis. Gene expression analysis was carried out between the 2 samples. For miRNA array, the TaqMan Array Human MicroRNA Card Set v3.0 was used.
Matrigel invasion assay
Matrigel invasion assays were performed using Biocoat Matrigel invasion chambers (BD Biosciences). Briefly, 1.5×104 cells diluted in 500 μl of serum-free MEM were placed on top of the upper chamber of the Matrigel plate in triplicates. The lower chamber contained MEM supplemented with 20% FBS. Matrigel plates were incubated for 24 h at 37 °C. Hema3 stain set was used to stain the cells which migrated to the lower surface of the Matrigel filter (Fisher Scientific). Filters were glued on a microscope slide. Pictures from different fields where taken under the microscope and the stained cells were counted and statistically analyzed.
In vitro proliferation assay (MTT)
Five thousand cells were plated in each well of a ninety six well plates that were used in this experiment (12 repetitions for each sample). The cells that were plated were SB2 parental, SB2 SB2 ADAR1 knockdown, ADAR1 non-target control and SB2 ADAR1 knockdown + CPEB1 overexpression. The cells were cultured for 5 days in 10% FBS normal growth MEM medium. Cell growth was analyzed by the colorimetric MTT (3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay, which determines relative number of cells based on the conversion of MTT to formazan (which has a purple color) in viable cells. Each day after plating of the cells, MTT (Sigma) was added to each well in 1mg/ml concentration in PBS, 20 ul for each well. After addition of the MTT, 2 hr incubation period was applied in 37 C. Medium and MTT were removed from the wells and were replaced by 100ul of diametyl sulfoxide –DMSO (Sigma). After 10 minutes of incubation in room temp with DMSO the plate was read and quantified by measuring absorbance at 570 nm using Epoch BioTek plate reader. This procedure was repeated daily over 5 days in order to compare if there are differences between the proliferation rates of cell lines which were ADAR1 and CPEB1 manipulated using lentivirus stable transduction.
Quantitative real-time PCR
RNA (20 ng/μl) from the SB2 and C8161 cell lines was harvested using a mirVana kit (Ambion) according to the manufacturer's instructions. The RNA was then transcribed into cDNA using TaqMan reverse transcriptase reagents for general cDNA or a TaqMan miRNA reverse transcription kit and miR-455-5p -specific primer for miRNA-specific cDNA (Applied Biosystems). The primers and fluorescence probes were obtained from Applied Biosystems. Reaction components for reverse transcription-PCR and amplifications were described previously38. Amplifications were run in triplicates, and averages were obtained after normalization with 18s rRNA or RNU6 (Applied Biosystems). Data were expressed in -fold change.
Statistical analysis
Student's t test was used to evaluate the statistical significance of the in vitro and in vivo data, two-ways anova test was also used in the in vivo data. Values for tumor growth are given as a mean volume ± S.D., and p values < 0.05 were considered statistically significant.
GEO Accession Number
Full microarray data are deposit in NCBI GEO archives (GSE31963).
Supplementary Material
Acknowledgments
We thank Dr. Stefan Maas from NIH for kindly providing the ADAR1 A-to-I RNA editing hairpin loop luciferase reporter plasmid. We thank Dr. Woonyoung Choi for her help with the microRNA microarray. We thank Rupaimoole Rajesha for his technical help and support. These studies are supported by SINF, MDACC grant and by NIH skin cancer SPORE p50 CA093459.
Footnotes
Author Contributions: M.B.-E. conceived and supervised the project. E.S. and A.K.M. designed performed and analyzed most of the data R.R.B., T.K., L.H., M.E.V., G.V.-T., and A.S. performed experiments and analyzed data. H.J.L., S.J.K. and I.J.F. helped with the spontaneous animal model. C.I., A.G.R, K.-M.N., K.Z., D.B., S.J.M.J., I.B., M.M., Y.-Y.W., A.K.E., P.H., and J.E.G. performed sequencing and analysis. A.K.S., G.A.C., and G.M. helped with miR analysis.
