

Artificial Antigen Presenting Cells (aAPC) are engineered platforms for T cell activation and expansion. By replacing and recapitulating the functions performed by endogenous antigen presenting cells, aAPC serve as cost-effective alternatives to cellular APC for immunotherapy and as minimalist cell-free systems for studying T cell activation.
aAPC are synthesized by coupling necessary T-cell stimulating proteins, such as co-stimulatory proteins and Major Histocompability Complex (MHC)/Human Leukocyte Antigen (HLA) loaded with antigen of interest, to the surface of an appropriate solid support. aAPC have been built upon a wide variety of biocompatible platforms, including cultured cell lines, liposomes, and biodegradable polymer particles, and have been functionalized with a variety of proteins that deliver T cell activating signals [1].
These simplified systems provide the minimum necessary signals for T cell stimulation, leading to robust T cell activation and expansion. However, as reviewed in this issue and elsewhere [2,3], it is becoming increasingly clear that T cell-APC interactions are temporally and spatially complex, with dynamic changes in the lateral organization of surface receptors on both the T cell and APC. Membrane heterogeneity, receptor clustering, and activation-induced membrane rearrangements on several scales are part of a complex molecular machine that underscores T cell activation [4].
This complexity is both a challenge and an opportunity for the biomedical engineer. On one hand, it is precisely the complex molecular mechanisms that underlie T cell receptor function and enable its precision and sensitivity. On the other hand, it is becoming clear that engineered platforms meant to activate immunity are capturing only the most rudimentary interactions that occur during T cell activation.
Here, we review our current understanding of the biophysical and spatial aspects of the T cell-APC interaction and its application to aAPC design. In doing so, we demonstrate how insight into the nature of T cell activation by aAPC flows in both directions. Artificial platforms for T cell activation can serve as models to better understand the endogenous system, and this knowledge can, in turn, be adapted for improved translational platforms for immunotherapy.
1. The Signal 1+2 Paradigm in aAPC Design
A general paradigm for the design of aAPC has been to mimic endogenous T cell activation by selecting T cell activating signals that lead to optimal stimulation. In the healthy host, these are provided by endogenous APC such as macrophages, B cells and dendritic cells (DCs). In aAPC design, these same signals are generated by coupling purified or recombinant proteins to an aAPC platform that can then trigger responses from receptors on the T cell membrane. Studies of T cell activation by aAPC have demonstrated that two signals, termed Signal 1 and Signal 2, are minimally necessary to trigger robust expansion of highly functional T cells (Figure 1).
Figure 1. The Signal 1+2 Paradigm.
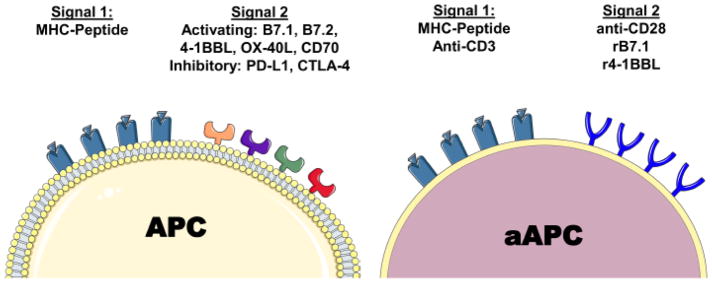
Endogenous APC present two necessary and sufficient signals for T cell activation. Signal 1 is cognate peptide presented in the context of MHC, whereas Signal 2 comprises numerous activating and inhibitory co-stimulatory ligands that bind receptors on T cells. aAPC are synthesized by coupling either specific MHC-peptide complexes or polyclonally activating anti-CD3 antibody as Signal 1, and either activating antibodies against co-stimulatory molecules such as CD28 or recombinant co-stimulatory molecules such as B7.1 (rB7.1).
1.1. Signal 1
Signal 1 is mediated by the interaction of TCR on the T cell with peptide presented by MHC on the APC. Peptide-bearing MHC preferentially interact with T cell receptors specific for one or several MHC-peptide combinations, and thus Signal 1 determines specificity of the T cell response for a given epitope. MHC-binding to TCR triggers activation of the TCR-associated CD3 signaling complex through as-of-yet incompletely understood mechanisms [5,6]. In aAPC design, Signal 1 can be provided by either MHC-peptide binding to TCR, or by engaging the CD3 complex directly with an anti-CD3 antibody (Figure 1).
Soluble Class I and Class II MHC proteins can be produced recombinantly and loaded with appropriate peptide for a variety of antigens of interest. The aAPC engineer must select an MHC allele and peptide that induce a T cell response against the antigen of interest. In humans, HLA-A2*01 has been most frequently studied, based on its high frequency among people of Northern European and American descent. In mice, Kb and Db alleles, as well as Ld, are frequently used based on their presence in the common laboratory strains C57BL6/J and Balb/c, respectively. Following stimulation, the yield and frequency of antigen-specific cells can be monitored using soluble, multimeric MHC reagents.
Alternatively, Signal 1 can be provided by an antibody against the CD3 signaling complex. A variety of activating CD3 antibodies are available, including the OTK3 clone in humans and 145-2C11 in mice. Importantly, activation via CD3 triggers non-specific expansion of all T cells, including regulatory T cells and cells reactive against irrelevant antigens; over time, this can result in preferential expansion of irrelevant cells and reduced activity against the target. Thus, for most applications, a source of T cells enriched for activity against the antigens of interest is desired. In cancer immunotherapy, tumor infiltrating lymphocytes can provide such a source of anti-tumor activity [7], or antigen-specific cells can be purified from Peripheral Blood Mononuclear Cells or other polyclonal sources by HLA-tetramer-based enrichment prior to polyclonal expansion [8]. Unfortunately, anti-CD3/anti-CD28 aAPC have only shown the ability to expand and sustain CD4 [9] but not CD8 T cell cultures [10,11] without additional feeder cell support, making them an appropriate choice only when CD4 cells are required, or when the additional cost and labor associated with culturing autologous feeder cells can be tolerated.
1.2 Signal 2
Signal 2, the co-stimulatory signal, is a series of interactions between receptors on the APC and T cell surface that modify TCR signaling by providing both activating and/or inhibitory signals. The prototypical interaction, between B7.1/B7.2 on the APC and CD28 on the T cell surface, leads to optimal T cell expansion. If Signal 1 is engaged in the absence of Signal 2 in vitro, CD4+ T cells enter a state of anergy, in which T cell proliferation and effector function after re-stimulation are limited [12]. Ineffective Signal 2 stimulation can also lead to the development of suppressive T cells, and the balance between anergy and regulatory development is an area of active study.
Several activating Signal 2 interactions have been identified, including the B7 family proteins B7.1 and B7.2, and their cognate T cell receptor CD28; and the tumor necrosis factor receptor (TNFR) family ligands OX-40L, CD70, 4-1BBL, which interact with OX-40, CD27, and 4-1BB on the T cell, respectively. Certain Signal 2 interactions can also be inhibitory to T cell expansion and effector function, such as the interaction of CTLA-4 with B7.1 or PD-1 with PD-L1 (B7.H1) or PD-L2 (B7-DC)[13].
In aAPC design, Signal 2 can be provided by coupling one or more of the aforementioned APC receptors to the surface of an aAPC platform (Figure 1). In practice, activating antibodies against co-stimulatory T cell receptors, such as the anti-mouse CD28 antibody 37.51, have been shown to be an effective replacement for more costly recombinant APC proteins [14]. While the downstream signaling mechanisms of each co-stimulatory receptor vary, engagement by certain “activating” antibody clones appears to be a near-universal mechanism for triggering co-stimulatory receptor activation.
Minimally, the addition of anti-CD28 antibody is required to design effective aAPC that induce robust T cell proliferation [15,16] and maintain the full complement of T cell effector functions [16,17]. Under certain conditions of extremely strong Signal 1 activation, robust expansion can be observed with Signal 1 alone [18], but the precise phenotypic characteristics of such T cells and their effectiveness in immunotherapy have not been described. On the other hand, many platforms instead rely on multiple co-stimulatory signals delivered simultaneously [19].
The precise signals delivered by each Signal 2 are likely to differ, and thus the choice of Signal 2 protein may be an important parameter in optimizing aAPC for a given application. For example, 4-1BBL may be more effective than anti-CD28 as an activating signal for memory CD8 T cell expansion [20,21], or even synergize with simultaneously presented anti-CD28 [22]. The role of inhibitory signals has been explored to a lesser extent; the addition of a PD-L1 to aAPC does not appear to decrease T cell proliferation [23], but may have a role in shaping the subsequent response.
1.3 Signal 3
In addition to the classic description of Signal 1+2, Signal 3 has been coined as a catch-all term for a variety of soluble signals released by APC that influence T cell activation and development. These include lymphotrophic cytokines such as IL-2, IL-7, and IL-15; inflammatory signals such as TNF-α; and cytokines which modulate T cell development such as TGF-β, IL-12, IL-4, and IL-5. The precise effect of each of these signals is beyond the scope of this review, but the cytokine milieu released by APC before and during T cell activation is a critical determinant of T cell development after activation [24–26].
During in vitro T cell stimulation by aAPC, the addition of exogenous IL-2 is required for robust T cell expansion [27–29]. Alternatively, proliferation can be supported by a variety of common gamma chain cytokines, such as IL-7, IL-15, and IL-21, which may have the further benefit of maintaining T cell replicative potential and inducing memory formation compared to IL-2 [30,31]. These cytokines have been added to aAPC-stimulated cultures to generate cells more amenable to tumor immunotherapy [32]. IL-12 and Type 1 interferons added to culture can support proliferation at low to intermediate antigen doses, and the development of full effector function at any antigen dose [33–35].
