

Abstract
Background
Microarray data must be normalized because they suffer from multiple biases. We have identified a source of spatial experimental variability that significantly affects data obtained with Cy3/Cy5 spotted glass arrays. It yields a periodic pattern altering both signal (Cy3/Cy5 ratio) and intensity across the array.
Results
Using the variogram, a geostatistical tool, we characterized the observed variability, called here the spotting effect because it most probably arises during steps in the array printing procedure.
Conclusions
The spotting effect is not appropriately corrected by current normalization methods, even by those addressing spatial variability. Importantly, the spotting effect may alter differential and clustering analysis.
Background
Microarray technology is probably the most successful in the area of functional genomics. Biologists use it to analyze gene expression at the genome scale by comparing the levels of messenger RNAs present in matched biological samples, for example grown under contrasted conditions or with different genetic configurations. Microarray data can be used for differential analysis, to identify genes whose expression strongly depends on the nature of the samples, as well as for clustering analysis, to identify coexpressed genes. Microarray data show a high level of variability. Some of this variability is relevant because it corresponds to the differential expression of genes. But, unfortunately, a large portion results from undesirable biases introduced during the many technical steps of the experimental procedure. Several sources of experimental noise have already been addressed, such as dye or fluorophore, fluorescence level or print-tips and statistical methods have been proposed to normalize data according to the related effects ([1,2]).
In this paper, we describe an experimental bias and use statistical methods to investigate the distribution of the signal across the microarray area. We use the variogram to analyze spatial dissimilarities between spots on the slide. Although spatial signal distribution across the slide has already been studied ([3,4]), the bias we report here has never before been explicitly characterized. We also present two experiments that give clues about the nature of the spotting effect, and finally we investigate the possibility to correct the effect and the efficiency of usual normalization procedures to do it.
Analyzed datasets are produced by using glass arrays and the two-color labeling strategy by which two conditions are compared directly. In these experiments, mRNA samples are collected from case and reference cells. The two corresponding cDNA samples are synthesized and labeled with either the Cy3 (green) or Cy5 (red) fluorophore and are mixed and hybridized simultaneously to a single array. For each DNA feature (representing a gene) printed and bound on the array, the fluorescence emitted by the hybridized labeled cDNA is measured in the Cy3 and Cy5 channels. Both fluorescence measurements are compared to define the relative gene expression in case versus reference cells. We designate these two values G and R and define the signal and intensity associated with a given gene as follows:
• the signal associated with a gene is the logarithm of the ratio R/G. This quantity is used to identify differentially expressed genes;
• the intensity is defined as the logarithm of the product R × G (or log(R × G)/2).
The goal of normalization is to correct the signal for experimental bias. Most existing normalization procedures do not specifically correct for potential spatial effects. The few that do only consider sources of variation that are restricted locally. For instance, the print-tip effect acts as a block effect, where the blocks are defined by the cluster of spots printed on the array with the same print-tip ([1]). The goal of this study is to determine whether normalization that corrects for additional spatial effects is necessary or whether current normalization models are sufficient.
Results
Our first test case is a self-hybridized microarray printed with Arabidopsis thaliana PCR-amplified cDNA sequences. In a self-hybridization microarray experiment, no gene should appear to be differentially expressed (R/G = 1) and the observed variability results from experimental effects. Also, no particular spatial pattern of intensity or signal is expected unless the DNA features are arranged on the array with respect to their type (for instance, transcribed versus intergenic regions as in [5]), which is not the case for this Arabidopsis microarray. Self-hybridization experiments have already proved to be efficient in detecting systematic biases ([1]). The Arabidopsis slide self-hybridization results show two spatial effects (Figure 1). First, the overall signal decreases from left to right. Second, the signal is arranged in a periodic pattern: sets of high signal vertical lines alternate with sets of low signal vertical lines. For practical reasons, rows in blocks are represented as vertical lines in Figures 1 and 5. Intensity values are structured according to a similar periodic pattern (data not shown).
