

Abstract
Aim
Activation of hepatic stellate cells and development of chronic inflammation are two key features in the progression of hepatic fibrosis. We have shown that in vitro activated stellate cells increase their expression of CXCL12 as well as the receptor CXCR4 and that receptor engagement promotes a profibrogenic phenotype. Furthermore, injury promotes increased hepatic expression of CXCL12 and a massive infiltration of CXCR4-expressing leukocytes, granulocytes, and myeloid cells. The primary site of inflammatory cell accumulation is around the CXCL12-rich portal tracts and within fibrotic septae, indicating a role for CXCR4 during injury. In order to characterize the relevance of the CXCR4/CXCL12 chemokine axis during hepatic injury we inhibited the axis using AMD3100, a CXCR4 small molecule inhibitor, in models of chronic and acute liver injury.
Methods
Mice were subjected to acute and chronic CCl4 liver injury with and without AMD3100 administration. The degree of liver injury, fibrosis, and the composition of the intrahepatic inflammatory response were characterized.
Results
Treatment of mice with AMD3100 in the chronic CCl4 model of liver injury led to an increase in hepatic inflammation and fibrosis with a specific increase in intrahepatic neutrophils. Furthermore, in an acute model of CCl4 induced liver injury, AMD3100 led to an increase in the number of intrahepatic neutrophils and a trend towards worse necrosis.
Conclusions
Together, this data suggests that inhibition of the CXCR4/CXCL12 chemokine axis is injurious through modulation of the hepatic inflammatory response and that this axis may serve a protective role in liver injury.
Keywords: CXCR4, CXCL12, Hepatic Stellate Cells, Liver Fibrosis
Introduction
Chemokines are small molecular weight proteins (8-13 kD) initially identified by their ability to provide migratory cues to inflammatory cells 1. Their expression is up-regulated in nearly all forms of tissue injury promoting immune cell infiltration and activation. The contribution of chemokines to the progression of liver disease is well established. Mice deficient in chemokine receptors CCR1, CCR5 2, CCR2 3, 4, and CCR7 5, demonstrate a decrease in the extent of injury in both chronic fibrotic and acute necrotic liver injury. Alternatively, some chemokine axes appear to be protective and their loss leads to worse outcomes in models of liver disease 6, 7.
Knowledge about the specific role of CXCL12 in chronic inflammatory and fibrotic diseases is fragmentary. Together with its primary receptor, CXCR4, the CXCL12 axis plays a major role in immune and hematopoietic cell egress and mobilization during homeostatic and injury conditions. CXCL12 levels are increased in many diseases including: pulmonary fibrosis 8, systemic lupus erythematosus 9, rheumatoid arthritis 10, multiple sclerosis 11, and inflammatory bowel disease 12. In these diseases, CXCL12 recruits subtypes of inflammatory cells that are injurious. Specifically, in pulmonary fibrosis, inhibition of the CXCL12/CXCR4 axis with either a CXCL12 neutralizing antibody, or AMD3100, a CXCR4 small molecule inhibitor, leads to a decrease in the number of bone marrow-derived fibrocytes, and overall better outcome 8. Additionally, administration of AMD3100 in certain models of myocardial infarction decreased the extent of tissue scarring and improved cardiac function 13, 14. Given these findings, the potential therapeutic benefit of AMD3100 has been discussed15.
CXCL12 expression has been detected both in the normal and injured liver though its function remains largely unknown. It is expressed by biliary epithelial cells 16, hepatic stellate cells 17, and hepatic sinusoidal endothelial cells 18. In patients with liver disease, CXCL12 protein levels are increased in both the liver and plasma and correlate with the extent of fibrosis 16. With hepatic injury there is an infiltration of CXCR4-expressing immune cells including lymphocytes, granulocytes, and monocytes primarily around the CXCL12-rich periportal regions, indicating the importance of this axis. The function of each of these cell populations is only now being uncovered, but they already show competing roles in fibrosis and its resolution 19-22.
Activation of hepatic stellate cells (HSCs) and development of chronic inflammation are two key features driving the progression of hepatic fibrosis. We have shown that in vitro, HSC activation drives expression of both CXCL12 and its receptor CXCR4 and that recombinant CXCL12 promotes a pro-fibrogenic phenotype 17. Therefore, hepatic CXCL12 may play multiple roles during injury by recruiting inflammatory cells and driving stellate cell activation. In this study we use AMD3100, a well-established small molecule inhibitor of CXCR4 to study the role of the CXCL12/CXCR4 axis in chronic fibrotic and acute necrotic models of liver injury.