There may be a further benefit to delivery of Signal 3 directly from aAPC rather than addition to culture, which could increase local cytokine concentration at the T cell-aAPC interface, spatially co-localize all 3 T cell stimulatory signals, and deliver cytokines to the appropriate site after in vivo administration. Thus far, application of Signal 3 in aAPC design has been limited by the capabilities of aAPC platforms. However, recent developments in design of aAPC based on biodegradable polymers, which can release encapsulated proteins in a spatially localized manner during hydrolytic degradation, have shown that IL-2 delivery from aAPC can significantly enhance T cell proliferation in vitro [36–38]. This “paracrine” delivery was ten-fold more effective in inducing T cell expansion than the same overall dose of IL-2 in the culture media [37].
2. Fixing Signals to a Solid Substrate
aAPC are constructed by coupling T cell activating proteins described in the previous section to a suitable, biocompatible platform. While multimerized, soluble MHC can induce some degree of T cell activation [39–41], large numbers of MHC molecules are required per T cell [42–45]. In contrast, as few as 10 cognate MHC presented on an APC cell surface can be sufficient to induce activation [46,47]. Similarly, fixing MHC-peptide (pMHC) or anti-CD3 to a physical substrate, such as a latex bead, liposome, or microplate surface, significantly enhances the strength of T cell stimulation [40,48–52].
This solid substrate enhancement points to fundamental questions about the mechanism by which TCR is triggered by MHC, which despite recent advances remains an open area of study. Two models, here termed “mechanical triggering” and “rapid re-binding,” may explain this phenomenon. A third complementary model, spatial segregation, will be discussed in detail in subsequent sections.
During T cell activation, fluctuations of the APC membrane can generate a pico- to femtonewton force on the T cell membrane [53]. These forces can also be triggered by a timed series of active pushing and pulling processes on the T cell [54]. Mechanical forces in this range induced by micromanipulation of TCR by pMHC or clonotypic antibodies are sufficient to trigger downstream signaling responses in T cells [55,56], and mechanical signals between T cells and APC enhanced by shear flow triggered stronger T cell stimulation in vitro [57]. Thus, “mechanical triggering” of the TCR has been proposed as a model of TCR activation by MHC, and may play a significant role in signal transmission.
The fixing of soluble MHC to a solid substrate allows the transmission of mechanical forces from a cell or microparticle surface. This is reflected in the dependence of T cell activation on substrate rigidity, with the strongest activation of mouse T cells triggered by ligands presented on polymer substrates with elastic moduli greater than 10–200 kPa [58]. Interestingly, with human T cells, it was noted that elastic moduli significantly above this range impeded stimulation [59], a finding attributed to decreased bond lifetimes under greater loading force [60]. While TCR interactions with anti-CD3 antibody generated the primary traction forces mediated by T cells, engagement of CD28 enhances the TCR associated force via an intracellular signaling pathway mediated by PI3K [61].
Secondly, fixation of Signal 1 to a solid support constrains the receptor-ligand interaction to a planar cell-surface interface and allows for rapid re-binding of cell surface receptors. During physiological T cell/APC interactions, receptors are confined to the two-dimensional axis of cell-cell contact. In contrast, biophysical measurements of TCR-MHC interactions have traditionally been made by characterizing binding of soluble MHC dimers or antibodies measured either by flow cytometry or surface plasmon resonance, which involve a third degree of freedom. “Two dimensional” measurements that more closely re-capitulate TCR-MHC interactions at the cell-cell membrane can be made by assessing interactions of a T cell with an MHC-coated particle [62,63], similar in design to an aAPC. 2D affinity analysis has resulted in the identification of a wider range of cognate T cell ligands [64,65]. Measurements in two dimensions broadly agree with kinetic binding parameters measured in situ, which show large increases in both association (100-fold) and disassociation (4–12 fold) compared to measurements in solution, ultimately resulting in increased overall affinity [66].
This enhanced 2D affinity, and particularly enhanced on-rate from constrained MHC, has led to a “rapid re-binding” model, wherein strong pMHC agonists confined to the surface efficiently rebind the same or neighboring TCR within individual receptor nanoclusters [63,65–68]. Compared to on-rates driven primarily by MHC diffusion, re-binding that takes place in a constrained two-dimensional membrane is significantly enhanced. In this model, the assembly of downstream signaling ligands can tolerate brief loss of contact between TCR and MHC as long as the receptor is efficiently rebound. Thus, the ability of cognate MHC-peptide to trigger TCR activation depends not only on the kinetic off-rate, as had been proposed by earlier models [69], but also the on-rate-the ability of MHC to re-bind the same or neighboring [68,70] TCR.
MHC can be fixed to a number of different platforms, from easily cultured cell lines to biodegradable polymer scaffolds. While different platforms possess different mechanical and surface properties, the most commonly used substrates all share in common biocompatibility and ease of synthesis. The earliest platforms were APC-like cells based on cultured cell lines [71,72], particularly the K562 human erythromyeloid line [19,20,73–76] and the murine NIH/3T3 fibroblast line [77–79], engineered to express Signal 1 and Signal 2. Among the earliest cell-free platforms used for T cell activation were liposomes, spherical vesicles with an aqueous interior generated by self-assembly of amphiphilic phospholipids and cholesterol, also modified to express the relevant T cell-activating signals [50,51,80–83].
Synthetic substrates have the additional benefit of having tunable mechanical, geometrical, and biodegradable properties, either by adjusting the synthesis method or base material. Sepharose [84,85] and latex (polystyrene) beads [10,28,86–89] were the first synthetic bead-based platforms used in aAPC design, and have been instrumental as reductionist systems for studying basic aspects of T cell biology. Iron-dextran microparticles have also been extensively characterized as aAPC, both for translational applications of tumor-specific T cell expansion [15,90] and as a tool for the study of basic aspects of T cell antigen recognition and development [91–94]. Additionally, microparticles can be synthesized from a variety of biodegradable polymers, such as poly (lactic acid) (PLA), poly (glycolic acid) (PGA), and their co-polymer, poly (lactic-co-glycolic acid) (PLGA) and used for antigen-specific T cell activation [38,95], with significant biocompatibility advantages over non-degradable platforms such as polystyrene.
3. Microscale T Cell-aAPC Interactions
Thus far, the primary focus of aAPC design has been the selection of T cell stimulating signals and platforms to which they are coupled. However, it has become evident that receptor organization between the T cell and APC plays an additional important role in endogenous activation. Recapitulating and even enhancing this aspect of T cell activation represents a new frontier in optimal aAPC design. Here, we will briefly review relevant spatial considerations of natural T cell-APC interactions.
The idea that spatial organization of the TCR and accessory signaling proteins plays a critical role in T cell activation gained support with the discovery of the immune synapse [96], a micro-scale cell-cell interaction structure formed during activation of T cells by APC. During immune synapse formation, adhesion molecules such as LFA-1 migrate to the periphery of the T cell contact site, the peripheral supramolecular activation cluster (pSMAC), whereas TCR, CD3, and other signaling proteins are found in the central SMAC (cSMAC) [97] (Figure 2). TCR nanoclusters form at the periphery of contact and traverse toward the cSMAC [98] where they are subsequently degraded. However, the discovery of the synapse was soon followed by studies showing that synapse formation, which occurs minutes after T cell-APC contact, was preceded by strong TCR signaling from TCR nanoclusters and was thus only a piece of the T cell activation puzzle [99]. Today, it is appreciated that the nature of the synapse varies with the strength and duration of stimulus, the activation state of the T cell, and the nature of the APC [100]. Additionally, the synapse can perform numerous functions, from turning off signaling [101] to providing a cell-cell contact zone for exchange of cytokines and cytotoxic granules [102].
Figure 2. Scales of Organization.

T cell membrane organization at several scales contributes to signaling and activation. TCR are pre-clustered in 15–30 nm nanoclusters prior to T cell activation, particularly on the surface of previously activated or memory cells. Upon MHC binding, 35–70 nm signaling nanoclusters form that drive downstream signaling. These clusters migrate to the center of the T cell-APC contact site, forming the cSMAC, and are subsequently internalized and degraded. Adhesion molecules such as LFA-1 are distributed in the pSMAC, which together with the cSMAC forms the micron-scale immune synapse.
Fundamental insights into the nature of the synapse have been made using planar lipid bilayers, acellular T cell activation platforms similar in concept to aAPC (reviewed in [103]). Planar synthetic surfaces can be designed using numerous techniques, most commonly by inserting freely diffusible, GPI-anchored T cell activating proteins into lipid membranes coated onto glass slides. The flat contact surface allows the use of imaging techniques with high spatial and/or temporal resolution such as total internal reflection microscopy [104], as well as control over activation parameters such as ligand strength and density.
New surface fabrication techniques allow T cell activating ligands to be patterned in precisely controlled ways on planar bilayers, providing a tool to study the effect of microscale APC membrane organization on T cell activation. For example, the presence of anti-CD28 at the periphery of the T cell contact site on planar arrays containing anti-CD3, CD28, and ICAM-1 significantly enhanced IL-2 secretion and downstream signaling by mouse CD4 T cells [105]. Similarly, mechanically trapping TCR nanoclusters at the peripheral regions of a forming synapse significantly prolonged and strengthened signaling from TCR nanoclusters [106]. However, separation of anti-CD3 and anti-CD28 contact sites by several microns in human T cells curtailed co-stimulatory activity [107]. Thus, while existing aAPC fabrication techniques generally rely on randomly distributed ligands, these findings suggest it may be possible to fine-tune T cell activation from aAPC by patterning activating ligands in ways that mimic T cell-APC interaction. However, this will require the development of technologies for heterogeneous ligand distribution on particle surfaces [108,109] (Figure 3A).
Figure 3. Scales of Organization.

A. Studies from patterned lipid bilayers suggest that T cells preferentially activate against surfaces with MHC-peptide (purple) forming the center of the contact site, and anti-CD28 (orange) distributed in the periphery. In contrast, most aAPC are synthesized with uncontrolled protein distribution
B. Ellipsoid rather than spherical micro-aAPC provide a greater surface area and decreased surface curvature for optimal T cell engagement, leading to enhanced antigen-specific T cell activation and proliferation.
