

Highlights
-
•
Metagenomics can detect any known viruses in clinical samples.
-
•
We compared different viral metagenomic library preparation methods using a biological reagent consisting of 25 human viral pathogens.
-
•
Our study will assist in the development of efficient applications of viral metagenomics.
Keywords: Viral metagenomics, Next generation sequencing, Virus pathogen discovery, Diagnosis, Method
Abstract
Unbiased metagenomic sequencing holds significant potential as a diagnostic tool for the simultaneous detection of any previously genetically described viral nucleic acids in clinical samples. Viral genome sequences can also inform on likely phenotypes including drug susceptibility or neutralization serotypes. In this study, different variables of the laboratory methods often used to generate viral metagenomics libraries were compared for their abilities to detect multiple viruses and generate full genome coverage. A biological reagent consisting of 25 different human RNA and DNA viral pathogens was used to estimate the effect of filtration and nuclease digestion, DNA/RNA extraction methods, pre-amplification and the use of different library preparation kits on the detection of viral nucleic acids. Filtration and nuclease treatment led to slight decreases in the percentage of viral sequence reads and number of viruses detected. For nucleic acid extractions silica spin columns improved viral sequence recovery relative to magnetic beads and Trizol extraction. Pre-amplification using random RT-PCR while generating more viral sequence reads resulted in detection of fewer viruses, more overlapping sequences, and lower genome coverage. The ScriptSeq library preparation method retrieved more viruses and a greater fraction of their genomes than the TruSeq and Nextera methods. Viral metagenomics sequencing was able to simultaneously detect up to 22 different viruses in the biological reagent analyzed including all those detected by qPCR. Further optimization will be required for the detection of viruses in biologically more complex samples such as tissues, blood, or feces.
1. Introduction
Viral metagenomics characterizes the genetic composition of viral particles enriched from environmental or biological samples. Because of low viral nucleic acid concentrations, genetic characterization first requires non-specific nucleic acid amplification followed by DNA sequencing. Sequencing can be performed by plasmid subcloning and Sanger sequencing or using highly parallel sequencing technologies also known as next generation or deep sequencing (Allander et al., 2001, Capobianchi et al., 2013, Delwart, 2007, Delwart, 2013, Mokili et al., 2012, Tang and Chiu, 2010). Viral metagenomics or sequencing of nucleic acids from enriched viral particle preparations has been frequently applied for viral discovery (Chiu, 2013, Delwart, 2012, Lipkin and Firth, 2013), resulting in the genetic characterization of numerous human and animal viruses (Allander et al., 2005, Allander et al., 2007, De Vlaminck et al., 2013, Grard et al., 2012, Handley et al., 2012, Kapoor et al., 2009, Kapoor et al., 2013, Li et al., 2009, Li et al., 2011, Li et al., 2013a, Li et al., 2013b, Quan et al., 2010, Wylie et al., 2012) but its non-specific nature also endows it with potential as a universal virus detection assay. Viral metagenomics can also be used for detection of contaminating viruses in biological products such as human and animal vaccines (Farsang and Kulcsar, 2012, McClenahan et al., 2011, McClenahan et al., 2014, Onions et al., 2011, Victoria et al., 2010). The mutation profile and genetic stability of attenuated vaccine viruses such as oral polio vaccine and influenza vaccines can also be tracked by deep sequencing (Bidzhieva et al., 2014, Neverov and Chumakov, 2010).
As the cost of massively parallel DNA sequencing continues to fall (Loman et al., 2012, Metzker, 2010, Rizzo and Buck, 2012), the limitations imposed by data transfer and bioinformatics analyses become more significant requiring increasing computing power. Until the cost of sequencing and bioinformatics become so insignificant that every molecule of nucleic acid in biological samples can be economically sequenced and analyzed, the sensitivity of viral metagenomics for virus detection remains dependent on the methods used for enriching and amplifying viral nucleic acids and the extent of deep sequencing applied to the resulting DNA library.
Virus particle enrichment steps include sample homogenization, filtration, ultra-centrifugation, and nuclease digestion of contaminating host and bacterial nucleic acids that dominate in biological samples, in order to reduce the amount of background nucleic acids. Non-specific nucleic acid amplification is also necessary to generate sufficient quantity of DNA in the proper format (flanked by appropriate sequences) for input into different deep sequencing platforms. In this study, a biological reagent generated by mixing numerous human DNA and RNA viral pathogens was used to compare virus particle enrichment, extraction, random amplification, and DNA library preparation methods to compare laboratory methods for viral detection and genetic characterization using deep sequencing.
2. Materials and methods
2.1. Sample
A biological reagent, expected to contain 25 human pathogenic viruses belonging to two DNA and seven RNA viral families was assembled from clinical specimens and egg- and cell culture-passaged viruses (Table 1 ). Before pooling, the clinical specimen, allantoic fluid, and cell culture supernatant were tested positive for corresponding virus. The presence of each virus in the pool was then tested using qPCR using primer pairs targeting different genomes. The qPCR threshold cycles (Ct) are shown in Table 1. Six of the expected 25 viruses were not detected by qPCR. Viral concentrations based on qPCR are approximate since quantified viral nucleic acids were not used to generate standard viral concentration curves. Multiple aliquots of the reagent were frozen and stored at −80 °C and shipped on dry ice.