Methods
Mouse Models of Fibrosis
All experiments were performed on C57Bl/6 male mice purchased from NCI (Bethesda Maryland). For induction of fibrosis using the chronic CCl4 model mice received 100μL of 10% CCl4 diluted in corn oil 3X a week for four weeks. Mice were sacrificed 48 hours after the last injection. Acute liver injury was induced by a single injection of 100μL of 25% CCl4 diluted in corn oil and sacrificed as indicated. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Mount Sinai School of Medicine (Permit Numbers: LA10-00059 and SP10-00363).
Administration of AMD3100
AMD3100 was administered either by continuous subcutaneous infusion with an osmotic pump (Alzet Osmotic Pumps) or by once weekly by I.P. injection. Osmotic pumps, which deliver 0.11μL/hr, were loaded with 100μL of 83.3mg/mL AMD3100 in PBS. Mice were anesthetized and a small incision and pocket made on the dorsal side. The pump was inserted and the incision closed with a staple. The total daily dose of AMD3100 administered over 24 hours was 200μg. For I.P. injections, mice received 200μg of AMD3100 in 100μL PBS. Mice that received a single dose of CCl4 for acute liver injury were additionally treated with or without a single injection of 100μg AMD3100 12 hours after CCl4 administration.
Analysis of liver function enzymes
Peripheral blood was collected via retro-orbital bleeding and immediately mixed with 0.5mM EDTA to prevent coagulation. 500μL blood was transferred into a Microtainer Plasma Separator (BD, Franklin Lakes, NJ), centrifuged according to manufactures recommendations and plasma stored at −80°C until analysis. Analysis was performed at the Mount Sinai Medical Center Clinical Laboratory.
Histological scoring
At time of sacrifice mouse livers were perfused with 10mL PBS and half the right lobe harvested and fixed in 10% formalin for 24hr and embedded in paraffin. Histological scoring of murine livers was performed by an expert hepatobilliary pathologist. All parameters are scored on a scale of 0-4. For each mouse, 10 high power fields were assessed.
Sirius red staining
Five-micron sections were stained for collagen with Sirius red (0.1% solution, diluted in picric acid, both from Sigma, St. Louis, MO). Percent fibrosis was assessed based on 36 10X-fields from Sirius red stained liver sections per animal in a blinded fashion. Each field was analyzed using a computerized Bioquant® morphometric system. Overall fibrosis was assessed by intensity of Sirius red staining.
Immunohistochemistry
Formalin-fixed, paraffin-embedded liver tissue obtained mice were deparaffinized, rehydrated, incubated with DAKO (Carpinteria, CA) Peroxidase block for 5 minutes followed by incubation with DAKO Serum Free Protein Block at room temperature for 10 minutes. Rabbit polyclonal anti-α smooth muscle actin antibody (Abcam, Cambridge, MA, catalog #ab-56984) at a dilution of 1:500 was used for overnight incubation at 4°C. Both primary isotype control antibody and secondary antibody with no primary antibody were used as negative controls. Slides were washed three times and revealed using DAKO Envision Plus System. Nuclei were stained with hematoxylin and percent α-SMA staining was assessed based on 25 100X-fields per animal in a blinded fashion. Each field was analyzed using a computerized Bioquant® morphometric system.
RNA isolation and quantitative real-time PCR
Murine livers were collected and stored in RNAlater (Qiagen, Valencia, CA) until time of use. RNA was isolated by homogenization and purification in TRIzol reagent (Invitrogen, Grand Island, NY) followed by RNAeasy clean-up with on-column DNA digestion (Qiagen). Reverse transcription was performed using RNA to cDNA EcoDry Premix Double Primed Kit (Clontech, Mountain View, CA) and quantitative real-time PCR performed using iScriptTM One-Step RT-PCR Kit with SYBR® Green (Bio-Rad, Hercules, CA). Quantification was performed by normalizing each Ct value to GAPDH and comparing ΔCt values between groups. Values are expressed either as absolute ΔCt or fold increase of experimental to control group. Primer sequences in Table 1.
Table 1.