Figure 1.
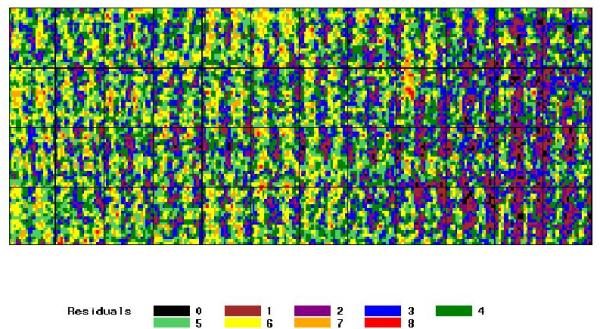
Spatial distribution of the signal for the self-hybridized Arabidopsis slide Each pixel represents the uncorrected log-ratio of the median Cy5 (635 nm) and Cy3 (532 nm) channel fluorescence measurements, associated to a printed DNA feature. Background is not represented. The picture is not a re-plot of the original image captured during the scanning process. Labels correspond to the 9-quantiles of the signal distribution.
Figure 5.
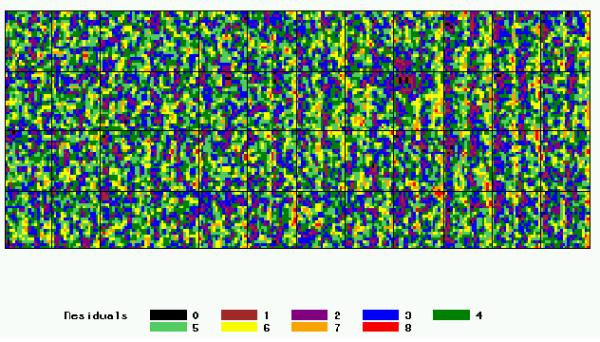
Distribution of the residuals (i.e. corrected signal) after reference normalization, for the Arabidopsis slide Labels correspond to the 9-quantiles of the residuals distribution.
To gain further insight in the data structure, we plotted the signal and intensity variograms for the Arabidopsis slide (figures 2 and 3), respectively. In both, the abscissa is the distance d between two points expressed as the number of rows that separates them, and the ordinate is the value of the variogram for a given distance calculated with formula (2) given in section Methods. When the observed value for each spot is independent from the value of all other spots at any given distance d, the variogram is a straight horizontal line. When a correlation exists only between closely neighboring spots (for example, because of local distortions of the slide), the curve will start at low V(d) values for small distances, and reach a higher horizontal plateau because the correlation disappears as d increases. Figures 2 and 3 highlight a different pattern: a given spot in a given row is similar to spots that are N = 10, 20, 30... rows apart. Spots that are 10 rows apart are particularly similar, which can be explained because spots in row N+10 are the duplicates of spots in row N within each block. Yet, this duplication does not explain the resemblance between spots distant by multiples of 10 rows higher than 10. This is probably due to the fact that all similar DNA features N × 10 rows apart from each other are printed in the same step (see Methods). The same pattern can also be observed in columns (data not shown). For convenience only, the observed periodic bias will be called the "spotting effect".
Figure 2.
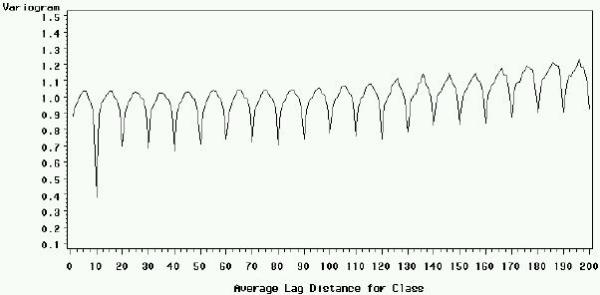
Variogram of the signal by row for the Arabidopsis slide, before normalization
Figure 3.
