

Abstract
Background
We have compared 38 isolates of the SARS-CoV complete genome. The main goal was twofold: first, to analyze and compare nucleotide sequences and to identify positions of single nucleotide polymorphism (SNP), insertions and deletions, and second, to group them according to sequence similarity, eventually pointing to phylogeny of SARS-CoV isolates. The comparison is based on genome polymorphism such as insertions or deletions and the number and positions of SNPs.
Results
The nucleotide structure of all 38 isolates is presented. Based on insertions and deletions and dissimilarity due to SNPs, the dataset of all the isolates has been qualitatively classified into three groups each having their own subgroups. These are the A-group with "regular" isolates (no insertions / deletions except for 5' and 3' ends), the B-group of isolates with "long insertions", and the C-group of isolates with "many individual" insertions and deletions. The isolate with the smallest average number of SNPs, compared to other isolates, has been identified (TWH). The density distribution of SNPs, insertions and deletions for each group or subgroup, as well as cumulatively for all the isolates is also presented, along with the gene map for TWH.
Since individual SNPs may have occurred at random, positions corresponding to multiple SNPs (occurring in two or more isolates) are identified and presented. This result revises some previous results of a similar type. Amino acid changes caused by multiple SNPs are also identified (for the annotated sequences, as well as presupposed amino acid changes for non-annotated ones). Exact SNP positions for the isolates in each group or subgroup are presented. Finally, a phylogenetic tree for the SARS-CoV isolates has been produced using the CLUSTALW program, showing high compatibility with former qualitative classification.
Conclusions
The comparative study of SARS-CoV isolates provides essential information for genome polymorphism, indication of strain differences and variants evolution. It may help with the development of effective treatment.
Keywords: SARS-CoV complete genome, isolates, genome sequence variation, SNP, classification, phylogenetics
Background
Severe Acute Respiratory Syndrome (SARS) is a new infectious disease reported first in the autumn of 2002 and diagnosed for the first time in March 2003 [1]. It is still a serious threat to human health and SARS coronavirus (CoV) has been associated with the pathogenesis of SARS according to Koch's postulate [2].
Significant research efforts have been made into investigation of the SARS-CoV genome sequence, aimed at establishing its origin and evolution to help eventually in preventing or curing the disease it causes. Although the task is a hard one, it opens up the opportunity, amongst others, for comparative investigation of different SARS-CoV isolates aimed at identification of genome regions properties expressing different levels of sequence polymorphism [3-8].
The genome of SARS-CoV consists of a single positive RNA strand approximately 30 Kb in length, consisting of about 10 open reading frames (ORF), and about 10 intergenic regions (IGRs). The first two overlapping ORFs at the 5' end encompass two-thirds of the genome, while the rest of the ORFs at the 3' end account for the remaining third.
We investigated 38 isolates of the SARS-CoV complete genome (two pairs of which were identical), sequenced and published by October 31st 2003 (with updated revisions up to February 20th, 2004). Sequences were taken from the PubMed NCBI Entrez site [9] in gbk and fasta formats (Table 1). The main goal was twofold: first, to analyze and compare nucleotide sequences, to identify SNPs positions, insertions and deletions, and second, to group them according to sequence similarity, eventually pointing to phylogeny of SARS-CoV isolates.
Table 1.
List of the SARS-CoV complete genome isolates investigated. Included are isolates' labels, IDs, accession numbers, length in nucleotides, dates of revisions considered and countries and sources of isolates.