Quantitative RT-PCR Primer Sequences.
| Primer Name | Primer Sequence (5’----3’) |
|---|---|
| GAPDH-Forward GAPDH-Reverse |
CAATGACCCCTTCATTGACC GATCTCGCTCCTGGAAGATG |
| α-SMA-Forward α-SMA-Reverse |
TCCTCCCTGGAGAAGAGCTAC TATGGTGGTTTCGTGGATGC |
| Collagen 1α1-Forward Collagen 1α1-Reverse |
GTCCCTGAAGTCAGCTGCATA TGGGACAGTCCAGTTCTTCAT |
| CXCL12-Forward CXCL12-Reverse |
GCTCTGCATCAGTGACGGTA AGATGCTTGACGTTGGCTCT |
| CXCL12-V1-Forward CXCL12-V1-Reverse |
CAACAGACAAGTGTGCATTGACCCG ATGTCAGCCTTCCTCGGGGGT |
| CXCL12-V2-Forward CXCL12-V2-Reverse |
ACAGACAAGTGTGCATTGACCCGA ACCTCTCACATCTTGAGCCTCTTGT |
| CXCL12-V3-Forward CXCL12-V3-Reverse |
TAGTTCCCCGCTTCCTGCCGATG CCCGAAGAGGGAGGAGCGAGTT |
Peripheral blood and liver leukocyte flow cytometry
Peripheral blood was collected via retro-orbital bleeding and immediately mixed with 0.5mM EDTA to prevent coagulation. Red blood cells were lysed by 3 washes in RBC lysis buffer, filtered, and washed in PBS/FBS followed by staining with antibodies for FACS analysis. For liver infiltrating leukocyte isolation, at the time of sacrifice the livers were perfused with 10mL PBS through the portal vein, carefully removed, and gallbladder excised. A portion of the liver was cut, weighed, chopped into small pieces and incubated in 0.01% Collagenase type IV (Sigma) at 37°C for 25 minutes. Liver tissue was then passed through a 70μm cell strainer and washed twice with PBS/FBS. Cells were resuspended in a 40% Percoll (GE Healthcare) and loaded onto a 70%/40% Percoll gradient and centrifuged at 2400 RPM at 24°C for 25 minutes with no acceleration or brake. The top layer of hepatocytes was removed and the liver infiltrating leukocytes located at the interface collected and washed 2X in PBS/FBS and cells counted. The following antibodies were used: rat anti-mouse CD45-APC-Cy7, clone: 30-F11 (BD Pharmigen), rat anti-mouse Ly-6G-Alexa Fluor 488, clone: RB6-8C5 (eBiosciences), rat anti-mouse Ly-6G-PE, clone: 1A8 (BD Pharmigen), rat anti-mouse TER-119-Biotin, clone: TER-119 (eBiosciences), rat anti-mouse CD3-Biotin, clone: 145-2C11 (eBiosciences), rat anti-mouse CD45R-Biotin, clone: RA3-6B2 (eBiosciences), rat anti-mouse NKp46-eFluor 450, clone: 29A1.4 (eBiosciences), rat anti-mouse CD11b-PerCP-Cy5.5, clone: M1/70 (eBiosciences), rat anti-mouse CD11b-Biotin, clone: M1/70 (BD Pharmigen), rat anti-mouse CD45R-APC-eFluor780, clone: RA3-6B2 (eBiosciences), rat anti-mouse Sca-1-APC, clone: D7 (eBiosciences), rat anti-mouse c-Kit-PE-Cy7, clone: 2B8 (eBiosciences). Multiparameter analyses of stained cell suspensions were performed on an LSRII (BD) and analyzed with FlowJo software (Tree Star, Inc).
Statistics
All results are expressed as the mean ± standard deviation. Statistical significance was tested using an unpaired Student's t and P < 0.05 indicated a significant difference.
Results
Increased hepatic expression of CXCL12 and CXCR4 in murine models of liver fibrosis
To determine whether there is an increase in protein and mRNA expression of CXCR4 and CXCL12 during murine liver injury we performed quantitative RT-PCR and Western blot analysis. In mice that received chronic CCl4 injections there was a 16-fold increase in CXCR4 transcript (Fig. 1A) and a substantial increase in CXCR4 (Fig. 1B) and CXCL12 (Fig. 1C) protein expression by Western blot. Despite the increase in CXCL12 protein, there was no change in the transcript levels (Fig. 1D). To determine if there are changes in the three known murine CXCL12 splice variants, we designed primers that could distinguish between them (Table 1). No changes were observed in any of the splice variants for CXCL12 in CCl4 treated livers (Fig. 1E).
Figure 1. Increased CXCL12 and CXCR4 expression in the chronic CCl4 model of liver fibrosis.