Table 1.
Summary of viruses contained in the multi-viral mix.
| Group | Family | Envelope | Species/type | Abbrev. | Genome (kb) | Ct | Sample origin |
|---|---|---|---|---|---|---|---|
| dsDNA | Adenoviridae | No | Adenovirus 2 | AdV2 | 35.9 | 29.71 | 293 cell culture |
| Adenovirus 41 | AdV41 | 34.2 | ND | Clinical specimen | |||
| Herpesviridae | Yes | Human herpesvirus 3 | HHV3 | 124.8 | 29.02 | MeWo cell culture | |
| Human herpesvirus 4 | HHV4 | 171.7 | 31.27 | B95-8 cell culture | |||
| Human herpesvirus 5 | HHV5 | 233.7 | 28.95 | MRC5 cell culture | |||
| Human herpesvirus 2 | HHV2 | 154.7 | 32.48 | MRC5 cell culture | |||
| Human herpesvirus 1 | HHV1 | 151.2 | 30.59 | MRC5 cell culture | |||
| dsRNA | Reoviridae | No | Rotavirus A | RVA | 18.5 | 24.49 | Clinical specimen |
| ssRNA (+) | Astroviridae | No | Astrovirus | AstV | 6.8 | 30.53 | Clinical specimen |
| Caliciviridae | No | Norovirus GI | NV GI | 7.6 | ND | Clinical specimen | |
| Norovirus GII | NV GII | 7.5 | ND | Clinical specimen | |||
| Sapovirus C12 | SaV C12 | 7.5 | 33.37 | Clinical specimen | |||
| Coronaviridae | Yes | Coronavirus 229E | CoV 229E | 27.2 | 36.48 | MRC5 cell culture | |
| Picornaviridae | No | Parechovirus 3 | HPeV3 | 7.2 | 29.35 | LLC-MK2 cell culture | |
| Rhinovirus A39 | HRV A39 | 7.1 | 31.16 | MRC5 cell culture | |||
| Coxsackievirus B4 | CVB4 | 7.4 | 30.72 | Hep-2 cell culture | |||
| ssRNA (−) | Orthomyxoviridae | Yes | Influenza B virus | IFV B | 14.2 | ND | Egg passage |
| Influenza A virus H1N1 | IFV A H1N1 | 13.2 | 32.02 | Egg passage | |||
| Influenza A virus H3N2 | IFV A H3N2 | 13.6 | ND | Egg passage | |||
| Paramyxoviridae | Yes | Metapneumovirus A | HMPV A | 13.3 | 31.86 | LLC-MK2 cell culture | |
| Respiratory syncytial virus A2 | RSV A2 | 15.2 | 34.33 | Hep-2 cell culture | |||
| Parainfluenzavirus 1 | PIV1 | 15.5 | 34.43 | PRF5 cell culture | |||
| Parainfluenzavirus 2 | PIV2 | 15.7 | 33.87 | PRF5 cell culture | |||
| Parainfluenzavirus 3 | PIV3 | 15.4 | ND | PRF5 cell culture | |||
| Parainfluenzavirus 4 | PIV4 | 17.4 | 31.83 | PRF5 cell culture | |||
ND: not detectable (Ct value >37).
2.2. Filtration and nuclease treatment
The viral multiplex reagent (200 μl) was centrifuged at 12.000 × g for 5 min at 8 °C and the supernatant filtered through a 0.45 μM filter (Millipore, Billerica, MA, USA) to remove possible host cellular debris and bacteria. The filtrate was treated with a nuclease mixture of 14U turbo DNase (Ambion, Life Technologies, Grand Island, NY, USA), 3U Baseline-ZERO (Epicentre, Chicago, IL, USA) and 20U RNase One (Promega, Madison, WI, USA) in 1× DNase buffer (Ambion, Life Technologies, Grand Island, NY, USA) at 37 °C for 1.5 h to reduce background nucleic acids from the host cells and bacteria. Viral nucleic acids protected from digestion by viral capsids, were then extracted from ∼200 μl resulting solutions by different methods (Victoria et al., 2008, Victoria et al., 2009).
2.3. Extraction methods
Three methods were used to extract the viral nucleic acids: QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany), Maxwell 16 Viral Total Nucleic Acid Purification Kit (Promega, Madison, WI, USA), and Trizol Reagent (Life Technologies, Grand Island, NY, USA). For Qiagen column and Maxwell bead extraction, viral nucleic acids were purified according to the manufacture's instruction. For Trizol extraction, viral RNA and DNA were isolated separately following the manufacture's protocol. The elution volume was 60 μl of nuclease free water.