C. Nanoscale aAPC, less than 100 nm in diameter, have recently been shown to be capable of activating T cells. These nano-aAPC (orange) can “sense” nanoscale TCR distribution, as they preferentially bind to activated T cells which have more clustered TCR (purple) than naive cells. This leads to higher avidity for aAPC binding, but fewer aAPC bound to each T cell, since each aAPC binds multiple TCR. This effect enhanced activation of activated cells by nano-aAPC, whereas this preference was not observed with micro-aAPC.
More effective aAPC mediated stimulation may also be possible by mimicking the micro-scale, cytoskeletal membrane rearrangements that occur at the T cell-APC interface. T cells have a sensitive leading edge that makes primary contact with APC, triggering a cell-cell interaction program [110] that leads to cellular flattening and an augmented area of surface contact [111]. Simultaneously, cytoskeletal rearrangements in the APC drive membrane polarization [112] and increased surface contact.
In contrast, most synthetic aAPC platforms are spherical, a shape which is easily synthesized using standard chemical synthesis procedures such as double-emulsion of PLGA [113]. This geometry not only fails to recapitulate APC membrane dynamics observed during activation, but also minimizes the surface area and maximizes the curvature at the T cell-aAPC interface. Recently, we utilized a novel particle fabrication method to control microparticle geometry with defined MHC dose and density [114]. Compared to spherical aAPC, ellipsoidal aAPC with increased aspect ratio and surface area preferentially engaged cognate T cells, with T cells observed to favor engaging aAPC along their long axis (Figure 3B). This interaction was reflected in enhanced T cell expansion and in vivo tumor killing activity after stimulation by ellipsoidal aAPC and highlights the importance of geometry considerations in aAPC design. Similar synthesis techniques could be used to mimic additional membrane geometries relevant to T cell-APC interactions, such as membrane protrusions/lamellopodia that enhance close membrane apposition [115].
Particle geometry also influences uptake by the reticuloendothelial system, an important consideration for aAPC that are directly administered in vivo. Ellipsoidal particles with a long characteristic axis show reduced phagocytosis [116–119]. Interestingly, receptor-mediated internalization of antibody coated non-spherical particles is significantly enhanced, suggesting the interaction of geometry and uptake may be pathway dependent [120]. These contrasting properties of identically shaped aAPC may be used to their advantage to prolong circulation time where receptor-mediated internalization is preferred, such as in drug delivery applications. In either case, in vitro uptake studies may not mimic in vivo behavior; adsorption of complement and other serum proteins significantly alter trafficking and clearance characteristics of in vivo administered aAPC.
4. Nanoscale Clustering and Nanoscale aAPC
Upon engagement with cognate antigen, TCR microclusters, estimated to be 35–70 nm in diameter and containing 7–20 TCR [121], form at the periphery of the T cell-APC contact site (first described in [122] and reviewed in [2]). These clusters are enriched for signaling molecules such as Lck, Zap-70, Lat, and SLP76, suggesting that they are directly involved in T cell signaling. Microcluster formation precedes but is associated with synapse formation, as both involve a cortical F-actin flow that leads to inward migration of TCR.
Spatial heterogeneity in membrane organization and TCR clustering can be detected even in the absence of stimulation by cognate antigen. Using immunoprecipitation and immunoblotting techniques, Fernandez-Miguel et al. demonstrated that the αβ T cell receptor can exist as a multivalent structure on naive T cells, composed of at least two TCR and a higher order CD3 stoichiometry [123]. TCR likely exist in several distinct monovalent and multivalent forms prior to antigen engagement [124], with the number of TCR in a cluster prior to activation ranging from a single receptor to 20 [125] within clusters that are 15–30 nm in diameter [126]. Although both TCR clusters formed prior to and after antigen engagement are generally less than 100 nm in diameter, the former have been termed “nanoclusters,” and the later, “microclusters.” The precise relationship between these structures has not been fully elucidated; however, it is possible that TCR nanoclusters concatenate to form the larger activation microclusters that are observed by TIRF [121]. Thus, receptor organization at both micro- and nanoscales play a central role in T cell activation (Figure 2).
In parallel, spatial heterogeneity of MHC on the APC surface mirrors the T cell membrane. Near-field scanning techniques have resolved protein-rich “patches” of MHC with radii between 70 and 600 nm on the surface of resting APC [127], containing approximately 25–125 MHC each. During antigen processing, MHC bearing peptides derived from a given pathogen are deposited as a cluster on the APC membrane [128–130], generating peptide-specific clusters that facilitate interaction with peptide-specific TCR. Subsequently, during T cell activation, MHC move in concert with their binding partners to form clusters in the cSMAC zone of the APC, with adhesion molecules such as ICAM-1 migrating to the pSMAC. These pre-formed clusters and subsequent movements are controlled by the intricate interactions of lipid and protein domains, such as lipid rafts and tetraspanin domains in the membrane [131], as well as protein interactions with the actin cytoskeleton [131].
Spatial rearrangements may serve as a critical piece of the mechanism behind T cell triggering after engagement by cognate MHC [132]. A common element in spatial activation models is kinase concentration and phosphatase exclusion – phosphorylating proteins are concentrated in the region that contains their substrate, whereas dephosphorylating proteins are excluded, tipping the local balance in favor of signal transduction. Consistent with this hypothesis, it was recently shown that close apposition between the T cell and APC membrane drove exclusion of proteins such as the phosphatase CD45, whereas the binding energy between TCR and pMHC was sufficient to keep these proteins within the central contact area [5]. This mechanism was sufficient for T cell signaling in a reconstituted system. Furthermore, new high-resolution imaging techniques have demonstrated nanoscale co-localization of TCR and downstream signaling proteins, which is enhanced after cognate pMHC binding [121]. Furthermore, spatial co-localization may play a complementary role with other activation models, including induction of conformational changes in the TCR-CD3 complex that initiate downstream activation [133].
Receptor clustering prior to ligand engagement can be a higher order mechanism which enhances binding and sensitivity. For example, clustering of chemo-receptors on E coli. significantly lowers the threshold and enhances the dynamic range of chemotactic responses [134]. Preliminary evidence suggests that such clustering may be partially responsible for the high degree of T cell sensitivity to antigen; for example, only multivalent but not monovalent TCR/CD3 complexes were phosphorylated after stimulation with a low dose of antigen [125,135]. Enhanced clustering after activation may also partially explain the increased sensitivity of previously activated T cells to antigen [136,137], acting in concert with changes in downstream signaling [138]. Similarly, clustering MHC on an APC surface augments T cell recognition [139]. Thus, receptor organization likely plays a significant role in the exquisite sensitivity and specificity of the TCR-MHC interaction, motivating its careful consideration in aAPC design.
4.1. MHC Valency and Density
With the importance of MHC clustering and spatial organization in endogenous T cell activation, it is no surprise that this parameter has been carefully explored for the design of artificial T cell activation platforms. Soluble MHC monomers, separated from their natural context in the APC membrane, have low, micromolar affinity for cognate TCR. This prompted the development of multimeric MHC constructs such as dimers and tetramers, which enhance overall binding avidity [140,141]. T cell triggering by these soluble multimeric MHC is highly dependent on intramolecular distances between MHC within the protein construct, with shorter intramolecular cross-linkers being significantly more effective than longer cross-linkers, and a significant decrease in stimulatory activity occurring at a distance of approximately 8 nm [142].
By analogy, while monomeric MHC coupled to aAPC microspheres can trigger T cell activation [48,88], studies with multivalent MHC suggest that multivalency induces stronger responses [49,143]. However, no comprehensive examination between such constructs has been performed. Furthermore, multivalent MHC constructs can have other biophysical disadvantages: commonly used MHC tetramers have been coupled aAPC [144–146], but their rigid tetrahedral geometry orients a fraction of MHC molecules toward the particle surface and consequently away from the interacting T cell.
A similar effect to MHC multivalency may be mediated by controlling density of monomeric MHC proteins on an APC surface. Antigen density on APC membranes is known to affect subsequent T cell response [147–150]. This principle may also apply to aAPC design, although antigen presented on aAPC is often presented at supraphysiologically high doses compared to the small numbers of cognate MHC-peptides that trigger T cell activation.
On planar lipid arrays functionalized with anti-CD3 antibody in a controlled and homogenous fashion, anti-CD3 spacing of less than 70 nm is required to observe T cell activation [151]. Using peptide-MHC “corrals” patterned on lipid arrays, the Groves group demonstrated that thresholds for T cell triggering are determined by the number of activating ligands available to individual TCR clusters, not the total MHC available to the entire cell [152]. A similar density threshold is observed for monomeric MHC presented on spherical microparticles, and the addition of more beads at a sub-threshold density is not sufficient to overcome this effect [10,49]. Additionally, the density of MHC may control the avidity of the resulting cultured T cells, with an inverse relationship between T cell avidity and MHC density. Higher avidity T cells result from aAPC with a lower MHC density, likely via clonal competition, and high density aAPC increase activation of low-avidity clones [153]. Thus, if high avidity T cell responses are desired, investigators must titrate density to achieve a balance between T cell quantity and quality.
Furthermore, there may be advantages to manufacturing aAPC with heterogenous or clustered protein signals which mimic the distribution of MHC on endogenous APC membranes. One study used anti-CD3, -CD28, and -LFA-1 monoclonal antibodies pre-clustered on liposomes using neutravidin rafts to efficiently activate MART-1 specific CD8 T cells [50], although the precise role of clustering in enhancing T cell responses could not be clearly defined. In a separate liposome-based study, CD4 stimulation was significantly stronger from aAPC presenting clustered compared to unclustered MHC [154]. A high density (5–8 nm separating distances) of clustered non-cognate MHC presented on the surface of nano-sized quantum dots is required to enhance binding and activation by cognate pMHC [155], a finding that may explain conflicting results obtained with the “pseudo-dimer” theory, which states that numerous weak binding events with non-cognate MHC augment less frequent strong binding events mediated by cognate MHC.