Increased hepatic CXCR4 and CXCL12 expression in mice with CCl4 induced liver fibrosis by quantitative RT-PCR (A) for CXCR4 and by Western blot for CXCR4 (B) and CXCL12 (C) demonstrates increased CXCR4 mRNA and CXCR4 and CXCL12 protein expression. No changes are seen in CXCL12 mRNA total (D) or splice variants (E) suggesting post-transcriptional regulation. *P<.05.
Continuous inhibition of the CXCL12/CXCR4 axis with AMD3100 leads to an increase in fibrosis and intrahepatic neutrophils in the chronic CCl4 models of liver fibrosis
Inhibition of the CXCR4/CXCL12 axis has been shown to decrease the extent of fibrosis in mouse models of pulmonary fibrosis. We therefore determined whether AMD3100 would have a similar effect in hepatic fibrosis. In the chronic CCl4 model, mice that received a continuous infusion of AMD3100 showed a significant increase in hepatic collagen content and fibrosis as measured by Sirius red staining (Figs. 2A & B). In accordance with the increased fibrosis, there was an increase in hepatic α-smooth muscle actin (α-SMA) staining by immunohistochemistry in the mice receiving CCl4 and AMD3100 over CCl4 alone (Figs. 2C & D), suggesting an increase in stellate cell activation. Similarly, transcript levels of collagen 1α1 (Fig. 2E) and α-SMA (Fig. 2F) were increased ~2 fold in the mice receiving AMD3100. Although not statistically significant a similar trend toward increased fibrosis was seen in the bile duct ligation model of hepatic fibrosis (data not shown).
Figure 2. Inhibition of the CXCR4/CXCL12 chemokine axis with continuous infusion of AMD3100 in the chronic CCl4 model of liver fibrosis promotes hepatic fibrosis.

Mice received 4 weeks of CCl4 with continuous infusion of AMD3100 or PBS by osmotic pumps. Increased fibrosis is seen in AMD3100 treated mice by Sirius red (A) and α-SMA (C) staining. Morphometric analysis was used to quantify the area of Sirius red (B) and α-SMA (D). Quantitative RT-CR further demonstrated an increase in collagen 1α1 (E) and α-SMA (F) transcript levels. Representative images shown, n=6, *P<.05.
As AMD3100 promotes immune cell egress from the bone marrow, we analyzed peripheral blood and intrahepatic inflammatory cells after AMD3100 administration. FACS analysis of the peripheral blood did not demonstrate a change in circulating leukocytes after administration of CCl4 alone (Fig. 3A, column 1 vs. 2), however the expected increase in circulating leukocytes due to AMD3100 was observed both in the control (Fig. 3A, column 1 vs. 3, 5×106 vs. 12×106 leukocytes/mL) and CCl4 treated groups (Fig. 3A, column 2 vs. 4, 6×106 vs. 13×106 leukocytes/mL), with the greatest changes seen in the neutrophil population (Fig. 3B).
Figure 3. Inhibition of the CXCR4/CXCL12 chemokine axis with continuous infusion of AMD3100 in the chronic CCl4 model of liver fibrosis promotes liver inflammation.

FACS analysis of circulating leukocytes showed that AMD3100 increased circulating inflammatory cells in both oil and CCl4 treated mice (A), with a specific increase in neutrophil populations (B). Additionally, isolated liver infiltrating inflammatory cells showed an increase in the total number of CD45-positive cells/gram of liver tissue (C) with a similar increase in the absolute number of neutrophils (D) in mice receiving AMD3100 both in oil and CCl4 treated mice. n=6-12, *P<0.05, **P<.01, ***P<.001.
Within the liver, no changes in inflammation were observed by liver histological analysis (data not shown); however, FACS analysis revealed a significant increase in the absolute number of CD45-positive liver infiltrating immune cells per gram of liver in mice receiving AMD3100; both in the oil treated (Fig. 3C, column 1 vs. 3, 1.4×106 vs. 2.4×106) and CCl4 treated groups (Fig. 3C, column 2 vs. 4, 2.5×106 vs. 4×106) again with a specific increase in the total number neutrophils (Fig. 3D) similar to the peripheral blood.
Once weekly administration of AMD3100 does not lead to any changes in the chronic CCl4 model of liver fibrosis
In models of myocardial ischemia administration of AMD3100 after injury leads to divergent outcomes depending on the dosing regimen 13, 14. Chronic augmentation of the CXCR4 axis with AMD3100 leads to an increased area of infarction and an expanded scar area while a single dose of AMD3100 shortly after induction of injury leads to increased myocardial vascularity, reduced fibrosis, and an overall increase in cardiac function. The single AMD3100 injection increased circulating endothelial progenitor cells, thought to be important in the revascularization of the myocardium after injury 14.