2.4. Pre-amplification to enrich the viral nucleic acids
Viral cDNA synthesis was performed by incubation of 10 μl extracted viral nucleic acids with 100 pmol of a primer containing a fixed 18 bp sequence plus a random nonamer at the 3′ end (GCCGACTAATGCGTAGTCNNNNNNNNN) at 85 °C for 2 min. Then, 200U SuperScript III reverse transcriptase (Invitrogen, Waltham, MA, USA), 0.5 mM of each deoxynucloside triphosphate (dNTP), 10 mM dithiothreitol, and 1× first-strand extension buffer were added to the mixture and incubated at 25 °C for 10 min, followed by 50 °C incubation for 1 h. The 2nd strand DNA synthesis was performed by incubation with 50 pmol of random primer at 95 °C for 2 min, 4 °C for 2 min, and then with 5U Klenow Fragment (New England Biolabs, Ipswich, MA, USA) at 37 °C for 1 h. The resulting products were either put into library preparation directly or PCR amplified by using 5 μl of the RT-Klenow DNA products and 2.5 μM primer consisting of the fixed 18 bp portion of the random primer (GCCGACTAATGCGTAGTC) with 1U AmpliTaq Gold DNA polymerase (Life Technologies, Grand Island, NY, USA), 2.5 mM MgCl2, 0.2 mM dNTPs, and 1× PCR Gold buffer in a reaction volume of 50 μl. Temperature cycling was performed as follows: 1 cycle of 95 °C for 5 min, 30 cycles of denaturing at 95 °C for 30 s, 55 °C for 30 s, 72 °C for 1.5 min (Victoria et al., 2008, Victoria et al., 2009). An additional extension for 10 min at 72 °C was added to the end of the run. The PCR products were purified twice by Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, United States) with 0.8:1 ratio of beads to sample.
2.5. NGS library preparation methods
Three NGS library preparation methods were tested using Nextera XT DNA Sample Preparation Kit (Illumina, San Diego, CA, USA), TruSeq RNA Sample Prep Kit v2 (Illumina, San Diego, CA, USA), and ScriptSeq v2 RNA-Seq Library Preparation Kit (Epicentre, Chicago, IL, USA) according to the manufacture's protocols with minor modifications. The DNA input for Nextera library preparations was either the RT and Klenow DNA polymerase dsDNA products or that same product following PCR amplification products (Table 2 ). For TruSeq library preparation, the same pre-amplification products nucleic acids were used as input DNA. For ScriptSeq library preparation, 10 μl of extracted viral nucleic acids were used directly as input and recommended PCR cycles were increased to 25 and 35 to obtain sufficient products for sequencing. The quality of the libraries was assessed by 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and the quantification was estimated by KAPA Library Quant Kit (Kapa Biosystems, Wilmington, MA, USA) real time PCR following the manufacturer's instructions.
Table 2.
Methods outline (two MiSeq runs were included).
| Lib ID | Filter | Nuclease | Extraction | Pre-amp. | Lib. prep. | Run 1 | Run 2 |
|---|---|---|---|---|---|---|---|
| N1 | Yes | Yes | Maxwell viral | Yes | Nextera | X | |
| N230 | Yes | Yes | Maxwell viral | Yes | Nextera | X | |
| N231 | Yes | Yes | Maxwell viral | Yes | Nextera | X | |
| N12 | Yes | Yes | Maxwell viral | No | Nextera | X | |
| N2 | Yes | No | Maxwell viral | Yes | Nextera | X | |
| N3 | No | No | Maxwell viral | Yes | Nextera | X | |
| N227 | No | No | Maxwell viral | Yes | Nextera | X | |
| N221 | No | No | Maxwell viral | Yes | TruSeq | X | |
| N225 | No | No | Maxwell viral | No | ScriptSeq25 | X | |
| N226 | No | No | Maxwell viral | No | ScriptSeq35 | X | |
| N32 | No | No | Maxwell viral | No | Nextera | X | |
| N4 | No | Yes | Maxwell viral | Yes | Nextera | X | |
| N42 | No | Yes | Maxwell viral | No | Nextera | X | |
| N5 | Yes | Yes | QIAamp viral | Yes | Nextera | X | |
| N232 | Yes | Yes | QIAamp viral | Yes | Nextera | X | |
| N233 | Yes | Yes | QIAamp viral | Yes | Nextera | X | |
| N6 | No | No | Trizol | Yes | Nextera | X |
2.6. NGS sequencing and sequence data analyses
The resulting libraries of single-stranded DNA fragments were sequenced using the MiSeq Illumina platform and in two multiplexed runs were performed by using 2 × 250 cycle MiSeq Reagent Kit v2 (Illumina, San Diego, CA, USA). The different treatments and library preparation methods (Table 2) were distinguished using unique barcodes/indices.