Further application of heterogeneous MHC clustering on aAPC will require the development of biocompatible, readily synthesized platforms with spatially controlled receptor patterning [109]. For example, the Little group recently reported a technique based on interfacial condensation of a liquid mask to create microspheres with “patchy” protein islets [156].
4.2. Nanoparticle aAPC Can Activate T cells and Sense Membrane Organization
Studies on synthetic bead-based aAPC have largely focused on the development of cell-sized, micro-scale aAPC in order to better mimic T cells interaction with antigen presenting cells. This choice is theoretically reinforced by microscale T cell-APC interactions described above. In fact, early studies by the Mescher group suggested only bead-based aAPC larger than 2 microns in diameter are able to induce T cell proliferation [88,157]. Steenblock et al.[38] demonstrated that polymer-based nanoparticles were much less efficient than microbeads in inducing short-term functional responses, with no reported proliferation. However, recent discoveries of TCR organization at the nanoscale reignited interest in developing nano-sized aAPC particles, which might theoretically be able to interact with individual TCR nanoclusters to stimulate T cells and “sense” TCR distribution.
Several publications have now demonstrated that nanoparticles less than 100 nm in diameter can trigger robust T cell activation. For example, our group developed “nano-aAPC” by coupling dimeric MHC-immunoglobulins and anti-CD28 to the surface of iron-dextran nanoparticles. These nano-aAPC are able to induce robust T cell expansion in vitro and have significant advantages over micron-sized particles. Unlike microparticles, nanoparticles of approximately 50–100 nm diameter can be transported by lymphatics to the lymph nodes [158,159], thus gaining access to a larger pool of T cells. Enhanced drainage of 50–100 nm aAPC compared to micron sized aAPC was observed in our hands [160], although even smaller nanoparticles may be required for optimal lymphatic transport [161]. In addition, nanoscale delivery vehicles may preferentially accumulate in tumors through the enhanced permeability and retention effect which is attributed to the tumor’s tendency toward leaky vasculature and poorly organized lymphatic drainage [162,163]. By delivering an immunostimulatory signal in situ, aAPC in the tumor microenvironment may address one of the most prominent hurdles in cancer immunotherapy, the immunosuppressive tumor microenvironment [164].
In addition to inducing antigen-specific T cell activation, nano-aAPC binding to TCR nanoclusters can be used to gain qualitative information on TCR clustering of T cells in various physiological states, such as naive and activated cells. In particular, TCR undergo a state-dependent, persistent increase in the extent of TCR clustering days to weeks after activation. Binding assays using dimeric MHC-Ig fusion proteins demonstrated that T cells activated four days previously showed enhanced binding of low concentrations of MHC, with a high degree of cooperativity [136,140]. Since this effect was not observed with monovalent MHC, the authors determined that a higher degree of clustering led to the enhanced binding of MHC dimers. Persistent TCR clusters on activated cells was directly visualized using electron [137] and k-Space Image Correlation microscopy [165], with a distinct increase in clustering noted as compared to naive cells.
Differences in TCR clustering modify the T cell’s ability to bind to and be activated by cognate pMHC. CD3 functionalized nanoparticles preferentially augment the expansion of antigen-experienced but not naive T cells that are concurrently stimulated with endogenous APC [166]. Recent work by our group demonstrates that nano-aAPC favor binding to clusters which are characteristic of activated rather than naive CD8 T cells [167]; furthermore, by exploiting the paramagnetic properties of iron-dextran nanoparticles, we demonstrated that magnet induced clustering of the cell-bound nanoparticles, and consequently their associated TCR clusters, was sufficient to induce robust activation of otherwise poorly sensitive naive T cells. This technique can be used to boost nano-aAPC mediated activation of naive T cells, and suggests an additional mechanism by which T cells more efficiently detect cognate antigen subsequent to activation.
5. Applications of aAPC: Past and Future
Despite these insights into the complex interactions underscoring T cell activation, most aAPC designs remain straightforward applications of the Signal 1 + 2 paradigm, with T cell activating proteins fixed in an uncontrolled manner to spherical microscale particles. This choice reflects the ease and simplicity of developing such systems, but does not reflect the rapid pace at which our understanding of T cell-APC interactions is advancing.
aAPC have been utilized as therapeutic platforms primarily in one of two ways. The first, direct vaccination, involves the direct administration of aAPC into a host, where aAPC must traffic to and co-localize with host T cells. The earliest reports of this approach involved 5 μm diameter silica microbeads coated with MHC-antigen but no Signal 2, injected intraperitoneally into tumor-bearing mice [48]. However, no antigen-specific responses or tumor activity could be detected unless a cell-based tumor vaccine was co-administered, suggesting that aAPC could only boost an existing response. This platform was subsequently assessed in a Phase I trial of patients with disseminated melanoma [168], with 8/15 patients developing antigen-specific cytolytic T cells, but only one partial tumor response. In mice, iron-dextran micro-aAPC bearing dimeric MHC-peptide and anti-CD28 injected intravenously [15] can mediate regression of both subcutaneous melanoma and intravenous lung metastases. Similarly, latex particles administered both intravenously and subcutaneously generated robust antigen-specific T cell responses in mice, and subcutaneously administered microparticles mediated B16 melanoma rejection [146].
This approach is particularly relevant to disease states like cancer, where endogenous antigen-presenting mechanisms are defective [169–171] and therefore motivate a desire to replace or supplement the endogenous APC compartment. Within the tumor microenvironment, APC can go beyond dysfunction to active immune inhibition [172–174]. Thus, even if antigen is delivered in a vaccine platform to the host APC, it may not lead to T cell activation, motivating the use of aAPC in vivo.
Thus, induction of tumor-specific cytolytic T cells does not necessarily correlate with tumor regression in human studies, a finding at least partially attributed to the suppressive mechanisms of the tumor microenvironment. Direct vaccination with aAPC therefore has significant unexplored potential, particularly as immunomodulatory strategies such as “checkpoint blockade” are developed to temper these suppressive mechanisms that doomed the first generation of cancer vaccines [175]. Considerations of aAPC size and shape are likely to feature prominently in this approach, as they influence both the in vivo trafficking and subsequent T cell interactions of administered aAPC.
The second approach, adoptive immunotherapy, involves the generation of antigen-specific T cells in vitro and adoptive transfer into a patient or animal model. For example, adoptive transfer of large numbers of tumor specific T cells generated from melanoma cultures can mediate complete and durable regression of even large and metastatic tumors [7], and adoptive transfer of CMV-specific lymphocytes has been studied as a means of limiting immunosuppression post-transplant [14,176]. This approach has several advantages, including complete control of T cell culture environment during growth, and avoids the problem of attaining optimal aAPC biodistribution in vivo. On the other hand, in vitro T cell culture is costly and labor-intensive compared to direct aAPC administration, and can be described as designing a “new drug” for each patient.
Moving forward, aAPC have tremendous potential as a platform for inducing T cell expansion and for studying fundamental aspects of TCR organization and signaling. New breakthroughs in controlled micropatterning and shape manipulation on the nanoscale will allow for optimization of T cell activation mechanisms. aAPC can be produced for many patients in bulk and designed from easily manufactured, biocompatible platforms, creating off-the-shelf reagents that has significantly reduced cost compared to cellular APC. Adoptive T cell immunotherapy is limited in part by the need to generate large numbers (up to 1010) of tumor-specific T cells quickly and reliably [177–179], which motivates the application of more advanced, biophysical understanding of T cell activation to optimize T cell expansion. The most rapid and robust T cell proliferation may only be possible when micro- and nanoscale considerations are taken into account. Thus, regardless of the therapeutic strategy chosen, size, shape, scale and surface distribution should be carefully considered as elements of effective aAPC design.
Highlights.