We therefore hypothesized that a single weekly AMD3100 injection during chronic liver injury may be beneficial in hepatic regeneration by releasing bone marrow progenitor cells. Fibrosis was induced by either 3X-weekly CCl4 administration with injection of AMD3100 once weekly 12 hours after the last CCl4 injection. We saw no differences in either the extent of fibrosis (Figs. 4A & B) or in the absolute number of inflammatory cells within the liver (Fig. 4C).
Figure 4. Inhibition of the CXCR4/CXCL12 chemokine axis with once weekly injections of AMD3100 in the chronic CCl4 model has no effect on hepatic fibrosis and inflammation.
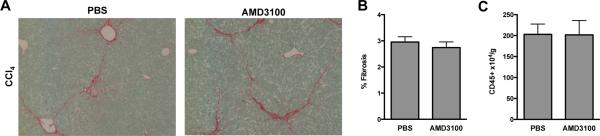
Mice received 4 weeks of CCl4 with once weekly injections of AMD3100. Administration of AMD3100 did not lead to any differences in the degree of fibrosis by Sirius red staining (A) quantified by morphometric analysis (B) or hepatic inflammation (C). Representative images shown, n=5.
AMD3100 is not protective in CCl4 induced acute hepatic injury
Bone marrow progenitor cells have the unique ability to attenuate the extent of injury in scenarios of massive hepatic insult. While the mechanism has not been elucidated, in nearly all forms of injury, delivery of bone marrow cell populations after injury promotes hepatic regeneration 23. Administration of G-CSF and AMD3100 after a sub-lethal dose of CCl4 attenuates the extent of injury with a concomitant increase in circulating CD34+ bone marrow progenitor cells 24. Given the rapid activity of AMD3100 and maximal bone marrow mobilization at 9 hours 25, as opposed to G-CSF which takes days to reach maximal mobilization 26, we tested whether AMD3100 alone could confer protection after administration of a sub-lethal CCl4 dose. Mice received 25μL CCl4 followed by 100μg AMD3100 12 hours later and were sacrificed between 36 and 144 hours after receiving CCl4.
There were no significant differences in hepatic injury measured by ALT and AST (Figs. 5A & B); however, by histological analysis there was a trend towards an increased degree of necrosis at 36 hours in mice administered AMD3100 (Figs. 5C & D). While there was only a trend of increased CD45+ inflammatory cells at 36 and 72 hours after CCl4 (Fig. 5E), there was a nearly 3-fold increase in the absolute number of liver infiltrating neutrophils at 36 hours in the AMD3100 treated group (Fig. 5F, 15×105 vs. 5×105 cells/g). Similarly, FACS analysis revealed an increase in the number of hematopoietic stem cells found in the liver at 36 hours, which was still present, yet not significant at 72 hours (Fig. 5G).
Figure 5. Inhibition of the CXCR4/CXCL12 chemokine axis with a single AMD3100 injection in an acute CCl4 model does not prevent injury and demonstrates a trend of increased hepatic inflammation.

Mice received a single injection of CCl4 followed by AMD3100 12 hours later and were sacrificed between 36 and 144 hours after the CCl4 injection. Serum ALT (A) and AST (B) levels showed no significant changes between AMD3100 and control treated groups. In mice treated with AMD3100 H&E images (C) and histological scoring for necrosis (D) showed a trend of increased necrosis at 36 hours, but not at any other time points. FACS analysis of intrahepatic leukocytes similarly showed a trend of increased inflammatory cells (E), with a nearly 3-fold increase of neutrophils at 36, but not 72 hours (F). FACS analysis for hematopoietic stem cells defined as CD45+, lineage negative, Sca-1 +, c-Kit + cells (LSK) showed a significant increase in the absolute number of LSK cells at 36 hours post-CCl4 injection in mice treated with AMD3100 (G). Representative images shown, n=3-6, *P<.05.
Discussion
Chemokines play a critical role in the progression of liver disease through modulation of hepatic immune cell infiltration and liver parenchymal cell activity 1. Despite the high levels of CXCL12 expression within the liver, its function is still largely unknown. Previous reports have demonstrated a potential protective role for CXCL12 during injury and regeneration through expansion of hepatic progenitor and oval cells 27-29 and we have shown that it can activate hepatic stellate cells 17.