Paired-end reads of 250 bp generated by MiSeq were demultiplexed using vendor software from Illumina. A virus discovery pipeline running on a 32-nodes Linux cluster was used to process the data. Usable reads were those remaining after applying the following filters. Human host reads and bacterial reads were subtracted by mapping the reads to human reference genome hg19 and bacterial RefSeq genomes release 59 using bowtie2 (Langmead and Salzberg, 2012). Low sequencing quality tails were trimmed using Phred quality score 10 as the threshold. Adaptor and primer sequences were trimmed using the default parameters of VecScreen. Reads were considered duplicates if bases at position 5 to 55 from 5′ end were identical. One random copy of duplicates was kept. The filtered reads were assembled using a de novo sequence assembler consists of SOAPdenovo2 (Luo et al., 2012), ABySS (Simpson et al., 2009), meta-Velvet (Namiki et al., 2012) and CAP3 (Huang and Madan, 1999). The assembled contigs and singlets were translated and aligned to a viral proteome database (consisting of all annotated full or near full viral genomes) using BLASTx. The significant hits to virus were then aligned to a non-virus-non-redundant (NVNR) universal proteome database using BLASTx. Hits with more significant E-value to NVNR than to virus were removed. The genome coverage of the target viruses were analyzed by Geneious 7 (Biomatters, San Francisco, CA, USA).
3. Results
3.1. Sequence data overview and normalization
Two MiSeq runs containing 9 and 7 barcodes generated ∼9 and ∼12 million reads respectively (Table 2). The raw sequence reads were demultiplexed and put through multiple quality filters, leaving a total of ∼ 6 million “usable” sequence reads, which were then de novo assembled separately for each barcode. The resulting contigs and singlets were then analyzed using BLAST search. In order to avoid misclassification a stringent E-value cutoff of 1 × 10−10 was used to identify viral sequences related to the 25 viruses expected in the NIBSC reagent and be considered virus hits. The efficiency of different treatments and library preparation methods in detecting these viruses are shown in Table 3 . Other viral hits were regarded as contamination from reagents, laboratory, and the biological samples in the viral pool (clinical specimens and bovine serum) including sequences from picobirnavirus, bocavirus and bovine virus diarrhea viruses. In N225 and N226 libraries 2–3% of all viral hits were to avian leucosis virus, originating from the reverse transcriptase in the ScriptSeq library preparation kit.
Table 3.
Heat map of viral reads for target viruses (E-value ≤ 1 × 10−10).

Note: The qPCR Ct values for all the viruses are given for information only and cannot be compared.
In order to normalize for the variable number of sequence reads with different barcode/index in the multiplexed libraries, a total of 150,000 raw sequence reads were taken randomly from each barcode for the subsequent analyses. The 150,000 raw sequence reads from each multiplexing library were deposited in NCBI's short read archive under accession number SRP051174.
3.2. Method repeatability
To investigate the method reproducibility, three treatment groups in which samples were independently processed in the same manners were compared (Table 3). Technical replicates were not performed for every combination of variables in Table 2.
Out of the 150,000 raw sequence reads, the method N1 and its two replicates, N3 and its one replicate, and N5 and its two replicates had similar percentage of usable and target viral reads (Fig. 1 and Table 3). The N1/N230/N231 group identified an average of 6.3 viruses (range 4–8), N3/N227 group identified an average of 12 viruses (range 11–13) and N5/N232/N233 group identified an average of 15.6 viruses (range 15–17) (Table 3). The genome coverage of the recovered viruses was also analyzed. The read numbers and genome coverage for human herpes virus 3 (HHV3) and rotavirus (RVA) are shown (Fig. 2 ), as these viruses were detected in all of the libraries with relatively high number of reads and represent a large dsDNA genome and a small ssRNA genome). RVA read numbers and genome coverage was higher and more reproducible than HHV3 (Fig. 2).
Fig. 1.
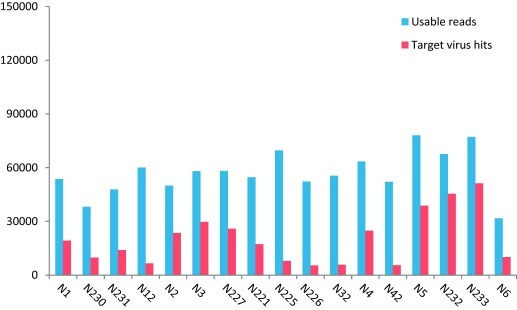
Usable reads and target virus hits in randomized subset data of 150,000 raw sequence reads.
Fig. 2.

Human herpesvirus 3 reads (E value ≤ 1 × 10−10) and genome coverage % (A) and rotavirus A reads (E value ≤ 1 × 10−10) and genome coverage % (B).
3.3. Filtration and nuclease treatment effect
Filtration and nuclease treatment is frequently used to enrich viral particles and reduce the background of host and bacterial genetic material (Allander et al., 2001, Thurber et al., 2009, Wommack et al., 2009). In this study, the effect of filtration and nuclease treatment on yield of viral sequence reads and number of viruses detected was estimated (Table 3).