Micro- and nanoscale interactions at the membrane shape T cell responses
Fixing T cell stimulating molecules to solid substrates induces robust activation
Valency and spatial arrangements of signaling ligands modify T cell activation
Artificial antigen presenting cell shape affects cell interaction and biodistribution
Future aAPC design will incorporate biophysical insights
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Oelke M, Schneck JP. Overview of a HLA-Ig based “Lego-like system” for T cell monitoring, modulation and expansion. Immunol Res. 2010;47:248–56. doi: 10.1007/s12026-009-8156-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dustin ML, Groves JT. Receptor signaling clusters in the immune synapse. Annu Rev Biophys. 2012;41:543–56. doi: 10.1146/annurev-biophys-042910-155238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie J, Tato CM, Davis MM. How the immune system talks to itself: the varied role of synapses. Immunol Rev. 2013;251:65–79. doi: 10.1111/imr.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grakoui a. The Immunological Synapse: A Molecular Machine Controlling T Cell Activation. Science (80-) 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [Google Scholar]
- 5.James JR, Vale RD. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature. 2012;487:64–9. doi: 10.1038/nature11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith-Garvin JE, Koretzky Ga, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Restifo NP, Dudley ME, Rosenberg Sa. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–81. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Labarrière N, Gervois N, Bonnin A, Bouquié R, Jotereau F, Lang F. PBMC are as good a source of tumor-reactive T lymphocytes as TIL after selection by Melan-A/A2 multimer immunomagnetic sorting. Cancer Immunol Immunother. 2008;57:185–95. doi: 10.1007/s00262-007-0361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB, et al. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol. 1997;159:5921–30. [PubMed] [Google Scholar]
- 10.Deeths M, Mescher M. B7-1-dependent co-stimulation results in qualitatively and quantitatively different responses by CD4+ and CD8+ T cells. Eur J Immunol. 1997:598–608. doi: 10.1002/eji.1830270305. [DOI] [PubMed] [Google Scholar]
- 11.Laux I, Khoshnan a, Tindell C, Bae D, Zhu X, June CH, et al. Response differences between human CD4(+) and CD8(+) T-cells during CD28 costimulation: implications for immune cell-based therapies and studies related to the expansion of double-positive T-cells during aging. Clin Immunol. 2000;96:187–97. doi: 10.1006/clim.2000.4902. [DOI] [PubMed] [Google Scholar]
- 12.Fathman CG, Lineberry NB. Molecular mechanisms of CD4+ T-cell anergy. Nat Rev Immunol. 2007;7:599–609. doi: 10.1038/nri2131. [DOI] [PubMed] [Google Scholar]
- 13.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8:467–77. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- 14.Riddell SR, Greenberg PD. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J Immunol Methods. 1990;128:189–201. doi: 10.1016/0022-1759(90)90210-m. [DOI] [PubMed] [Google Scholar]
- 15.Ugel S, Zoso A, De Santo C, Li Y, Marigo I, Zanovello P, et al. In vivo administration of artificial antigen-presenting cells activates low-avidity T cells for treatment of cancer. Cancer Res. 2009;69:9376–84. doi: 10.1158/0008-5472.CAN-09-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hombach A, Sent D, Schneider C, Heuser C. Receptors CD28 Costimulation Is Required for Interleukin 2 Secretion and Receptor-mediated T-Cell Proliferation but Does Not Affect Receptor-mediated Target Cell. Cancer Res. 2001;61:1976–1982. [PubMed] [Google Scholar]
- 17.Mescher MF, Popescu FE, Gerner M, Hammerbeck CD, Curtsinger JM. Activation-induced non-responsiveness (anergy) limits CD8 T cell responses to tumors. Semin Cancer Biol. 2007;17:299–308. doi: 10.1016/j.semcancer.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luxembourg aT, Brunmark A, Kong Y, Jackson MR, Peterson Pa, Sprent J, et al. Requirements for stimulating naive CD8+ T cells via signal 1 alone. J Immunol. 1998;161:5226–35. [PubMed] [Google Scholar]
- 19.Butler MO, Lee J-S, Ansén S, Neuberg D, Hodi FS, Murray AP, et al. Long-lived antitumor CD8+ lymphocytes for adoptive therapy generated using an artificial antigen-presenting cell. Clin Cancer Res. 2007;13:1857–67. doi: 10.1158/1078-0432.CCR-06-1905. [DOI] [PubMed] [Google Scholar]
- 20.Sluijter BJR, van den Hout MFCM, Stam aGM, Lougheed SM, Suhoski MM, van den Eertwegh aJM, et al. 4-1BB-mediated expansion affords superior detection of in vivo primed effector memory CD8+ T cells from melanoma sentinel lymph nodes. Clin Immunol. 2010;137:221–33. doi: 10.1016/j.clim.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Snyder KM, Suhoski MM, Maus MV, Kapoor V, June CH, et al. 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J Immunol. 2007;179:4910–8. doi: 10.4049/jimmunol.179.7.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudolf D, Silberzahn T, Walter S, Maurer D, Engelhard J, Wernet D, et al. Potent costimulation of human CD8 T cells by anti-4-1BB and anti-CD28 on synthetic artificial antigen presenting cells. Cancer Immunol Immunother. 2008;57:175–83. doi: 10.1007/s00262-007-0360-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuertes Marraco Sa, Baumgaertner P, Legat A, Rufer N, Speiser DE. A stepwise protocol to coat aAPC beads prevents out-competition of anti-CD3 mAb and consequent experimental artefacts. J Immunol Methods. 2012;385:90–5. doi: 10.1016/j.jim.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 24.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–89. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richer MJ, Nolz JC, Harty JT. Pathogen-Specific Inflammatory Milieux Tune the Antigen Sensitivity of CD8+ T Cells by Enhancing T Cell Receptor Signaling. Immunity. 2012:1–13. doi: 10.1016/j.immuni.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–62. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Garlie N, LeFever A. T cells coactivated with immobilized anti-CD3 and anti-CD28 as potential immunotherapy for cancer. J Immunother. 1999;22:336–345. doi: 10.1097/00002371-199907000-00007. [DOI] [PubMed] [Google Scholar]
- 28.Curtsinger JM, Schmidt CS, Mondino A, Lins C, Kedl RM, Jenkins MK, et al. Inflammatory Cytokines Provide a Third Signal for Activation of Naive CD4 + and CD8 + T Cells. J Immunol. 1999;162:3256–3262. [PubMed] [Google Scholar]
- 29.Mescher MF, Curtsinger JM, Agarwal P, Casey Ka, Gerner M, Hammerbeck CD, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. 2006;211:81–92. doi: 10.1111/j.0105-2896.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 30.Yang S, Ji Y, Gattinoni L, Zhang L, Yu Z, Restifo NP, et al. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol Immunother. 2013;62:727–36. doi: 10.1007/s00262-012-1378-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111:5326–33. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butler MO, Friedlander P, Milstein MI, Mooney MM, Metzler G, Murray AP, et al. Establishment of antitumor memory in humans using in vitro-educated CD8+ T cells. Sci Transl Med. 2011;3:80ra34. doi: 10.1126/scitranslmed.3002207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–51. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curtsinger J, Valenzuela J. Cutting edge: type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005:8–13. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 35.Curtsinger J, Lins D. Signal 3 tolerant CD8 T cells degranulate in response to antigen but lack granzyme B to mediate cytolysis. J Immunol. 2005;175:4392–4399. doi: 10.4049/jimmunol.175.7.4392. [DOI] [PubMed] [Google Scholar]
- 36.Han H, Peng J-R, Chen P-C, Gong L, Qiao S-S, Wang W-Z, et al. A novel system of artificial antigen-presenting cells efficiently stimulates Flu peptide-specific cytotoxic T cells in vitro. Biochem Biophys Res Commun. 2011;411:530–5. doi: 10.1016/j.bbrc.2011.06.164. [DOI] [PubMed] [Google Scholar]
- 37.Steenblock ER, Fadel T, Labowsky M, Pober JS, Fahmy TM. An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response. J Biol Chem. 2011;286:34883–92. doi: 10.1074/jbc.M111.276329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steenblock ER, Fahmy TM. A comprehensive platform for ex vivo T-cell expansion based on biodegradable polymeric artificial antigen-presenting cells. Mol Ther. 2008;16:765–72. doi: 10.1038/mt.2008.11. [DOI] [PubMed] [Google Scholar]
- 39.Ge Q, Stone JD, Thompson MT, Cochran JR, Rushe M, Eisen HN, et al. Soluble peptide-MHC monomers cause activation of CD8+ T cells through transfer of the peptide to T cell MHC molecules. Proc Natl Acad Sci U S A. 2002;99:13729–34. doi: 10.1073/pnas.212515299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Kurlander RJ. Comparison of anti-CD3 and anti-CD28-coated beads with soluble anti-CD3 for expanding human T cells: differing impact on CD8 T cell phenotype and responsiveness to restimulation. J Transl Med. 2010;8:104. doi: 10.1186/1479-5876-8-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maile R, Wang B, Schooler W, Meyer a, Collins EJ, Frelinger Ja. Antigen-specific modulation of an immune response by in vivo administration of soluble MHC class I tetramers. J Immunol. 2001;167:3708–14. doi: 10.4049/jimmunol.167.7.3708. [DOI] [PubMed] [Google Scholar]
- 42.Appel H, Gauthier L, Pyrdol J, Wucherpfennig KW. Kinetics of T-cell Receptor Binding by Bivalent HLA-DR-Peptide Complexes That Activate Antgen-specific Human T-cells. J Biol Chem. 2000;275:312–321. doi: 10.1074/jbc.275.1.312. [DOI] [PubMed] [Google Scholar]