Here we show that in a murine model of fibrosis, there is an increase in CXCL12 and CXCR4 and that inhibition of this pathway with AMD3100 leads to increased fibrosis and hepatic inflammation. While CXCR4 regulation clearly occurs at the mRNA and protein levels, despite an increase in CXCL12 protein expression there is no change in the transcript levels of any of the murine splice variants indicating that it is not transcriptionally regulated as has been previously reported 29, 30.
We utilized AMD3100, a CXCR4 small molecule inhibitor to modulate the CXCL12/CXCR4 axis during liver injury. As AMD3100 has a very short half-life we used sub-cutaneous osmotic pumps to allow for continual infusion of AMD3100 31. In mice receiving AMD3100 there was an increase in the degree of fibrosis, stellate cell activation, and the number of circulating and hepatic inflammatory cells with a specific increase in neutrophils. While CXCL12 promotes stellate cell activation during liver injury treatment with AMD3100 augmented α-SMA expression suggestive of increased stellate cell activity.
Multiple factors converge on stellate cell activation including chemokines, growth factors, and direct cell-cell contact. While inhibition of the CXCL12/CXCR4 axis can mitigate stellate cells activation this effect may have been offset by the increased intrahepatic inflammation and stellate cell activation through a CXCR4-independent mechanism. AMD3100 has allosteric agonist function through CXCR7 32, which stellate cells express, however, there is no change in activation with AMD3100 treatment in stellate cells with knock down of CXCR7, precluding a role through CXCR7 (data not shown).
In models of myocardial infarction (MI) continuous administration of AMD3100 augments injury, while a single dose, post-MI, promotes egress of bone marrow endothelial progenitor cells, increased myocardial vascularization and better outcomes than controls 13, 14. As continuous AMD3100 delivery similarly promoted fibrosis in our models, we hypothesized that once weekly delivery of AMD3100 during the course of chronic CCl4-induced injury would be protective. AMD3100 however had no effect on the extent of fibrosis or inflammation and may be due to the minimal role of ischemic injury in the chronic CCl4 model. Consequently, these treatments may be beneficial during liver transplantation, where ischemia/reperfusion induced injury is a major sources hepatocyte damage and early organ failure 33, 34.
We next investigated a possible role of AMD3100 as a therapy for acute hepatic injury. Despite the increased survival of rats treated with G-CSF and AMD3100 24, AMD3100 mono-therapy after acute CCl4 injury did not reduce the extent of hepatic injury measured by ALT, AST, and necrosis. In fact, similar to our previous results, there was a trend of worsened injury and increased liver inflammatory cells. Interestingly, at 36 hours post-injury, there was a 3-fold increase in neutrophils, known to be injurious and a 2-fold increase in hematopoietic stem cells, known to be protective. As AMD3100 led to an increase in cell populations with opposing roles, the neutrophilic predominance may have offset any potential beneficial effects of the hematopoietic stem cells.
In all our models, AMD3100 led to an increase in hepatic inflammation. AMD3100 promotes CXCR4 expressing inflammatory cell egress and simultaneously inhibits their recruitment to sites of injury, potentially altering the composition of infiltrating cells and the hepatic response to injury. Furthermore, AMD3100 is now known to preferentially release neutrophils from the lungs, while inhibiting their return to the bone marrow for removal 35, potentially explaining the specific increase of intrahepatic neutrophils seen in mice treated with AMD3100. Interestingly, increased intrahepatic inflammation secondary to AMD3100 alone did not induce any injury response. Only together with CCl4 treatment did the increased inflammatory cell populations lead to overall worse injury.
In conclusion, treatment with AMD3100 in models of hepatic injury and fibrosis consistently worsened the extent of inflammation and injury. Together our data confirms that inhibition of the CXCR4/CXCL12 axis worsens liver disease by altering the intrahepatic inflammatory response and is not a viable therapeutic target for patients with liver fibrosis.
Acknowledgments
The authors would like to thank Dr. Feng Hong and Hsini Chou for their technical support. This work was supported by NIH grants DK-6047402, DK-071745, and R56DK-092128 (M.B.B.) and DK-090986 (Y.S.).
Footnotes
No conflicts of interest, financial or otherwise are declared by the authors.