With one variable changing and the other parameters fixed, the effect of filtration was first measured by comparing N1 or N2 (with filtration) vs. N4 or N3 (no filtration). N1 and N4 (no filtration) had 36% and 39% of target/usable virus reads respectively (Fig. 1). Filtration resulted in only a slight 3–4% decrease in the number of virus reads and in the number of viruses detected (3 fewer viruses) (Fig. 1 and Table 3). In a similar fashion when the effect of nuclease digestion was compared (N1 vs. N2 and N3 vs. N4), its use decreased the number of virus reads by 11–12% (Fig. 1) and a smaller number of viruses were detected (2 and 5 fewer viruses) (Table 3). The HHV3 detected in method N1 through N4 ranged from 836 to 2227 reads, covering 5–9% of the genome, while RVA had 4401–9004 reads, covering 62–88% of the genome. Overall only minor differences were seen among the groups with or without filtration and nuclease digestion (Fig. 2).
3.4. Extraction methods
The performance of three extraction methods using either Qiagen silica columns, Maxwell silica coated magnetic beads and Trizol (guanidinium thiocyanate–phenol–chloroform) on recovering viral sequence reads were compared (Table 3). The N5 method detected the highest percentage of the target virus reads (Fig. 1) and largest number of target viruses (n = 20) (Table 3). It also generated the highest genome coverage for both HHV3 and RVA (Fig. 2). Silica columns N5 and magnetic beads N1 had similar numbers of HHV3 reads (1600 vs. 1533) but the genome coverage differed greatly (70 vs. 5%) with the magnetic bead-extracted nucleic acids producing reads with considerable overlaps covering the same limited genomic regions (Fig. 3 ).
Fig. 3.

Effect of extraction method on HHV3 genome coverage. (A) Bead extraction N1 methods obtained 1533 HHV3 reads, covering 4.5% of the reference genome. (B) Silica column N5 method obtained 1600 reads, covering 69.6% of the reference genome. Assembly to reference genome JQ972913 was done with Geneious 7.0. Different scales are used to show depth of sequencing.
3.5. Pre-amplification
Unlike sequencing plentiful human or bacterial DNA or RNA, amplification is necessary to generate sufficient input DNA when sequencing minute, generally unquantifiable, amounts of viral nucleic acids. In this study, the effect of pre-amplification of viral nucleic acids using a random RT-PCR step on subsequent virus detection and genome coverage with the Nextera kit was evaluated. The effects of such pre-amplification were compared using three groups: N1 vs. N12; N3 vs. N32; N4 vs. N42 (Table 3).
The percentage of usable reads out of 150,000 raw reads in all six treatments were close (35–42%), but the target virus reads obtained by pre-amplification methods (N1, 36%; N3, 51%; N4, 39%) were much higher than non-pre-amplification methods (N12, 11%; N32, 10%; N42, 11%) (Fig. 1). Despite the higher number of virus reads, pre-amplification resulted in fewer viruses being detected (Table 3). The use of pre-amplification methods also resulted in lower genome coverage for HHV3 and similar coverage for RVA (Fig. 2). These results indicate that pre-amplification resulted largely in the generation of more biased genome coverage with near-duplicate reads accumulating over the same region decreasing viral genome coverage.
3.6. Library preparation methods
Different DNA library preparation kits are available commercially. Three such kits (Table 3). Nextera (N227) and TruSeq (N221) used as input pre-amplified DNA were compared. ScriptSeq used viral nucleic acids directly (without pre-amplification) with either 25 (N225) or 35 (N227) PCR cycles during the ScriptSeq library generation. All input nucleic acids were identically treated (without filtration and nuclease digestion). These four experiments generated 39%, 36%, 46% and 35% of usable reads. The target virus reads accounted for 45%, 32%, 11% and 10% of the usable reads (Fig. 1). Both ScriptSeq experiments (N225 and N226) detected 20 viruses, while Nextera (N227) and TruSeq (N221) identified 11 and 14 viruses respectively (Table 3). ScriptSeq also generated higher genome coverage using fewer virus reads for both HHV3 and RVA (Fig. 2). Thus, despite the lower number of viral reads, ScriptSeq methods were superior in retrieving target viruses and acquiring large segments of their viral genomes. TruSeq slightly exceeded Nextera in virus detection and genome coverage.
4. Discussion
Viral metagenomics provides an effective method to identify any known virus in biological samples and thus has great potential for use in clinical diagnostic and reference labs. The sensitivity of the approach depends on the depth of sequencing and the method used to generate the input DNA libraries. In this study, the impact of four steps used in viral metagenomic library preparations on the detection of different viruses and percentage of genome coverage were compared. A laboratory assembled biological reagent consisting of a mixture of 25 human viral pathogens was employed. This biological reagent, an important resource for the future comparisons of different viral detection methods, includes large and small viral particles with DNA or RNA viral genomes originating from cell culture supernatants or biological samples. The methodological steps compared were physical and enzymatic enrichment of viral particle-associated nucleic acids using filtration and nuclease digestion, nucleic acid extraction techniques, random RT-PCR pre-amplification, and different Illumina library preparation methods.