| Label | ID | Accession No. | Length | Revision date | Country/Source |
| 1. | TWH | Ap006557.1 | 29727 | 02-AUG-2003 | Taiwan: patient #01 |
| TWC2 | Ay362698.1 | 13-AUG-2003 | Taiwan: Hoping Hospital | ||
| 2. | TWC3 | Ay362699.1 | 29727 | 13-AUG-2003 | Taiwan: Hoping Hospital |
| 3. | TWK | Ap006559.1 | 29727 | 02-AUG-2003 | Taiwan: patient #06 |
| 4. | TWS | Ap006560.1 | 29727 | 02-AUG-2003 | Taiwan: patient #04 |
| 5. | TWY | Ap006561.1 | 29727 | 02-AUG-2003 | Taiwan: patient #02 |
| 6. | Urbani | Ay278741.1 | 29727 | 12-AUG-2003 | USA: Atlanta |
| 7. | TWJ | Ap006558.1 | 29725 | 02-AUG-2003 | Taiwan: patient #043 |
| 8. | TWC | Ay321118.1 | 29725 | 26-JUN-2003 | Taiwan, first fatal case |
| 9 | WHU | Ay394850.2 | 29728 | 12-JAN-2004 | China: Wuhan |
| 10. | TW1 | Ay291451.1 | 29729 | 14-MAY-2003 | Taiwan |
| 11. | Frankfurt 1 | Ay291315.1 | 29727 | 11-JUN-2003 | Germany: Frankfurt |
| 12. | FRA | Ay310120.1 | 29740 | 12-DEC-2003 | Germany: patient from Frankfurt |
| 13. | HKU-39849 | Ay278491.2 | 29742 | 29-AUG-2003 | China: Hong Kong |
| 14. | Tor2 | Ay274119.3 | 29751 | 16-MAY-2003 | Canada: Toronto, patient #2 |
| Nc_004718.3 | 06-FEB-2004 | Canada: Toronto, patient #2 | |||
| 15. | HSR 1 | Ay323977.2 | 29751 | 15-OCT-2003 | Italy |
| 16. | CUHK-Su10 | Ay282752.2 | 29736 | 17-NOV-2003 | China: Hong Kong |
| 17. | CUHK-W1 | Ay278554.2 | 29736 | 31-JUL-2003 | China: Hong Kong |
| 18. | GZ50 | Ay304495.1 | 29720 | 05-NOV-2003 | China: Hong Kong |
| 19. | AS | Ay427439.1 | 29711 | 21-OCT-2003 | Italy: Milan |
| 20. | Sin2500 | Ay283794.1 | 29711 | 12-AUG-2003 | Singapore |
| 21. | Sin2679 | Ay283796.1 | 29711 | 12-AUG-2003 | Singapore |
| 22. | Sin2774 | Ay283798.2 | 29711 | 02-OCT-2003 | Singapore |
| 23. | Sin2677 | Ay283795.1 | 29705 | 12-AUG-2003 | Singapore |
| 24. | Sin2748 | Ay283797.1 | 29706 | 12-AUG-2003 | Singapore |
| 25. | BJ01 | Ay278488.2 | 29725 | 01-MAY-2003 | China: Beijing |
| 26. | BJ02 | Ay278487.3 | 29745 | 05-JUN-2003 | China: Beijing |
| 27. | BJ03 | Ay278490.3 | 29740 | 05-JUN-2003 | China: Beijing |
| 28. | BJ04 | Ay279354.2 | 29732 | 05-JUN-2003 | China: Beijing |
| 29. | Taiwan TC1 | Ay338174.1 | 29573 | 28-JUL-2003 | Taiwan |
| 30. | Taiwan TC2 | Ay338175.1 | 29573 | 28-JUL-2003 | Taiwan |
| 31. | Taiwan TC3 | Ay348314.1 | 29573 | 29-JUL-2003 | Taiwan |
| 32. | GD01 | Ay278489.2 | 29757 | 18-AUG-2003 | China: Beijing |
| 33. | SZ3 | Ay304486.1 | 29741 | 05-NOV-2003 | China: Hong Kong |
| 34. | SZ16 | Ay304488.1 | 29731 | 05-NOV-2003 | China: Hong Kong |
| 35. | ZJ01 | Ay297028.1 | 29715 | 19-MAY-2003 | China: Beijing |
| 36. | ZMY 1 | Ay351680.1 | 29749 | 03-AUG-2003 | China: Guangdong |
According to the length of isolates (insertions and deletions) and the presence of SNPs, we classified them into three main groups with subgroups: "regular" isolates with no insertions or deletions (with different numbers of SNPs), isolates with "long insertions" and isolates with "many individual" insertions and deletions (with different positions of SNPs), which is close to phylogenetic analysis results.
Results and discussion
Genome polymorphism
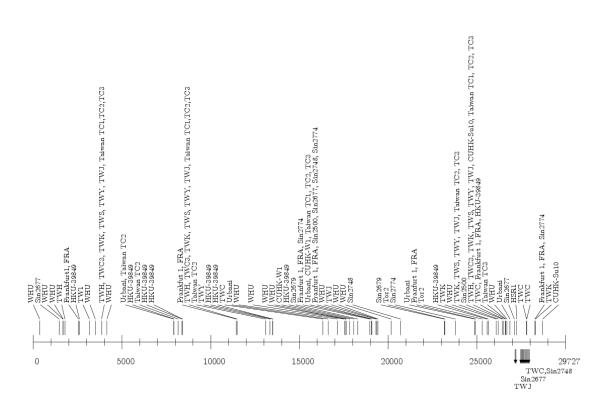
All the sequences are between 29573 and 29757 in length (Table 1), with a high degree of similarity (>99% pairwise). Still, they can be differentiated on the basis of sequence polymorphism (insertions and deletions), number and sites of SNPs [8]. Results of the comparison of genome primary structure of the analyzed isolates are given in Figure 1.