References
- 1.Saiman Y, Friedman SL. The role of chemokines in acute liver injury. Front Physiol. 2012;3:213. doi: 10.3389/fphys.2012.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seki E, De Minicis S, Gwak G, et al. CCR1 and CCR5 promote hepatic fibrosis in mice. The Journal of Clinical Investigatin. 2009;119:1858–70. doi: 10.1172/JCI37444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ajuebor MN, Hogaboam CM, Le T, Swain MG. C-C chemokine ligand 2/monocyte chemoattractant protein-1 directly inhibits NKT cell IL-4 production and is hepatoprotective in T cell-mediated hepatitis in the mouse. Journal of immunology. 2003 May 15;170:5252–9. doi: 10.4049/jimmunol.170.10.5252. [DOI] [PubMed] [Google Scholar]
- 4.Seki E, de Minicis S, Inokuchi S, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009 Jul;50:185–97. doi: 10.1002/hep.22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonacchi A, Petrai I, Defranco RM, et al. The chemokine CCL21 modulates lymphocyte recruitment and fibrosis in chronic hepatitis C. Gastroenterology. 2003 Oct;125:1060–76. doi: 10.1016/s0016-5085(03)01194-6. [DOI] [PubMed] [Google Scholar]
- 6.Zeremski M, Dimova R, Brown Q, Jacobson IM, Markatou M, Talal AH. Peripheral CXCR3-associated chemokines as biomarkers of fibrosis in chronic hepatitis C virus infection. J Infect Dis. 2009 Dec 1;200:1774–80. doi: 10.1086/646614. [DOI] [PubMed] [Google Scholar]
- 7.Zhang W, Fei Y, Gao J, Liu B, Zhang F. The role of CXCR3 in the induction of primary biliary cirrhosis. Clin Dev Immunol. 2011;2011:564062. doi: 10.1155/2011/564062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. 2007 Feb 2;353:104–8. doi: 10.1016/j.bbrc.2006.11.149. [DOI] [PubMed] [Google Scholar]
- 9.Harb R, Xie G, Lutzko C, et al. Bone marrow progenitor cells repair rat hepatic sinusoidal endothelial cells after liver injury. Gastroenterology. 2009 Aug;137:704–12. doi: 10.1053/j.gastro.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hashmi A, Hakim W, Kruglov E, et al. Adenosine inhibits cytosolic calcium signals and chemotaxis in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2007;292:G395–G401. doi: 10.1152/ajpgi.00208.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCandless EE, Piccio L, Woerner BM, et al. Pathological expression of CXCL12 at the blood-brain barrier correlates with severity of multiple sclerosis. Am J Pathol. 2008 Mar;172:799–808. doi: 10.2353/ajpath.2008.070918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dotan I, Werner L, Vigodman S, et al. CXCL12 is a constitutive and inflammatory chemokine in the intestinal immune system. Inflamm Bowel Dis. 2010 Apr;16:583–92. doi: 10.1002/ibd.21106. [DOI] [PubMed] [Google Scholar]
- 13.Dai S, Yuan F, Mu J, et al. Chronic AMD3100 antagonism of SDF-1alpha-CXCR4 exacerbates cardiac dysfunction and remodeling after myocardial infarction. J Mol Cell Cardiol. 2010 Oct;49:587–97. doi: 10.1016/j.yjmcc.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jujo K, Hamada H, Iwakura A, et al. CXCR4 blockade augments bone marrow progenitor cell recruitment to the neovasculature and reduces mortality after myocardial infarction. Proceedings of the National Academy of Sciences of the United States of America. 2010 Jun 15;107:11008–13. doi: 10.1073/pnas.0914248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jantunen E, Lemoli RM. Preemptive use of plerixafor in difficult-to-mobilize patients: an emerging concept. Transfusion. 2012 Apr;52:906–14. doi: 10.1111/j.1537-2995.2011.03349.x. [DOI] [PubMed] [Google Scholar]
- 16.Wald O, Pappo O, Safadi R, Dagan-Berger M, Beider K. Involvement of the CXCL12/CXCR4 pathway in the advanced liver disearse that is associated with hepatitis C virus or hepatitis B virus. Eur J Immunol. 2004;34:1164–74. doi: 10.1002/eji.200324441. [DOI] [PubMed] [Google Scholar]
- 17.Hong F, Tuyama A, Lee T, et al. Hepatic Stellate Cells Express Functional CXCR4: Role in Stromal Cell-Derived Factor 1a mediated Stellate Cell Activation. Hepatology. 2009;49:2055–67. doi: 10.1002/hep.22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sawitza I, Kordes C, Reister S, Haussinger D. The niche of stellate cells within rat liver. Hepatology. 2009 Nov;50:1617–24. doi: 10.1002/hep.23184. [DOI] [PubMed] [Google Scholar]