Viral enrichment methods including filtration and nuclease treatment slightly decreased the number of virus reads and number of viruses identified. The result with the NIBSC reagent may be due the possible retention of viruses or virus aggregate on the 0.45 μm filters or the release of nuclease-sensitive viral nucleic acids from virions in the reagent solution. Unlike the sample analyzed in this study, the use of filtration and nuclease treatment are considered useful to reduce background of host and bacterial DNA from biologically more complex clinical samples such as tissue, plasma, feces, and respiratory secretions (Allander et al., 2001, Delwart, 2007). The lower efficiency of the Trizol extraction method may be due to the loss of nucleic acids during the interphase collection and precipitation steps. The silica columns yielded the best results despite the known release of background nucleic acids from these columns (Lysholm et al., 2012, Naccache et al., 2013, Naccache et al., 2014). Pre-amplification, while increasing the number of viral reads actually decreased genome coverage and number of viruses detected. This observation is due to the generation of many similar reads over the same limited genome region reducing overall coverage. Thus, non-specific pre-amplification (prior to the PCR required to incorporate Illumina primers) is best avoided when the input DNA (from the initial RT and Klenow steps) is in sufficient quantity for direct library preparation using Illumina kits. Lastly the use of the ScriptSeq kits detected more viruses with greater genome coverage than the Nextera and TruSeq kits despite generating fewer viral reads. This result may also reflect the fewer PCR cycles used in the use of the TruSeq kit resulting in fewer duplicate reads covering the same genome region. The maximum number of viruses detected was 20 out of 25 (N225 and N226) when analyzing a normalized 150,000 reads or 22/25 (N226) viruses when all ∼2 million usable reads were included (three and four reads for IFN A H3N2 and PIV3 respectively were detected with the complete dataset).
Few studies have measured the efficiency of viral metagenomics relative to PCR. A study by Greninger et al. (2010) compared Illumina deep sequencing to RT-PCR for the detection of influenza A virus H1N1 in nasopharyngeal swabs. It was shown that deep sequencing detected H1N1 at titers near the detection limit of specific RT-PCR particularly when samples were first treated with DNase prior to extraction, and the percentage of sequence reads was correlated with virus titer (Greninger et al., 2010). Here, all viruses quantifiable by qPCR with Ct <37 as well as 4 of the 6 viruses not detectable by qPCR were detected by deep sequencing. This result indicates that deep sequencing is a highly sensitive method with the added advantage of simultaneously detecting all known viruses. Further studies will be required to compare the sensitivity of viral metagenomics to that of highly optimized clinical PCRs. The sensitivity of viral metagenomics for any specific virus target will also be affected by the quantity of other remaining nucleic acids, both non-viral and from other viruses, which may vary widely between clinical samples.
Defining limits of detection in different types of clinical samples and the ability to quantify viral loads based on viral read numbers will require further studies including appropriate viral spiking studies into biologically more complex samples including tissues, plasma, feces, or respiratory secretions. Because different random amplification methods may show different efficiencies with different types of viral nucleic acids (circular, linear, ss, ds, RNA or DNA genomes) the use of multiple amplification methods may be required to optimize sensitivity for the wide range of possible viral nucleic acids.
The use of deep sequencing for viral diagnostic purposes will also benefit from specimen type-specific protocols to maximally enrich viral nucleic acids. Spiking of diverse biological samples with the multi-virus reagent analyzed – in this study should allow the sensitivity of different methods to be more readily compared.
Acknowledgements
The study was supported by the Blood Systems Research Institute and NIH R01 HL083254 to Dr. Delwart. Work at the National Institute for Biological Reagents and Control was supported by the UK Department of Health. We are grateful to Dr. Kathryn Doris for provision of qPCR data.