Figure 1.
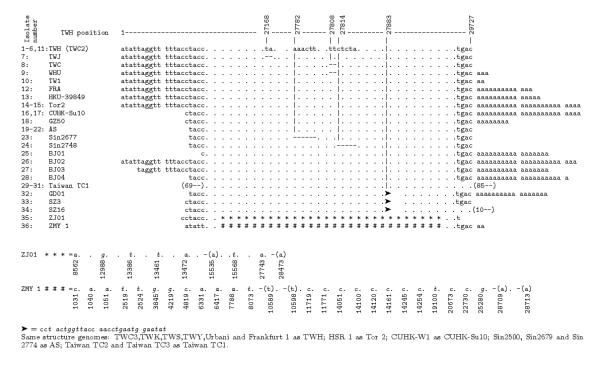
Comparison of nucleotide structures of SARS-CoV complete genome isolates. Insertions are denoted as emphasized (italic) and 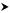 , deletions by minus sign ("-"). Positions are given in relation to the TWH isolate. The two isolates with a large number of individual insertions (ZJ01, ZMY 1) are given separately, with exact positions of insertions and deletions.
, deletions by minus sign ("-"). Positions are given in relation to the TWH isolate. The two isolates with a large number of individual insertions (ZJ01, ZMY 1) are given separately, with exact positions of insertions and deletions.
Analysis of genomic polymorphism of the isolates resulted in the following facts
I) Some of the isolates are nucleotide-identical or almost identical. There are two pairs of nucleotide-identical isolate sequences: (TWH, TWC2) and Tor2 (with accession numbers Ay274119, Nc_004718). Therefore, instead of 38, we consider the dataset to contain 36 isolates. Further, the isolate TWC3 differs in just one position with TWH (see table in additional file 1), which is about randomly expected [11]. Isolates Frankfurt 1 and FRA are identical up to the poly-"a" of length 13 present at the 3' end of FRA (Figure 1).
II) Similarity analysis showed that a significant number of isolates have the same length (29727 bases), the same beginning and ending subsequences (that seem to be exact starts and ends of the complete SARS-CoV genome up to the poly-"a" at the 3' end), thus forming a kind of referent group; these are the isolates TWH, TWC3, TWK, TWS, TWY, Urbani, Frankfurt 1 (Figure 1). The fully sequenced isolate TWH then has been chosen as the referent isolate for sequence comparisons since its average number of SNPs compared to other isolates is the smallest. For example, TWH and Urbani have an average number of SNPs 15.7 and 17.6 respectively for all the isolates, and 5.7 and 10.5 respectively for the referent group. For SNPs see the tables in the additional files 1 and 2.
III) Most isolates, compared to TWH, are shorter at the 5'end (e.g., Sin2500, Sin2679, Sin2774, Sin2677, Sin2748, AS), have various length poly-"a" strings at the 3' end (e.g., Tor2, HSR1, FRA, BJ02, TW1, HKU-39489, WHU), or both (BJ01, BJ03, BJ04, CUHK-W1, CUHK-Su10). Three of the isolates, Taiwan TC1, Taiwan TC2, Taiwan TC3, have both starting and ending deletions (at the 5' end 69, at the 3' end 85 nucleotides). Several isolates (e.g. TWJ, TWC, Sin2677, Sin2748) have some short deletions inside the sequence (Figure 1).
IV) There is a group of isolates that have significant length insertions (29 nucleotides) inside the sequence. These are the isolates GD01, SZ3, SZ16. A significant number of individual insertions have been identified in ZJ01 and ZMY 1 isolates (Figure 1, additional files 3,4,5).
Among the SNP contents of isolates, there is a significant difference in the number of SNPs for different pairs of isolates. For TWH as the referent isolate, this number varies from 1 to 80 SNPs. Isolates may be classified into three groups based on the number of SNPs with TWH (Figure 2):
Figure 2.