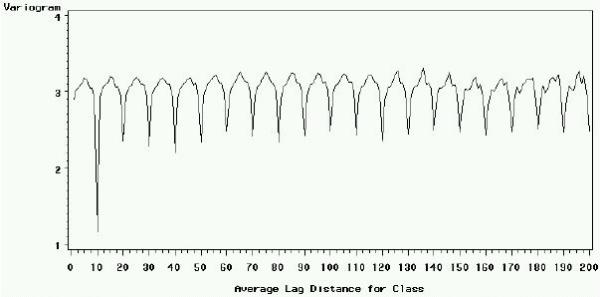
Variogram of the intensity (log(R × G)) by row for the Arabidopsis slide
Detection of the spotting effect in multiple microarray datasets
To determine whether the spotting effect is particular to the presented Arabidopsis slide or a common experimental bias in spotted microarrays, we studied twelve slides provided by three European or Canadian Laboratories and five slides available in the public Stanford MicroArray Database http://genome-www5.stanford.edu/MicroArray/SMD/. Results are described for one slide from the first set (Tor270, see Table 1) and two from the second (Lieb3727, [5] and Zhu473, [6]) as representative samples of our analysis. All slides were used for transcription profile comparisons or clustering analysis, except for the Arabidopsis slide that was a self-hybridization experiment and the Lieb3727 slide that was a chromatin immunoprecipitation microarray experiment (ChIP-chip). All were printed with PCR amplicons using two different robots (Microgrid II and ChipWriters) and according to various spotting designs: 16, 32 or 48 print-tip heads, duplicate prints in rows or columns, side by side, or far apart. Table 1 provides a summarized description of the microarrays for which results are presented below.
Table 1.
Characteristics of the datasets studied
| Lab. or Database | Robot | Print-tips Heads | Cols × Rows/Block | Nber of Rep. |
| Arabidopsis (URGV) | (Microgrid II) | 48 | 21 × 20 | row N and N + 10 |
| UHN Toronto (Tor270) | ChipWriters | 32 | 24 × 25 | col N and N + 1 |
| SMD Zhu473 | ? | 16 | 24 × 24 | ? |
| SMD Lieb3727 | ? | 32 | 24 × 21 | None |
The variograms in Figure 4 show that the raw signal is periodically structured according to rows in the different microarray datasets (25, 24 and 21 rows per block), stressing the prevalence of spotting effect biases and the need for correction through normalization procedures. We also computed the Type III Mean Squares (MSq) values associated with the print-tips, spotting and intensity-per-block effects, according to the following model:
Figure 4.
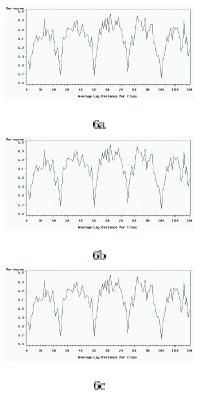
Variogram for the raw signal by row (A) Tor270 slide; (B) Zhu473 slide; (C) Lieb3727 slide. No normalization is performed.
![]()
![]()
where Ys is the signal measured at spot s, and 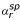 , βbl and Ints the mean spotting row, block and intensity effects, respectively. Spotting row effect means that only one parameter is estimated for all the rows spotted at the same time. For example, in the self-hybridized Arabidopsis slide dataset the row effect is the same for the rows 1,11, ..., 231.
, βbl and Ints the mean spotting row, block and intensity effects, respectively. Spotting row effect means that only one parameter is estimated for all the rows spotted at the same time. For example, in the self-hybridized Arabidopsis slide dataset the row effect is the same for the rows 1,11, ..., 231.
Type III MSq values measure variability due to a factor after adjustment for the other factors in the model and point out which effects need specific correction (see [7]). In summary, for the 18 slides analyzed, MSq values of the spotting effect were 10-fold lower to 4-fold higher than intensity-per-block MSq values and were equal to 10-fold higher than the print-tips effect MSq values. This result confirms that the spotting effect is present in many experiments and at least as important as other documented sources of variability.