- 43.Abastado JP, Lone YC, Casrouge a, Boulot G, Kourilsky P. Dimerization of soluble major histocompatibility complex-peptide complexes is sufficient for activation of T cell hybridoma and induction of unresponsiveness. J Exp Med. 1995;182:439–47. doi: 10.1084/jem.182.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stone JD, Cochran JR, Stern LJ. T-cell activation by soluble MHC oligomers can be described by a two-parameter binding model. Biophys J. 2001;81:2547–57. doi: 10.1016/S0006-3495(01)75899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abdel HR, O’Herrin SMO, Lebowitz MS, Srikrishnan A, Bieler J, Schneck J, et al. Potent T Cell Activation with Dimeric Class II Ligand: The Role of CD4 Coreceptor. J Exp Med. 1998;188:4–11. doi: 10.1084/jem.188.9.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irvine DJ, Purbhoo Ma, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–9. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
- 47.Ma Z, Sharp Ka, Janmey Pa, Finkel TH. Surface-anchored monomeric agonist pMHCs alone trigger TCR with high sensitivity. PLoS Biol. 2008;6:e43. doi: 10.1371/journal.pbio.0060043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rogers J, Mescher MF. Augmentation of in vivo cytotoxic T lymphocyte activity and reduction of tumor growth by large multivalent immunogen. J Immunol. 1992;149:269–76. [PubMed] [Google Scholar]
- 49.Motta I, Lone YC, Kourilsky P. In vitro induction of naive cytotoxic T lymphocytes with complexes of peptide and recombinant MHC class I molecules coated onto beads: role of TCR/ligand density. Eur J Immunol. 1998;28:3685–95. doi: 10.1002/(SICI)1521-4141(199811)28:11<3685::AID-IMMU3685>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 50.Zappasodi R, Di Nicola M, Carlo-Stella C, Mortarini R, Molla A, Vegetti C, et al. The effect of artificial antigen-presenting cells with preclustered anti-CD28/-CD3/-LFA-1 monoclonal antibodies on the induction of ex vivo expansion of functional human antitumor T cells. Haematologica. 2008;93:1523–34. doi: 10.3324/haematol.12521. [DOI] [PubMed] [Google Scholar]
- 51.Herrmann S, Mescher M. The requirements for antigen multivalency in class I antigen recognition and triggering of primed precursor cytolytic T lymphocytes. J Immunol. 1986:2816–2825. [PubMed] [Google Scholar]
- 52.Lamers C. Optimization of culture conditions for activation and large-scale expansion of human T lymphocytes for bispecific antibody-directed cellular immunotherapy. Int J Cancer. 1992;979:973–979. doi: 10.1002/ijc.2910510623. [DOI] [PubMed] [Google Scholar]
- 53.Henry N, Hivroz C. Early T-cell activation biophysics. HFSP J. 2009;3:401–11. doi: 10.2976/1.3254098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Husson J, Chemin K, Bohineust A, Hivroz C, Henry N. Force generation upon T cell receptor engagement. PLoS One. 2011;6:e19680. doi: 10.1371/journal.pone.0019680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y-C, Chen B-M, Wu P-C, Cheng T-L, Kao L-S, Tao M-H, et al. Cutting Edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J Immunol. 2010;184:5959–63. doi: 10.4049/jimmunol.0900775. [DOI] [PubMed] [Google Scholar]
- 56.Kim ST, Takeuchi K, Sun Z-YJ, Touma M, Castro CE, Fahmy A, et al. The alphabeta T cell receptor is an anisotropic mechanosensor. J Biol Chem. 2009;284:31028–37. doi: 10.1074/jbc.M109.052712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lim TS, Mortellaro A, Lim CT, Hämmerling GJ, Ricciardi-castagnoli P. Mechanical Interactions between Dendritic Cells and T Cells Correlate with T Cell Responsiveness. J Immunol. 2012;187:258–265. doi: 10.4049/jimmunol.1100267. [DOI] [PubMed] [Google Scholar]
- 58.Judokusumo E, Tabdanov E, Kumari S, Dustin ML, Kam LC. Mechanosensing in T Lymphocyte Activation. Biophysj. 2012;102:L5–L7. doi: 10.1016/j.bpj.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Connor RS, Hao X, Shen K, Bashour K, Akimova T, Hancock WW, et al. Substrate rigidity regulates human T cell activation and proliferation. J Immunol. 2012;189:1330–9. doi: 10.4049/jimmunol.1102757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Evans E. Probing the relation between force-lifetime-and chemistry in single molecular bonds. Annu Rev Biophys Biomol …. 2001;30:105–28. doi: 10.1146/annurev.biophys.30.1.105. [DOI] [PubMed] [Google Scholar]
- 61.Bashour KT, Gondarenko A, Chen H, Shen K, Liu X, Huse M, et al. CD28 and CD3 have complementary roles in T-cell traction forces. Proc Natl Acad Sci U S A. 2014;111:2241–6. doi: 10.1073/pnas.1315606111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang N, Huang J, Edwards L, Liu B. Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity. 2011;34:13–23. doi: 10.1016/j.immuni.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, et al. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464:932–6. doi: 10.1038/nature08944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sabatino JJ, Huang J, Zhu C, Evavold BD. High prevalence of low affinity peptide-MHC II tetramer-negative effectors during polyclonal CD4+ T cell responses. J Exp Med. 2011;208:81–90. doi: 10.1084/jem.20101574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu C, Jiang N, Huang J, Zarnitsyna VI, Evavold BD. Insights from in situ analysis of TCR-pMHC recognition: response of an interaction network. Immunol Rev. 2013;251:49–64. doi: 10.1111/imr.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huppa JB, Axmann M, Mörtelmaier Ma, Lillemeier BF, Newell EW, Brameshuber M, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–7. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aleksic M, Dushek O, Zhang H, Shenderov E, Chen J-L, Cerundolo V, et al. Dependence of T cell antigen recognition on T cell receptor-peptide MHC confinement time. Immunity. 2010;32:163–74. doi: 10.1016/j.immuni.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dushek O, Das R, Coombs D. A role for rebinding in rapid and reliable T cell responses to antigen. PLoS Comput Biol. 2009;5:e1000578. doi: 10.1371/journal.pcbi.1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rudolph MG, Stanfield RL, Wilson Ia. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–66. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 70.Valitutti S, Müller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide–MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 71.Kondo S, Demachi-Okamura A, Hirosawa T, Maki H, Fujita M, Uemura Y, et al. An HLA-modified ovarian cancer cell line induced CTL responses specific to an epitope derived from claudin-1 presented by HLA-A*24:02 molecules. Hum Immunol. 2013;74:1103–10. doi: 10.1016/j.humimm.2013.06.030. [DOI] [PubMed] [Google Scholar]
- 72.Sasawatari S, Tadaki T, Isogai M, Takahara M, Nieda M, Kakimi K. Efficient priming and expansion of antigen-specific CD8+ T cells by a novel cell-based artificial APC. Immunol Cell Biol. 2006;84:512–21. doi: 10.1111/j.1440-1711.2006.01462.x. [DOI] [PubMed] [Google Scholar]
- 73.Friedman KM, Devillier LE, Feldman Sa, Rosenberg Sa, Dudley ME. Augmented lymphocyte expansion from solid tumors with engineered cells for costimulatory enhancement. J Immunother. 2011;34:651–61. doi: 10.1097/CJI.0b013e31823284c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maus MV, Thomas AK, Leonard DGB, Allman D, Addya K, Schlienger K, et al. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat Biotechnol. 2002;20:143–8. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 75.Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci Transl Med. 2011;3:83ra41. doi: 10.1126/scitranslmed.3001809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Singh H, Figliola MJ, Dawson MJ, Olivares S, Zhang L, Yang G, et al. Manufacture of Clinical-Grade CD19-Specific T Cells Stably Expressing Chimeric Antigen Receptor Using Sleeping Beauty System and Artificial Antigen Presenting Cells. PLoS One. 2013;8:e64138. doi: 10.1371/journal.pone.0064138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dupont J, Latouche J-B, Ma C, Sadelain M. Artificial antigen-presenting cells transduced with telomerase efficiently expand epitope-specific, human leukocyte antigen-restricted cytotoxic T cells. Cancer Res. 2005;65:5417–27. doi: 10.1158/0008-5472.CAN-04-2991. [DOI] [PubMed] [Google Scholar]
- 78.Latouche JB, Sadelain M. Induction of human cytotoxic T lymphocytes by artificial antigen-presenting cells. Nat Biotechnol. 2000;18:405–9. doi: 10.1038/74455. [DOI] [PubMed] [Google Scholar]
- 79.Papanicolaou Ga, Latouche J-B, Tan C, Dupont J, Stiles J, Pamer EG, et al. Rapid expansion of cytomegalovirus-specific cytotoxic T lymphocytes by artificial antigen-presenting cells expressing a single HLA allele. Blood. 2003;102:2498–505. doi: 10.1182/blood-2003-02-0345. [DOI] [PubMed] [Google Scholar]
- 80.Herrmann SH, Weinberger O, Burakoff J, Mescher MF. Analysis of the Two-Signal Requirement for Precursor Cytolytic T Lymphocyte Activation. J Immunol. 1982;128:1968–1974. [PubMed] [Google Scholar]
- 81.Prakken B, Wauben M, Genini D, Samodal R, Barnett J, Mendivil a, et al. Artificial antigen-presenting cells as a tool to exploit the immune “synapse”. Nat Med. 2000;6:1406–10. doi: 10.1038/82231. [DOI] [PubMed] [Google Scholar]
- 82.Haveman LM, Bierings M, Klein MR, Beekman JM, de Jager W, Kuis W, et al. Selection of perforin expressing CD4+ adenovirus-specific T-cells with artificial antigen presenting cells. Clin Immunol. 2013;146:228–39. doi: 10.1016/j.clim.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 83.De La Peña H, Madrigal Ja, Rusakiewicz S, Bencsik M, Cave GWV, Selman A, et al. Artificial exosomes as tools for basic and clinical immunology. J Immunol Methods. 2009;344:121–32. doi: 10.1016/j.jim.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 84.Crispe I, Bevan M, Staerz U. Selective activation of Lyt 2+ precursor T cells by ligation of the antigen receptor. Nature. 1985;317:627–629. doi: 10.1038/317627a0. [DOI] [PubMed] [Google Scholar]
- 85.Manger B, Weiss a, Weyand C, Goronzy J, Stobo JD. T cell activation: differences in the signals required for IL 2 production by nonactivated and activated T cells. J Immunol. 1985;135:3669–73. [PubMed] [Google Scholar]
- 86.Curtsinger JM, Lins DC, Mescher MF. CD8 + Memory T Cells (CD44 high, Ly-6C +) Are More Sensitive than Naive Cells (CD44 low, Ly-6C −) to TCR/CD8 Signaling in Response to Antigen. J Immunol. 1998;160:3236–3243. [PubMed] [Google Scholar]
- 87.Curtsinger J, Deeths MJ, Pease P, Mescher MF. Artificial cell surface constructs for studying receptor-ligand contributions to lymphocyte activation. J Immunol Methods. 1997;209:47–57. doi: 10.1016/s0022-1759(97)00146-4. [DOI] [PubMed] [Google Scholar]