Structural tree for SARS-CoV isolates. The tree is based on qualitative analysis of sequence variation of 36 isolates.
1. with less than 15 (TWC3, TWK, TWS, TWY, Urbani, TWJ, TWC, TW1, Tor2, HSR1, CUHK-Su10, AS, Sin2500, Sin2679, Sin2774, Sin2677, Sin2748, Taiwan TC1, Taiwan TC2, Taiwan TC3, Frankfurt1, FRA, HKU-39849, CUHK-W1),
2. between 15 and 30 (WHU, GZ50, BJ01-BJ04, ZJ01),
3. with equal to or greater than 30 SNPs (GD01, SZ3, SZ16, ZMY 1).
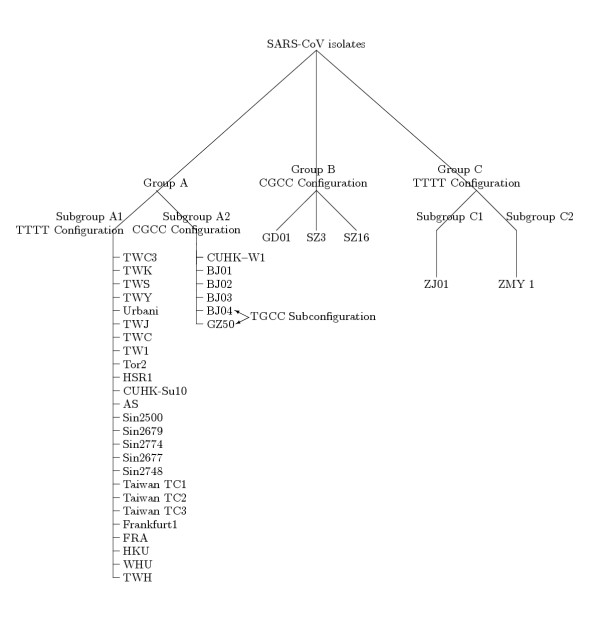
Finally, besides the number, there are differences in positions of SNPs (potential mutation sites). In order to avoid nucleotide changes that probably arose during propagation of the virus in cell culture and sequencing, Figure 3 represents positions (on the relative scale of all isolates and on TWH scale) where two or more SNPs occurred, not taking into consideration isolates with long insertions (GD01, SZ3 and SZ16). The positions of multiple SNPs of these three isolates, similar as far as these three are concerned, are highly different from all the others and are represented in Figure 4. These results coincide with those published in Marra et al's paper [4] for Urbani and Tor2 isolates, but differ from those published in Ruan's paper [8] for the 14 isolates therein analyzed (Sin-group, BJ-group, Tor2, Urbani, CUHK-W1, HKU-39849, GD01), which were obviously based on different revisions of the PubMed NCBI Entrez database [9]; lengths of the sequences Tor2, CUHK-W1, GD01, BJ01-BJ04 differ from the revisions we analyzed and consequently in some nucleotides and the number of base changes at given positions. Differences include the following positions (based upon Urbani and TWH SARS-CoV): 2601 (Tor2 T instead of C, BJ04 T instead of missing base), 7919 (BJ03 C instead of T), 8559 (BJ04 T instead of A), 8572 (BJ01 T instead of G, GD01 G instead of T), 9404 (BJ04 T instead of missing base), 9479 (BJ04 T instead of missing base), 9854 (BJ04 T instead of missing base), 19838 (GD01 G instead of A), 21721 (GD01, BJ01, A instead of missing base, BJ04 G instead of missing base), 22222 (BJ04 C instead of N), 27243 (GD01 T instead of C, BJ03 T instead of N), 29279 (all A's). The results obtained also differ from Hsueh et al. [12] regarding nucleotides in HKU-39849 isolate on positions 7746, 9404, 9479, 17564, 17846, 19064, 21721, 22222, 27827.
Figure 3.
Positions with two or more SNPs in A and C groups with amino acid changes. Positions are represented on the relative scale of all the isolates and on the TWH scale. Isolates from group B have not been counted, since their positions of SNPs while coordinated among them, are highly different from all the others. SNPs are in bold type. Proteins associated with SNPs are represented based on TWH annotation. IDs of annotated isolates are in grey boxes. Positions of SNPs causing amino acid changes, together with amino acid and their properties' change [16] are in grey. Legend of A. Ac. properties: Hp:hydrophobic, Ar:aromatic, Ap:aliphatic, P:polar, NCh: negative charged, PCh:positive charged, S: small, T:tiny
Figure 4.