Nature of the spotting effect
The spotting effect could be explained in different ways. The amount of material deposited on or bound to the slide and the shape of DNA spots can be affected by multiple factors, such as the time during which the print-tips are soaked in the DNA source microtiter plates, the time during which the print-tips touch the slides, the speed at which the print-tips move, the concentration and the salinity of the DNA solutions, the temperature and the relative humidity of the arrayer printing cabinet, and the physicochemical characteristics of the print-tips and of the glass surface. With most spotting robots, the printing of high density arrays containing thousands of features lasts for hours and subtle changes in spotting conditions may, therefore, alter all these factors. For example, DNA solutions may evaporate over time. In that regard, the spotting effect may be related to the "time-of-print" effect reported in [8].
Alternatively, the spotting effect may reflect DNA source plate variations because all DNA features printed simultaneously originate from the same plate. To test this hypothesis, we analyzed results from two microarray experiments for which the plate effect was controlled. In the first experiment, slides were printed with a unique 384-well-plate containing human cDNA amplicons and hybridized to targets prepared from RNA isolated from primary CD4+ T cell. In the second experiment, slides were printed with oligonucleotides of 70 bases, synthesized according to a different chemistry, and hybridized to cDNA from various developmental stages of Plasmodium falciparum. The oligonucleotides have the same length and are resuspended in solutions showing a narrow concentration range. In both cases, the spotting effect was greatly reduced (data not shown). This observation suggests that the plate effect is a major component of the spotting effect.
Spotting effect and normalization
Many authors have already pointed out the dangers of systematic normalization procedures. It is important to determine the conditions in which the correction of the spotting effect is appropriate and to verify that no biological effect can be confounded with the experimental biases corrected by the normalization. Taking into consideration the association between spotting and plate effects described above, three cases can be considered.
1. Probes are arranged according to their biological characteristics, for instance intergenic regions separated from transcription units, or genes expected to be differentially expressed grouped together in particular plates. In this case, it is impossible to distinguish between a significant plate effect due to coexpression of genes belonging to the same class, or due to technical artifacts.
2. Probes are arranged according to their chromosomal order. Such structure may lead to significant differences between plates if genes with similar expression profiles are spatially clustered in the genome (silent neighboring genes in heterochromatic regions, for example). Such spatial clustering has been recently observed in several organisms ([9,10]) and may affect many others.
3. Probes are randomly distributed among plates. Most human array experiments verify this hypothesis. The results presented in Section 4 prove that this configuration does not cause the spotting effect to disappear.
A normalization procedure to correct the effect is advisable only in the last case because, in the first two, regardless of the importance of the spotting bias, the correction would unavoidably alter the biological information contained in the data. Thus, the effect can considerably affect the conclusions of experiments corresponding to the first two cases. In particular, results of experiments studying gene similarity or the relationship between relative chromosomal position and coexpression could be essentially twisted, as also pointed out by Balazsi et al. in [11].
Assuming that the experiment of interest corresponds to the third case, one has to investigate whether a specific normalization for the spotting effect is needed or if standard normalization is sufficient. We present here the consequences of the normalization procedure proposed by Yang et al. in [1], one of the most widely used methods in the microarray community, on the self-hybridized Arabidopsis cDNA array data. Only results obtained with background-corrected signals and the global loess normalization procedure are presented. The analysis performed on background-uncorrected data, or with the print-tip loess normalization procedure gave similar results.
The Arabidopsis slide signal normalized with the reference procedure (residual) still shows a periodic pattern as illustrated in Figure 5 and calculated with the variogram (Figure 6). This observation indicates that the bias introduced by the spotting effect is not fully corrected. According to our experience, other usual normalization procedures do not perform an efficient correction of the spotting effect.
Figure 6.