References
- Allander T., Andreasson K., Gupta S., Bjerkner A., Bogdanovic G., Persson M.A., Dalianis T., Ramqvist T., Andersson B. Identification of a third human polyomavirus. J. Virol. 2007;81:4130–4136. doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allander T., Emerson S.U., Engle R.E., Purcell R.H., Bukh J. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proc. Natl. Acad. Sci. U. S. A. 2001;98:11609–11614. doi: 10.1073/pnas.211424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allander T., Tammi M.T., Eriksson M., Bjerkner A., Tiveljung-Lindell A., Andersson B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc. Natl. Acad. Sci. U. S. A. 2005;102:12891–12896. doi: 10.1073/pnas.0504666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidzhieva B., Zagorodnyaya T., Karagiannis K., Simonyan V., Laassri M., Chumakov K. Deep sequencing approach for genetic stability evaluation of influenza A viruses. J. Virol. Methods. 2014;199:68–75. doi: 10.1016/j.jviromet.2013.12.018. [DOI] [PubMed] [Google Scholar]
- Capobianchi M.R., Giombini E., Rozera G. Next-generation sequencing technology in clinical virology. Clin. Microbiol. Infect. 2013;19:15–22. doi: 10.1111/1469-0691.12056. [DOI] [PubMed] [Google Scholar]
- Chiu C.Y. Viral pathogen discovery. Curr. Opin. Microbiol. 2013;16:468–478. doi: 10.1016/j.mib.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vlaminck I., Khush K.K., Strehl C., Kohli B., Luikart H., Neff N.F., Okamoto J., Snyder T.M., Cornfield D.N., Nicolls M.R., Weill D., Bernstein D., Valantine H.A., Quake S.R. Temporal response of the human virome to immunosuppression and antiviral therapy. Cell. 2013;155:1178–1187. doi: 10.1016/j.cell.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delwart E. Animal virus discovery: improving animal health, understanding zoonoses, and opportunities for vaccine development. Curr. Opin. Virol. 2012;2:344–352. doi: 10.1016/j.coviro.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delwart E. A roadmap to the human virome. PLoS Pathog. 2013;9:e1003146. doi: 10.1371/journal.ppat.1003146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delwart E.L. Viral metagenomics. Rev. Med. Virol. 2007;17:115–131. doi: 10.1002/rmv.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farsang A., Kulcsar G. Extraneous agent detection in vaccines – a review of technical aspects. Biologicals. 2012;40:225–230. doi: 10.1016/j.biologicals.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grard G., Fair J.N., Lee D., Slikas E., Steffen I., Muyembe J.J., Sittler T., Veeraraghavan N., Ruby J.G., Wang C., Makuwa M., Mulembakani P., Tesh R.B., Mazet J., Rimoin A.W., Taylor T., Schneider B.S., Simmons G., Delwart E., Wolfe N.D., Chiu C.Y., Leroy E.M. A novel rhabdovirus associated with acute hemorrhagic fever in central Africa. PLoS Pathog. 2012;8:e1002924. doi: 10.1371/journal.ppat.1002924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greninger A.L., Chen E.C., Sittler T., Scheinerman A., Roubinian N., Yu G., Kim E., Pillai D.R., Guyard C., Mazzulli T., Isa P., Arias C.F., Hackett J., Schochetman G., Miller S., Tang P., Chiu C.Y. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS ONE. 2010;5:e13381. doi: 10.1371/journal.pone.0013381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handley S.A., Thackray L.B., Zhao G., Presti R., Miller A.D., Droit L., Abbink P., Maxfield L.F., Kambal A., Duan E., Stanley K., Kramer J., Macri S.C., Permar S.R., Schmitz J.E., Mansfield K., Brenchley J.M., Veazey R.S., Stappenbeck T.S., Wang D., Barouch D.H., Virgin H.W. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell. 2012;151:253–266. doi: 10.1016/j.cell.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Madan A. CAP3: a DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Li L., Victoria J., Oderinde B., Mason C., Pandey P., Zaidi S.Z., Delwart E. Multiple novel astrovirus species in human stool. J. Gen. Virol. 2009;90:2965–2972. doi: 10.1099/vir.0.014449-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Simmonds P., Cullen J.M., Scheel T.K., Medina J.L., Giannitti F., Nishiuchi E., Brock K.V., Burbelo P.D., Rice C.M., Lipkin W.I. Identification of a pegivirus (GB virus-like virus) that infects horses. J. Virol. 2013;87:7185–7190. doi: 10.1128/JVI.00324-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Deng X., Linsuwanon P., Bangsberg D., Bwana M.B., Hunt P., Martin J.N., Deeks S.G., Delwart E. AIDS alters the commensal plasma virome. J. Virol. 2013;87:10912–10915. doi: 10.1128/JVI.01839-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., McGraw S., Zhu K., Leutenegger C.M., Marks S.L., Kubiski S., Gaffney P., Dela Cruz F.N., Jr., Wang C., Delwart E., Pesavento P.A. Circovirus in tissues of dogs with vasculitis and hemorrhage. Emerg. Infect. Dis. 2013;19:534–541. doi: 10.3201/eid1904.121390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Shan T., Wang C., Cote C., Kolman J., Onions D., Gulland F.M., Delwart E. The fecal viral flora of California sea lions. J. Virol. 2011;85:9909–9917. doi: 10.1128/JVI.05026-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Victoria J., Kapoor A., Blinkova O., Wang C., Babrzadeh F., Mason C.J., Pandey P., Triki H., Bahri O., Oderinde B.S., Baba M.M., Bukbuk D.N., Besser J.M., Bartkus J.M., Delwart E.L. A novel picornavirus associated with gastroenteritis. J. Virol. 