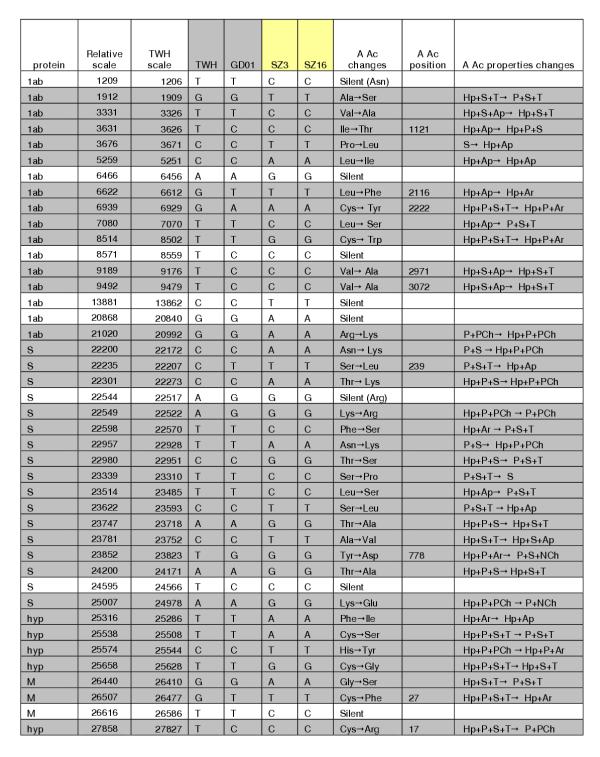
Positions with two or more SNPs in B group with amino acid changes. Only SNPs in B group isolates, regarding TWH, have been counted. The same notation is applied as in Figure 3.
Additional file 1,2,3,4,5 represent SNPs for all the isolates in all five groups, whether they occur in ORFs or IGR (for annotated isolates), as well as the number of SNPs in ORFs and SNPs in IGR, per isolate. The total number of SNPs is 312 (only 2 in IGRs: TWH positions 27812 for the isolate Taiwan TC3 and 27827 for the isolates BJ01 and CUHK-W1). The average number of SNPs per isolate is 15.7 and significant difference from the average shows TWC3 (just 1 SNP) and ZMY 1 (even 80).
Grouping of isolates
The isolates from the dataset considered may be classified according to their sequence polymorphism and SNP contents properties just described. At first, properties (III, IV) may result in three different groups (Figure 2):
A. "regular isolates" whose nucleotide structure is close to the referent group (different 5' and 3' ends, short deletion, individual insertion): TWH, TWC3, TWK, TWS, TWY, Urbani, TWJ, TWC, TW1, Tor2, HSR1, CUHK-Su10, AS, Sin2500, Sin2679, Sin2774, Sin2677, Sin2748, Taiwan TC1, Taiwan TC2, Taiwan TC3, WHU, Frankfurt1, FRA, HKU, CUHK-W1, GZ50 and BJ01-BJ04 (Figure 5, 6a)
Figure 5.
Density distribution of SNPs, insertions and deletions in the isolates of A1 group. SNPs are represented above the line, insertions below the line, upward oriented, and deletions below the line, downward oriented. The TWH scale is used. The same holds for Figures 6,7,8.
Figure 6.
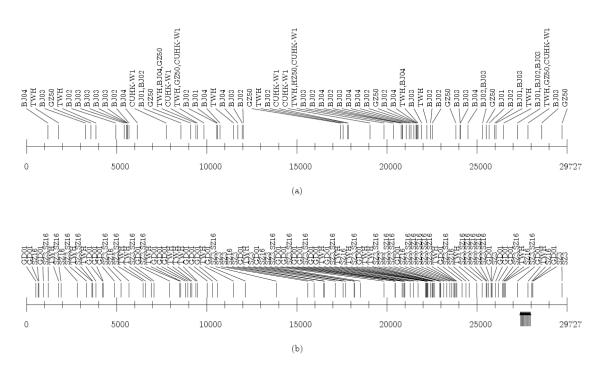
(a and b). Density distribution of SNPs, insertions and deletions in the isolates of A2 and B groups. In A2 group there are no insertions / deletions. In B group there are large insertions in GD01, SZ3 and SZ16 isolates.