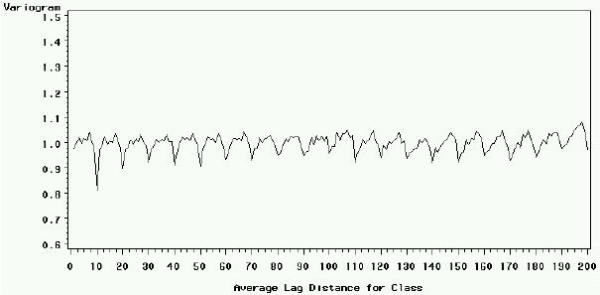
Variogram of the residuals (i.e. corrected signal) after reference normalization, for the Arabidopsis slide
Because of the strong pattern observed in rows, the spotting effect may be treated as row effect per block or as a global row effect across the slide. Preliminary results suggest that such models adequately correct the periodic spatial bias described in various microarray datasets. Row models are advantageous because they rely exclusively on the geometrical information that is embedded in the data and that is mandatory according to the "Minimum Information About a Microarray Experiment" (MIAME) guidelines. In contrast, plate origin information is very rarely available and is difficult to integrate into statistical analysis considering that successive technical steps usually take place in multi-titer plates of different formats (e.g. 96-, 384- and 1536-well plates) during spotted microarray DNA production.
Discussion
We have observed that transcription profiling datasets obtained with spotted glass microarrays and the two-color labeling strategy show a bias that leads to periodic patterns according to rows and columns of the array grid. These patterns affect the entire area and alter both signal and intensity. We propose that such patterns result from artifacts introduced during the DNA feature preparation into microtiter plates or the slide printing procedure because features spotted together yield the most similar signals.
Color swaps are now routinely included in microarray experimental designs to correct for labeling biases. They consist in repeated hybridizations in which the case and reference samples are labeled at least once with each of the Cy3 and Cy5 fluorophores. Preliminary analyses indicate that the spotting effect is reduced when raw data from opposite color swap hybridizations are combined (unpublished results). This observation is consistent with the fact that the spotting effect depends on the position of the spots on the slide and that the relative spot position remains the same from slide to slide in most setups. We suggest that the reduction of the spotting effect resulting from the combination of raw opposite color datasets may constitute additional justification for the inclusion of color swaps in microarray experiments.
We have shown that the variogram is an efficient tool to display spatial correlations between spots. Furthermore, it is possible to test the null hypothesis that no spatial correlation exists (for instance the Moran test described in [12]). Such tests could be performed together with the variogram analysis as part of the data normalization procedure to investigate the significance of observed spatial biases and to evaluate the need and efficiency of different correction methods.
Conclusions
We have proved that the spotting effect is statistically significant, is as important as other effects that are commonly corrected, and should be taken into account in normalization procedures. This effect is expected to increase the number of false positives and negatives in classical microarray studies. In differential analysis, some rows or columns may contain artificially high or low numbers of "differentially expressed genes". In clustering analysis, genes may be associated because of a similarity caused by the spotting effect.
Methods
Description of the first test slide
The Arabidopsis glass slide (Corniong GAP II) studied in section Results was hybridized with Cy3 and Cy5 labeled cDNA samples, both prepared from the same mRNA extracted from Arabidopsis flower buds (self-hybridization). The microarray structure consists of 4 × 12 blocks, each with 20 rows and 21 columns. cDNA sequences were spotted in duplicate, i.e. rows N and N+10 (for N = 1 to 10) in the same block were printed with the same series of amplicons. The robot printing head consisted of 48 (4 × 12) print-tips, each defining a block. During a single printing step, the robot printed 48 spots on a slide (each corresponding to a different DNA feature) distant by 20 rows in one direction and 21 columns in the other (the distance between print-tips); then, within a fraction of a second, the robot arm moved laterally and printed the duplicate spots 10 rows away before moving to the next slide. Once all slides were spotted with a given set of 48 duplicated amplicons, the robot washed all print-tips simultaneously, loaded them with the next set of 48 amplicons, and resumed printing. Each set was printed on all slides in approximatively 2 min and the entire procedure lasted 16 h for the 10000 duplicated cDNAs.