- 19.Wintermeyer P, Cheng CW, Gehring S, et al. Invariant natural killer T cells suppress the neutrophil inflammatory response in a mouse model of cholestatic liver damage. Gastroenterology. 2009 Mar;136:1048–59. doi: 10.1053/j.gastro.2008.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Novobrantseva TI, Majeau GR, Amatucci A, et al. Attenuated liver fibrosis in the absence of B cells. The Journal of clinical investigation. 2005 Nov;115:3072–82. doi: 10.1172/JCI24798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karlmark KR, Weiskirchen R, Zimmermann HW, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009 Jul;50:261–74. doi: 10.1002/hep.22950. [DOI] [PubMed] [Google Scholar]
- 22.Connolly MK, Bedrosian AS, Mallen-St Clair J, et al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. The Journal of clinical investigation. 2009 Nov;119:3213–25. doi: 10.1172/JCI37581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takami T, Terai S, Sakaida I. Stem cell therapy in chronic liver disease. Curr Opin Gastroenterol. 2012 May;28:203–8. doi: 10.1097/MOG.0b013e3283521d6a. [DOI] [PubMed] [Google Scholar]
- 24.Mark AL, Sun Z, Warren DS, et al. Stem cell mobilization is life saving in an animal model of acute liver failure. Ann Surg. 2010 Oct;252:591–6. doi: 10.1097/SLA.0b013e3181f4e479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broxmeyer HE, Orschell CM, Clapp DW, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005 Apr 18;201:1307–18. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stroncek DF, Clay ME, Herr G, et al. The kinetics of G-CSF mobilization of CD34+ cells in healthy people. Transfus Med. 1997 Mar;7:19–24. doi: 10.1046/j.1365-3148.1997.d01-75.x. [DOI] [PubMed] [Google Scholar]
- 27.Zheng D, Oh SH, Jung Y, Petersen BE. Oval cell response in 2-acetylaminofluorene/partial hepatectomy rat is attenuated by short interfering RNA targeted to stromal cell-derived factor-1. Am J Pathol. 2006 Dec;169:2066–74. doi: 10.2353/ajpath.2006.060211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mavier P, Martin N, Couchie D, Preaux AM, Laperche Y, Zafrani ES. Expression of stromal cell-derived factor-1 and of its receptor CXCR4 in liver regeneration from oval cells in rat. Am J Pathol. 2004 Dec;165:1969–77. doi: 10.1016/S0002-9440(10)63248-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsuchiya A, Imai M, Kamimura H, et al. Increased Susceptibility to Severe Chronic Liver Damage in CXCR4 Conditional Knock-Out Mice. Dig Dis Sci. 2012 Jun 5; doi: 10.1007/s10620-012-2239-8. [DOI] [PubMed] [Google Scholar]
- 30.Migliaccio AR, Martelli F, Verrucci M, et al. Altered SDF-1/CXCR4 axis in patients with primary myelofibrosis and in the Gata1 low mouse model of the disease. Experimental hematology. 2008 Feb;36:158–71. doi: 10.1016/j.exphem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Clercq E, Yamamoto N, Pauwels R, et al. Highly potent and selective inhibition of human immunodeficiency virus by the bicyclam derivative JM3100. Antimicrob Agents Chemother. 1994 Apr;38:668–74. doi: 10.1128/aac.38.4.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Molecular pharmacology. 2009 May;75:1240–7. doi: 10.1124/mol.108.053389. [DOI] [PubMed] [Google Scholar]
- 33.Martinez-Mier G, Toledo-Pereyra LH, McDuffie JE, Warner RL, Ward PA. Neutrophil depletion and chemokine response after liver ischemia and reperfusion. J Invest Surg. 2001 Mar-Apr;14:99–107. doi: 10.1080/08941930152024228. [DOI] [PubMed] [Google Scholar]
- 34.Zwacka RM, Zhang Y, Halldorson J, Schlossberg H, Dudus L, Engelhardt JF. CD4(+) T-lymphocytes mediate ischemia/reperfusion-induced inflammatory responses in mouse liver. The Journal of clinical investigation. 1997 Jul 15;100:279–89. doi: 10.1172/JCI119533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devi S, Wang Y, Chew WK, et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J Exp Med. 2013 Oct 21;210:2321–36. doi: 10.1084/jem.20130056. [DOI] [PMC free article] [PubMed] [Google Scholar]