- 88.Mescher MF. Surface contact requirements for activation of cytotoxic T lymphocytes. J Immunol. 1992;149:2402–5. [PubMed] [Google Scholar]
- 89.Tham EL, Jensen PL, Mescher MF. Activation of antigen-specific T cells by artificial cell constructs having immobilized multimeric peptide-class I complexes and recombinant B7-Fc proteins. J Immunol Methods. 2001;249:111–9. doi: 10.1016/s0022-1759(00)00335-5. [DOI] [PubMed] [Google Scholar]
- 90.Durai M, Krueger C, Ye Z, Cheng L, Mackensen A, Oelke M, et al. In vivo functional efficacy of tumor-specific T cells expanded using HLA-Ig based artificial antigen presenting cells (aAPC) Cancer Immunol Immunother. 2009;58:209–20. doi: 10.1007/s00262-008-0542-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ndhlovu ZM, Oelke M, Schneck JP, Griffin DE. Dynamic regulation of functionally distinct virus-specific T cells. Proc Natl Acad Sci U S A. 2010;107:1–6. doi: 10.1073/pnas.0915168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee JB, Oelke M, Ramachandra L, Canaday DH, Schneck JP. Decline of influenza-specific CD8+ T cell repertoire in healthy geriatric donors. Immun Ageing. 2011;8:6. doi: 10.1186/1742-4933-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pène J, Rahmoun M, Temmerman S, Yssel H. Use of anti-CD3/CD28 mAb coupled magnetic beads permitting subsequent phenotypic analysis of activated human T cells by indirect immunofluorescence. J Immunol Methods. 2003;283:59–66. doi: 10.1016/j.jim.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 94.Ndhlovu ZM, Angenendt M, Heckel D, Schneck JP, Griffin DE, Oelke M. Development of an artificial-antigen-presenting-cell-based assay for the detection of low-frequency virus-specific CD8(+) T cells in whole blood, with application for measles virus. Clin Vaccine Immunol. 2009;16:1066–73. doi: 10.1128/CVI.00365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Demento SL, Cui W, Criscione JM, Stern E, Tulipan J, Kaech SM, et al. Biomaterials Role of sustained antigen release from nanoparticle vaccines in shaping the T cell memory phenotype. Biomaterials. 2012:1–8. doi: 10.1016/j.biomaterials.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Monks CRF, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 97.Fooksman DR, Vardhana S, Vasiliver-Shamis G, Liese J, Blair Da, Waite J, et al. Functional anatomy of T cell activation and synapse formation. Annu Rev Immunol. 2010;28:79–105. doi: 10.1146/annurev-immunol-030409-101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25:117–127. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee K-H, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science (80-) 2002;295:1539–42. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 100.Dustin ML. T-cell activation through immunological synapses and kinapses. Immunol Rev. 2008;221:77–89. doi: 10.1111/j.1600-065X.2008.00589.x. [DOI] [PubMed] [Google Scholar]
- 101.Cemerski S, Das J, Giurisato E, Markiewicz Ma, Allen PM, Chakraborty AK, et al. The balance between T cell receptor signaling and degradation at the center of the immunological synapse is determined by antigen quality. Immunity. 2008;29:414–22. doi: 10.1016/j.immuni.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dustin ML, Long EO. Cytotoxic immunological synapses. Immunol Rev. 2010;235:24–34. doi: 10.1111/j.0105-2896.2010.00904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Irvine DJ, Doh J. Synthetic surfaces as artificial antigen presenting cells in the study of T cell receptor triggering and immunological synapse formation. Semin Immunol. 2007;19:245–54. doi: 10.1016/j.smim.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 104.Dustin ML. Visualizing immune system complexity. Sci Signal. 2009;2:mr4. doi: 10.1126/scisignal.266mr4. [DOI] [PubMed] [Google Scholar]
- 105.Shen K, Thomas VK, Dustin ML, Kam LC. Micropatterning of costimulatory ligands enhances CD4+ T cell function. Proc Natl Acad Sci U S A. 2008;105:7791–6. doi: 10.1073/pnas.0710295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mossman KD, Campi G, Groves JT, Dustin ML. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 2005;310:1191–3. doi: 10.1126/science.1119238. [DOI] [PubMed] [Google Scholar]
- 107.Bashour KT, Tsai J, Shen K, Lee J-H, Sun E, Milone MC, et al. Cross talk between CD3 and CD28 is spatially modulated by protein lateral mobility. Mol Cell Biol. 2014;34:955–64. doi: 10.1128/MCB.00842-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pawar AB, Kretzschmar I. Fabrication, assembly, and application of patchy particles. Macromol Rapid Commun. 2010;31:150–68. doi: 10.1002/marc.200900614. [DOI] [PubMed] [Google Scholar]
- 109.Sunshine JC, Green JJ. Nanoengineering approaches to the design of artificial antigen-presenting cells. Nanomedicine (Lond) 2013;8:1173–89. doi: 10.2217/nnm.13.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Negulescu Pa, Krasieva TB, Khan A, Kerschbaum HH, Cahalan MD. Polarity of T cell shape, motility, and sensitivity to antigen. Immunity. 1996;4:421–30. doi: 10.1016/s1074-7613(00)80409-4. [DOI] [PubMed] [Google Scholar]
- 111.Donnadieu E, Bismuth G, Trautmann A. Antigen recognition by helper T cells elicits a sequence of distinct changes of their shape and intracellular calcium. Curr Biol. 1994;4:584–95. doi: 10.1016/s0960-9822(00)00130-5. [DOI] [PubMed] [Google Scholar]
- 112.Al-Alwan MM, Rowden G, Lee TDG, West Ka. Cutting Edge: The Dendritic Cell Cytoskeleton Is Critical for the Formation of the Immunological Synapse. J Immunol. 2001;166:1452–1456. doi: 10.4049/jimmunol.166.3.1452. [DOI] [PubMed] [Google Scholar]
- 113.Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomater Sci Polym Ed. 2006;17:247–89. doi: 10.1163/156856206775997322. [DOI] [PubMed] [Google Scholar]
- 114.Sunshine JC, Perica K, Schneck JP, Green JJ. Particle shape dependence of CD8+ T cell activation by artificial antigen presenting cells. Biomaterials. 2014;35:269–77. doi: 10.1016/j.biomaterials.2013.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kumari S, Curado S, Mayya V, Dustin ML. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim Biophys Acta. 2014;1838:546–56. doi: 10.1016/j.bbamem.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gratton SEa, Ropp Pa, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci U S A. 2008;105:11613–8. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Champion Ja, Katare YK, Mitragotri S. Particle shape: a new design parameter for micro- and nanoscale drug delivery carriers. J Control Release. 2007;121:3–9. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Champion Ja, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A. 2006;103:4930–4. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yoo J-W, Mitragotri S. Polymer particles that switch shape in response to a stimulus. Proc Natl Acad Sci U S A. 2010;107:11205–10. doi: 10.1073/pnas.1000346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Barua S, Yoo J. Particle shape enhances specificity of antibody-displaying nanoparticles. Proc Natl Acad Sci. 2013;110:3270–3275. doi: 10.1073/pnas.1216893110/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1216893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lillemeier BF, Mörtelmaier Ma, Forstner MB, Huppa JB, Groves JT, Davis MM. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat Immunol. 2010;11:90–6. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bunnell SC. T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J Cell Biol. 2002;158:1263–1275. doi: 10.1083/jcb.200203043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fernández-Miguel G, Alarcón B. Multivalent structure of an αβT cell receptor. Proc Natl Acad Sci. 1999;96:1547–1552. doi: 10.1073/pnas.96.4.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Alarcón B, Swamy M, van Santen HM, Schamel WWa. T-cell antigen-receptor stoichiometry: pre-clustering for sensitivity. EMBO Rep. 2006;7:490–5. doi: 10.1038/sj.embor.7400682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schamel WWa, Arechaga I, Risueño RM, van Santen HM, Cabezas P, Risco C, et al. Coexistence of multivalent and monovalent TCRs explains high sensitivity and wide range of response. J Exp Med. 2005;202:493–503. doi: 10.1084/jem.20042155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sun ZJ, Kim ST, Kim IC, Fahmy A, Reinherz EL, Wagner G. Solution structure of the CD3ed ectodomain and comparison with CD3eg as a basis for modeling T cell receptor topology and signaling. PNAS. 2004;101:16867–16872. doi: 10.1073/pnas.0407576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hwang J, Gheber La, Margolis L, Edidin M. Domains in cell plasma membranes investigated by near-field scanning optical microscopy. Biophys J. 1998;74:2184–90. doi: 10.1016/S0006-3495(98)77927-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lu X, Gibbs JS, Hickman HD, David A, Dolan BP, Jin Y, et al. Endogenous viral antigen processing generates peptide-specific MHC class I cell-surface clusters. Proc Natl Acad Sci U S A. 2012;109:15407–12. doi: 10.1073/pnas.1208696109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bosch B, Heipertz EL, Drake JR, Roche Pa. Major histocompatibility complex (MHC) class II-peptide complexes arrive at the plasma membrane in cholesterol-rich microclusters. J Biol Chem. 2013;288:13236–42. doi: 10.1074/jbc.M112.442640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ferez M, Castro M, Alarcon B, van Santen HM. Cognate peptide-MHC complexes are expressed as tightly apposed nanoclusters in virus-infected cells to allow TCR crosslinking. J Immunol. 2014;192:52–8. doi: 10.4049/jimmunol.1301224. [DOI] [PubMed] [Google Scholar]
- 131.Vogt AB, Spindeldreher S, Kropshofer H. Clustering of MHC-peptide complexes prior to their engagement in the immunological synapse: lipid raft and tetraspan microdomains. Immunol Rev. 2002;189:136–51. doi: 10.1034/j.1600-065x.2002.18912.x. [DOI] [PubMed] [Google Scholar]