Variogram
The structure of the spatial distribution of the signal on a slide can be studied with a geostatistical tool called a variogram ([13,14]). In geostatistics, the variogram has been used to detect departure of stationarity in the data. In the microarray data analysis context, it represents a useful exploratory tool to study spatial correlations due to systematic biases. A variogram (also called semi-variogram) is defined (2), and estimated (3), for a distance d and a variable Y, as follows:
![]()
![]()
where N(d) is the set of all possible pairs of spots (si, sj) with a distance d between one another, and with |N(d)| the cardinal of N(d). As implied by expression (2), V(d) decreases when the number of similar points separated by the distance d increases.
Authors' contributions
TMH did the major part of the data analysis, and created all tables and figures. JJD and SR proposed the statistical methods and supervised their application in collaboration with TMH. FB completed all wet laboratory Arabidopsis microarray work supervised by PH, and they provided the initial dataset for testing. Tor270 experiment was performed by EC. EC and PH conducted the interpretation of the statistical results in the light of hardware experimental settings. The manuscript was written by PH and TMH. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
E.C. was supported by a fellowship from Agence Nationale de Recherches sur le SIDA
Contributor Information
Tristan Mary-Huard, Email: maryhuar@inapg.fr.
Jean-Jacques Daudin, Email: daudin@inapg.fr.
Stéphane Robin, Email: robin@inapg.fr.
Frédérique Bitton, Email: bitton@urgv.fr.
Eric Cabannes, Email: cabannes@pasteur.fr.
Pierre Hilson, Email: pihil@gengenp.rug.ac.be.
References
- Yang Y, Dudoit S, Luu P, Speed T. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nuclear Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J. Microarray data normalization and transformation. Nature Genet. 2002;32:496–501. doi: 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- Schuschhardt J, Beule D, Malik A, Wolski E, Eickhoff H, Lehrach H, Herzel H. Normalization strategies for cDNA microarrays. Nucleic Acids Res. 2000;28:e47. doi: 10.1093/nar/28.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman C, Jensen L, Jarmer H, Berka R, Gautier L, Nielser H, Saxild H, Nielsen C, Brunak S, Knudsen S. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biol. 2002;3:1–16. doi: 10.1186/gb-2002-3-9-research0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb J, Liu X, Botstein D, Brown P. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nature Genet. 2001;28:327–34. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- Zhu G, Spellman P, Volpe T, Brown P, Botstein D, Davis T, Futcher B. Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature. 2000;406:90–4. doi: 10.1038/35017581. [DOI] [PubMed] [Google Scholar]
- Searle S. Linear Models. New York: John Wiley & Sons, Inc; 1971. [Google Scholar]
- Ball C, Chen Y, Panavally S, Sherlock G, Speed T, Spellman P, Yang Y. In DNA Microarrays: A Molecular Cloning Manual. Cold Spring Harbor Press; 2003. Section 7: An introduction to microarray bioinformatics. [Google Scholar]
- Cohen B, Mitra R, Hughes J, Church G. A computational analysis of whole-genome expression data reveals chromosomal domains of gene espression. Nature Genet. 2000;26:183–186. doi: 10.1038/79896. [DOI] [PubMed] [Google Scholar]
- Spellman P, Rubin G. Evidence for large domains of similarly expressed genes in the Drosophila genome. J Biol. 2002;1:1–5. doi: 10.1186/1475-4924-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazsi G, Kay K, Barabasi A, Oltvai Z. Spurious spatial periodicity of co-expression in microarray data due to printing design. Nucleic Acids Res. 2003;31:4425–4433. doi: 10.1093/nar/gkg485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Carlin B, Gelfand A. Monographs on Statistics and Applied Probability. Chapman and Hall/CRC Press; 2004. Hierarchical Modeling and Analysis for Spatial Data. [Google Scholar]
- Jowett G. The Accuracy of systematic sampling from conveyor belts. Applied Statistics. 1952;1:50–59. [Google Scholar]
- Cressie A. Wiley series in probability. Wiley; 1997. Statistics for spatial data. [Google Scholar]