2009;83:12002–12006. doi: 10.1128/JVI.01241-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkin W.I., Firth C. Viral surveillance and discovery. Curr. Opin. Virol. 2013;3:199–204. doi: 10.1016/j.coviro.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman N.J., Misra R.V., Dallman T.J., Constantinidou C., Gharbia S.E., Wain J., Pallen M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., He G., Chen Y., Pan Q., Liu Y., Tang J., Wu G., Zhang H., Shi Y., Yu C., Wang B., Lu Y., Han C., Cheung D.W., Yiu S.M., Peng S., Xiaoqian Z., Liu G., Liao X., Li Y., Yang H., Wang J., Lam T.W. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysholm F., Wetterbom A., Lindau C., Darban H., Bjerkner A., Fahlander K., Lindberg A.M., Persson B., Allander T., Andersson B. Characterization of the viral microbiome in patients with severe lower respiratory tract infections, using metagenomic sequencing. PLoS ONE. 2012;7:e30875. doi: 10.1371/journal.pone.0030875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClenahan S., Uhlenhaut C., Krause P.R. Regulatory approaches for control of viral contamination of vaccines. PDA J. Pharm. Sci. Technol. 2011;65:557–562. doi: 10.5731/pdajpst.2011.00819. [DOI] [PubMed] [Google Scholar]
- McClenahan S.D., Uhlenhaut C., Krause P.R. Optimization of virus detection in cells using massively parallel sequencing. Biologicals. 2014;42:34–41. doi: 10.1016/j.biologicals.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Metzker M.L. Sequencing technologies – the next generation. Nat. Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Mokili J.L., Rohwer F., Dutilh B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012;2:63–77. doi: 10.1016/j.coviro.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naccache S.N., Greninger A.L., Lee D., Coffey L.L., Phan T., Rein-Weston A., Aronsohn A., Hackett J., Jr., Delwart E.L., Chiu C.Y. The perils of pathogen discovery: origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J. Virol. 2013;87:11966–11977. doi: 10.1128/JVI.02323-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naccache S.N., Hackett J., Jr., Delwart E.L., Chiu C.Y. Concerns over the origin of NIH-CQV, a novel virus discovered in Chinese patients with seronegative hepatitis. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E976. doi: 10.1073/pnas.1317064111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namiki T., Hachiya T., Tanaka H., Sakakibara Y. MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 2012;40:e155. doi: 10.1093/nar/gks678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neverov A., Chumakov K. Massively parallel sequencing for monitoring genetic consistency and quality control of live viral vaccines. Proc. Natl. Acad. Sci. U. S. A. 2010;107:20063–20068. doi: 10.1073/pnas.1012537107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onions D., Cote C., Love B., Toms B., Koduri S., Armstrong A., Chang A., Kolman J. Ensuring the safety of vaccine cell substrates by massively parallel sequencing of the transcriptome. Vaccine. 2011;29:7117–7121. doi: 10.1016/j.vaccine.2011.05.071. [DOI] [PubMed] [Google Scholar]
- Quan P.L., Wagner T.A., Briese T., Torgerson T.R., Hornig M., Tashmukhamedova A., Firth C., Palacios G., Baisre-De-Leon A., Paddock C.D., Hutchison S.K., Egholm M., Zaki S.R., Goldman J.E., Ochs H.D., Lipkin W.I. Astrovirus encephalitis in boy with X-linked agammaglobulinemia. Emerg. Infect. Dis. 2010;16:918–925. doi: 10.3201/eid1606.091536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo J.M., Buck M.J. Key principles and clinical applications of “next-generation” DNA sequencing. Cancer Prev. Res. (Phila.) 2012;5:887–900. doi: 10.1158/1940-6207.CAPR-11-0432. [DOI] [PubMed] [Google Scholar]
- Simpson J.T., Wong K., Jackman S.D., Schein J.E., Jones S.J., Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang P., Chiu C. Metagenomics for the discovery of novel human viruses. Future Microbiol. 2010;5:177–189. doi: 10.2217/fmb.09.120. [DOI] [PubMed] [Google Scholar]
- Thurber R.V., Haynes M., Breitbart M., Wegley L., Rohwer F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009;4:470–483. doi: 10.1038/nprot.2009.10. [DOI] [PubMed] [Google Scholar]
- Victoria J.G., Kapoor A., Dupuis K., Schnurr D.P., Delwart E.L. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathog. 2008;4:e1000163. doi: 10.1371/journal.ppat.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victoria J.G., Kapoor A., Li L., Blinkova O., Slikas B., Wang C., Naeem A., Zaidi S., Delwart E. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J. Virol. 2009;83:4642–4651. doi: 10.1128/JVI.02301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victoria J.G., Wang C., Jones M.S., Jaing C., McLoughlin K., Gardner S., Delwart E.L. Viral nucleic acids in live-attenuated vaccines: detection of minority variants and an adventitious virus. J. Virol. 2010;84:6033–6040. doi: 10.1128/JVI.02690-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wommack K.E., Williamson K.E., Helton R.R., Bench S.R., Winget D.M. Methods for the isolation of viruses from environmental samples. Methods Mol. Biol. 2009;501:3–14. doi: 10.1007/978-1-60327-164-6_1. [DOI] [PubMed] [Google Scholar]
- Wylie K.M., Weinstock G.M., Storch G.A. Emerging view of the human virome. Transl. Res. 2012;160:283–290. doi: 10.1016/j.trsl.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]