- 132.Sherman E, Barr V, Manley S, Patterson G, Balagopalan L, Akpan I, et al. Functional nanoscale organization of signaling molecules downstream of the T cell antigen receptor. Immunity. 2011;35:705–20. doi: 10.1016/j.immuni.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Minguet S, Swamy M, Alarcón B, Luescher IF, Schamel WWa. Full activation of the T cell receptor requires both clustering and conformational changes at CD3. Immunity. 2007;26:43–54. doi: 10.1016/j.immuni.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 134.Bray D, Levin MD, Morton-Firth CJ. Receptor clustering as a cellular mechanism to control sensitivity. Nature. 1998;393:85–88. doi: 10.1038/30018. [DOI] [PubMed] [Google Scholar]
- 135.Schamel WWa. The stoichiometry of the T cell antigen receptor and its implications for the signal transduction mechanism. Signal Transduct. 2007;7:311–319. doi: 10.1002/sita.200600123. [DOI] [Google Scholar]
- 136.Fahmy TM, Bieler JG, Edidin M, Schneck JP. Increased TCR avidity after T cell activation: a mechanism for sensing low-density antigen. Immunity. 2001;14:135–43. [PubMed] [Google Scholar]
- 137.Kumar R, Ferez M, Swamy M, Arechaga I, Rejas MT, Valpuesta JM, et al. Increased Sensitivity of Antigen-Experienced T Cells through the Enrichment of Oligomeric T Cell Receptor Complexes. Immunity. 2011;35:375–87. doi: 10.1016/j.immuni.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 138.Adachi K, Davis MM. T-cell receptor ligation induces distinct signaling pathways in naïve vs. antigen-experienced T cells. Proc Natl Acad Sci. 2010 doi: 10.1073/pnas.1017340108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fooksman DR, Grönvall GK, Tang Q, Edidin M. Clustering class I MHC modulates sensitivity of T cell recognition. J Immunol. 2006;176:6673–80. doi: 10.4049/jimmunol.176.11.6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fahmy TM, Bieler JG, Schneck JP. Probing T cell membrane organization using dimeric MHC-Ig complexes. J Immunol Methods. 2002;268:93–106. doi: 10.1016/s0022-1759(02)00203-x. [DOI] [PubMed] [Google Scholar]
- 141.Altman J, Moss P, Goulder P, Barouch DH, McHeyzer-Williams M, Bell J, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science (80-) 1996;274:94–96. [Google Scholar]
- 142.Cebecauer M, Guillaume P, Mark S, Michielin O, Boucheron N, Bezard M, et al. CD8+ cytotoxic T lymphocyte activation by soluble major histocompatibility complex-peptide dimers. J Biol Chem. 2005;280:23820–8. doi: 10.1074/jbc.M500654200. [DOI] [PubMed] [Google Scholar]
- 143.Oelke M, Maus MV, Didiano D, June CH, Mackensen A, Schneck JP. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nat Med. 2003;9:619–24. doi: 10.1038/nm869. [DOI] [PubMed] [Google Scholar]
- 144.Jiang X, Lu X, Liu R, Zhang F, Zhao H. HLA Tetramer Based Artificial Antigen-Presenting Cells Efficiently Stimulate CTLs Specific for Malignant Glioma. Clin Cancer Res. 2007;13:7329–34. doi: 10.1158/1078-0432.CCR-07-1025. [DOI] [PubMed] [Google Scholar]
- 145.Maus M, Riley JL, Kwok WW, Nepom GT, June CH. HLA tetramer-based artificial antigen-presenting cells for stimulation of CD4+ T cells. Clin Immunol. 2003;106:16–22. doi: 10.1016/S1521-6616(02)00017-7. [DOI] [PubMed] [Google Scholar]
- 146.Shen C, Cheng K, Miao S, Wang W, He Y, Meng F, et al. Latex bead-based artificial antigen-presenting cells induce tumor-specific CTL responses in the native T-cell repertoires and inhibit tumor growth. Immunol Lett. 2013;150:1–11. doi: 10.1016/j.imlet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 147.Gottschalk Ra, Hathorn MM, Beuneu H, Corse E, Dustin ML, Altan-Bonnet G, et al. Distinct influences of peptide-MHC quality and quantity on in vivo T-cell responses. Proc Natl Acad Sci. 2012:1–6. doi: 10.1073/pnas.1119763109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bullock TNJ, Mullins DW, Engelhard VH. Antigen density presented by dendritic cells in vivo differentially affects the number and avidity of primary, memory, and recall CD8+ T cells. J Immunol. 2003;170:1822–9. doi: 10.4049/jimmunol.170.4.1822. [DOI] [PubMed] [Google Scholar]
- 149.Gottschalk Ra, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med. 2010;207:1701–1711. doi: 10.1084/jem.20091999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Leignadier J, Labrecque N. Epitope density influences CD8 memory T cell differentiation. PLoS One. 2010;5:e13740. doi: 10.1371/journal.pone.0013740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Delcassian D, Depoil D, Rudnicka D, Liu M, Davis DM, Dustin ML, et al. Nanoscale ligand spacing influences receptor triggering in T cells and NK cells. Nano Lett. 2013;13:5608–14. doi: 10.1021/nl403252x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Manz BN, Jackson BL, Petit RS, Dustin ML, Groves J. T-cell triggering thresholds are modulated by the number of antigen within individual T-cell receptor clusters. Proc Natl Acad Sci U S A. 2011;108:9089–94. doi: 10.1073/pnas.1018771108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Walter S, Herrgen L, Schoor O, Wernet D, Bühring H, Stevanovic S. Cutting Edge: Predetermined Avidity of Human CD8 T Cells Expanded on Calibrated MHC/Anti-CD28-Coated Microspheres. J Immunol. 2003;171:4974–4978. doi: 10.4049/jimmunol.171.10.4974. [DOI] [PubMed] [Google Scholar]
- 154.Giannoni F, Barnett J, Bi K. Clustering of T cell ligands on artificial APC membranes influences T cell activation and protein kinase C θ translocation to the T cell plasma membrane. J Immunol. 2005;174:3204–3211. doi: 10.4049/jimmunol.174.6.3204. [DOI] [PubMed] [Google Scholar]
- 155.Anikeeva N, Gakamsky D, Schøller J, Sykulev Y. Evidence that the density of self peptide-MHC ligands regulates T-cell receptor signaling. PLoS One. 2012;7:e41466. doi: 10.1371/journal.pone.0041466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kamalasanan K, Jhunjhunwala S, Wu J, Swanson A, Gao D, Little SR. Patchy, anisotropic microspheres with soft protein islets. Angew Chem Int Ed Engl. 2011;50:8706–8. doi: 10.1002/anie.201101217. [DOI] [PubMed] [Google Scholar]
- 157.Steenblock E, Wrzesinski S, Flavell R, Fahmy T. Antigen presentation on artificial acellular substrates: modular systems for flexible, adaptable immunotherapy. Expert Opin Biol Ther. 2009;9:451–464. doi: 10.1517/14712590902849216. [DOI] [PubMed] [Google Scholar]
- 158.Cai S, Yang Q, Bagby TR, Forrest ML. Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Adv Drug Deliv Rev. 2011;63:901–08. doi: 10.1016/j.addr.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008;38:1404–13. doi: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 160.Perica K, De León Medero A, Durai M, Chiu YL, Bieler JG, Sibener L, et al. Nanoscale Artificial Antigen Presenting Cells for T Cell Immunotherapy. Nanomedicine. 2013;10:119–29. doi: 10.1016/j.nano.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25:1159–64. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 162.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 163.Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target. 2007;15:457–64. doi: 10.1080/10611860701539584. [DOI] [PubMed] [Google Scholar]
- 164.Rabinovich Ga, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Boyle S, Kolin DL, Bieler JG, Schneck JP, Wiseman PW, Edidin M. Quantum Dot Fluorescence Characterizes the Nanoscale Organization of T Cell Receptors for Antigen. Biophys J. 2011;101:L57–L59. doi: 10.1016/j.bpj.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lo Y-C, Edidin Ma, Powell JD. Selective activation of antigen-experienced T cells by anti-CD3 constrained on nanoparticles. J Immunol. 2013;191:5107–14. doi: 10.4049/jimmunol.1301433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Perica K, Tu A, Richter A, Bieler JG, Edidin M, Schneck JP. Magnetic Field-Induced T Cell Receptor Clustering by Nanoparticles Enhances T Cell Activation and Stimulates Antitumor Activity. ACS Nano. 2014;8:2252–2260. doi: 10.1021/nn405520d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mitchell MS. Phase I Trial of Large Multivalent Immunogen Derived from Melanoma Lysates in Patients with Disseminated Melanoma. Clin Cancer Res. 2004;10:76–83. doi: 10.1158/1078-0432.CCR-0689-3. [DOI] [PubMed] [Google Scholar]
- 169.Ye F, Yu Y, Hu Y, Lu W, Xie X. Alterations of dendritic cell subsets in the peripheral circulation of patients with cervical carcinoma. J Exp Clin Cancer Res. 2010;29:78. doi: 10.1186/1756-9966-29-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Satthaporn S, Robins A, Vassanasiri W, El-Sheemy M, Jibril Ja, Clark D, et al. Dendritic cells are dysfunctional in patients with operable breast cancer. Cancer Immunol Immunother. 2004;53:510–8. doi: 10.1007/s00262-003-0485-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Della Bella S, Gennaro M, Vaccari M, Ferraris C, Nicola S, Riva a, et al. Altered maturation of peripheral blood dendritic cells in patients with breast cancer. Br J Cancer. 2003;89:1463–72. doi: 10.1038/sj.bjc.6601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hurwitz Aa, Watkins SK. Immune suppression in the tumor microenvironment: a role for dendritic cell-mediated tolerization of T cells. Cancer Immunol Immunother. 2012;61:289–93. doi: 10.1007/s00262-011-1181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ma Y, Shurin GV, Gutkin DW, Shurin MR. Tumor associated regulatory dendritic cells. Semin Cancer Biol. 2012;22:298–306. doi: 10.1016/j.semcancer.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Walter Ea, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–44. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 177.Mackensen A, Meidenbauer N, Vogl S, Laumer M, Berger J, Andreesen R. Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J Clin Oncol. 2006;24:5060–9. doi: 10.1200/JCO.2006.07.1100. [DOI] [PubMed] [Google Scholar]
- 178.Chapuis A, Ragnarsson G. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med. 2013;27 doi: 10.1126/scitranslmed.3004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dudley ME, Wunderlich J, Nishimura MI, Yu D, Yang JC, Topalian SL, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–73. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]