Competing Financial Interests: The authors declare no competing financial interest.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Miller AJ, Mihm MC., Jr Melanoma. The New England journal of medicine. 2006;355:51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- 3.Xie S, et al. Dominant-negative CREB inhibits tumor growth and metastasis of human melanoma cells. Oncogene. 1997;15:2069–2075. doi: 10.1038/sj.onc.1201358. [DOI] [PubMed] [Google Scholar]
- 4.Mobley AK, Braeuer RR, Kamiya T, Shoshan E, Bar-Eli M. Driving transcriptional regulators in melanoma metastasis. Cancer metastasis reviews. 2012;31:621–632. doi: 10.1007/s10555-012-9358-8. [DOI] [PubMed] [Google Scholar]
- 5.Jean D, Harbison M, McConkey DJ, Ronai Z, Bar-Eli M. CREB and its associated proteins act as survival factors for human melanoma cells. The Journal of biological chemistry. 1998;273:24884–24890. doi: 10.1074/jbc.273.38.24884. [DOI] [PubMed] [Google Scholar]
- 6.Melnikova VO, Bar-Eli M. Transcriptional control of the melanoma malignant phenotype. Cancer biology & therapy. 2008;7:997–1003. doi: 10.4161/cbt.7.7.6535. [DOI] [PubMed] [Google Scholar]
- 7.Melnikova VO, Mourad-Zeidan AA, Lev DC, Bar-Eli M. Platelet-activating factor mediates MMP-2 expression and activation via phosphorylation of cAMP-response element-binding protein and contributes to melanoma metastasis. The Journal of biological chemistry. 2006;281:2911–2922. doi: 10.1074/jbc.M508683200. [DOI] [PubMed] [Google Scholar]
- 8.Dobroff AS, et al. Silencing cAMP-response element-binding protein (CREB) identifies CYR61 as a tumor suppressor gene in melanoma. J Biol Chem. 2009;284:26194–26206. doi: 10.1074/jbc.M109.019836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertolotto C, et al. Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J Cell Biol. 1998;142:827–835. doi: 10.1083/jcb.142.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melnikova VO, et al. CREB inhibits AP-2alpha expression to regulate the malignant phenotype of melanoma. PLoS One. 2010;5:e12452. doi: 10.1371/journal.pone.0012452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venables JP. Unbalanced alternative splicing and its significance in cancer. BioEssays: news and reviews in molecular, cellular and developmental biology. 2006;28:378–386. doi: 10.1002/bies.20390. [DOI] [PubMed] [Google Scholar]
- 12.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes & development. 2010;24:2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallo A, Galardi S. A-to-I RNA editing and cancer: from pathology to basic science. RNA biology. 2008;5:135–139. doi: 10.4161/rna.5.3.6739. [DOI] [PubMed] [Google Scholar]
- 14.Levanon EY, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nature biotechnology. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 15.Laurencikiene J, Kallman AM, Fong N, Bentley DL, Ohman M. RNA editing and alternative splicing: the importance of co-transcriptional coordination. EMBO reports. 2006;7:303–307. doi: 10.1038/sj.embor.7400621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin Y, Zhang W, Li Q. Origins and evolution of ADAR-mediated RNA editing. IUBMB life. 2009;61:572–578. doi: 10.1002/iub.207. [DOI] [PubMed] [Google Scholar]
- 17.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annual review of biochemistry. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keegan LP, Gallo A, O'Connell MA. The many roles of an RNA editor. Nat Rev Genet. 2001;2:869–878. doi: 10.1038/35098584. [DOI] [PubMed] [Google Scholar]
- 19.Maas S, Rich A, Nishikura K. A-to-I RNA editing: recent news and residual mysteries. J Biol Chem. 2003;278:1391–1394. doi: 10.1074/jbc.R200025200. [DOI] [PubMed] [Google Scholar]
- 20.Hood JL, Emeson RB. Editing of neurotransmitter receptor and ion channel RNAs in the nervous system. Current topics in microbiology and immunology. 2012;353:61–90. doi: 10.1007/82_2011_157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wulff BE, Nishikura K. Modulation of microRNA expression and function by ADARs. Current topics in microbiology and immunology. 2012;353:91–109. doi: 10.1007/82_2011_151. [DOI] [PubMed] [Google Scholar]
- 22.Zinshteyn B, Nishikura K. Adenosine-to-inosine RNA editing. Wiley interdisciplinary reviews Systems biology and medicine. 2009;1:202–209. doi: 10.1002/wsbm.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heale BS, et al. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009;28:3145–3156. doi: 10.1038/emboj.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nemlich Y, et al. MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. The Journal of clinical investigation. 2013;123:2703–2718. doi: 10.1172/JCI62980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alon S, et al. Systematic identification of edited microRNAs in the human brain. Genome research. 2012;22:1533–1540. doi: 10.1101/gr.131573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Si K, et al. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse- specific long-term facilitation in aplysia. Cell. 2003;115:893–904. doi: 10.1016/s0092-8674(03)01021-3. [DOI] [PubMed] [Google Scholar]
- 27.Sasayama T, et al. Over-expression of Aurora-A targets cytoplasmic polyadenylation element binding protein and promotes mRNA polyadenylation of Cdk1 and cyclin B1. Genes to cells: devoted to molecular & cellular mechanisms. 2005;10:627–638. doi: 10.1111/j.1365-2443.2005.00870.x. [DOI] [PubMed] [Google Scholar]
- 28.Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO reports. 2007;8:763–769. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ota H, et al. ADAR1 Forms a Complex with Dicer to Promote MicroRNA Processing and RNA- Induced Gene Silencing. Cell. 2013;153:575–589. doi: 10.1016/j.cell.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang W, et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nature structural & molecular biology. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caldeira J, et al. CPEB1, a novel gene silenced in gastric cancer: a Drosophila approach. Gut. 2012;61:1115–1123. doi: 10.1136/gutjnl-2011-300427. [DOI] [PubMed] [Google Scholar]
- 32.Zhang JH, et al. Cytoplasmic polyadenylation element binding protein is a conserved target of tumor suppressor HRPT2/CDC73. Cell death and differentiation. 2010;17:1551–1565. doi: 10.1038/cdd.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levanon EY, et al. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic acids research. 2005;33:1162–1168. doi: 10.1093/nar/gki239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galeano F, et al. Human BLCAP transcript: new editing events in normal and cancerous tissues. International journal of cancer Journal international du cancer. 2010;127:127–137. doi: 10.1002/ijc.25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gommans WM, McCane J, Nacarelli GS, Maas S. A mammalian reporter system for fast and quantitative detection of intracellular A-to-I RNA editing levels. Analytical biochemistry. 2010;399:230–236. doi: 10.1016/j.ab.2009.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landen CN, Jr, et al. Cancer research. 2005;65:6910–6918. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- 38.Villares GJ, et al. Targeting melanoma growth and metastasis with systemic delivery of liposome-incorporated protease-activated receptor-1 small interfering RNA. Cancer Res. 2008;68:9078–9086. doi: 10.1158/0008-5472.CAN-08-2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.