B. isolates with "long insertions": GD01, SZ3 and SZ16 (Figure 6b) and
C. isolates with "many individual" insertions: ZJ01 and ZMY 1 (Figure 7a,7b).
Figure 7.
(a and b). Density distribution of SNPs, insertions and deletions in the isolates of C1, C2 groups.
Further, SNPs properties (1–3) may divide A group into A1 and A2, and C group into C1 and C2 subgroups:
A1. TWH, TWC3, TWK, TWS, TWY, Urbani, TWJ, TWC, TW1, Tor2, HSR1, CUHK-Su10, AS, Sin2500, Sin2679, Sin2774, Sin2677, Sin2748, Taiwan TC1, Taiwan TC2, Taiwan TC3, Frankfurt1, FRA, HKU and CUHK-W1 (Figure 5)
A2. WHU, BJ01-BJ04 and GZ50 (Figure 6a)
C1: ZJ01 (Figure 7a)
C2: ZMY 1 (Figure 7b)
Finally, the positions of SNPs will move CUHK-W1 from A1 into A2 group (more than 50% of common SNP positions) while WHU will move from A2 into A1 (less than 30% of common SNP positions), giving the final grouping of isolates presented as a structural tree (Figure 2):
A1. TWH, TWC3, TWK, TWS, TWY, Urbani, TWJ, TWC, TW1, Tor2, HSR1, CUHK-Su10, AS, Sin2500, Sin2679, Sin2774, Sin2677, Sin2748, Taiwan TC1, TC2, TC3, Frankfurt1, FRA, HKU and WHU (Figure 5 and the additional file 1)
A2. CUHK-W1, GZ50 and BJ01-BJ04 (Figure 6a and the additional file 2)
B. GD01, SZ3 and SZ16 (Figure 6b and the additional file 3)
C1. ZJ01 (Figure 7a and the additional file 4)
C2. ZMY 1 (Figure 7b and the additional file 5).
Although qualitative in nature, the structural tree turns out to be close to the quantitative grouping which is a basis for (computational) phylogenetic classification.
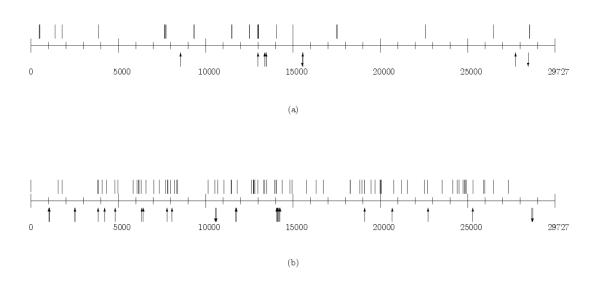
Tables in additional files 1,2,3,4,5 represent SNPs, insertions and deletions in groups A-C (see additional files 1 for isolates of A1 group, on the relative and TWH scale, additional files 2 for isolates of A2 group and TWH, on the TWH scale, additional files 3 for isolates of group B with TWH, and additional files 4,5 for isolates of C1, C2 groups, respectively). Figures 5,6,7 represent density distribution of SNPs, insertions and deletions on the TWH scale, for the same groups of isolates. Figure 8 represents the overall density distribution of SNPs, insertions and deletions for all the 36 isolates, along with the gene map for TWH (which is quite similar to gene maps of other isolates). Density distributions do not show regularities yet (with respect to the number of available sequences) that could provide for precise statistical characterization. Still, they exhibit crowding regions close to the 3' end which is also characterized by the presence of a number of proteins of unknown function.
Figure 8.
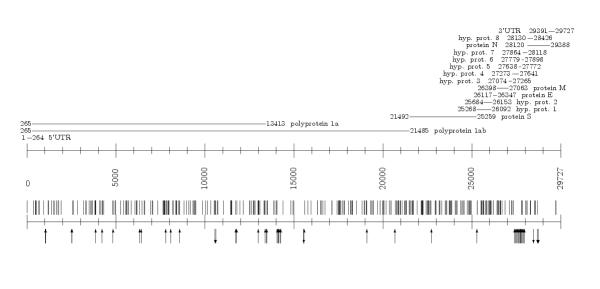
The overall density distribution of SNPs, insertions and deletions along with the gene map for TWH.
It can also be noted that the proposed grouping of 36 isolates, based on different criteria, still conserves the previous classification T-T-T-T / C-G-C-C [8]. All the isolates from groups A1 and C have T-T-T-T configuration, while all the isolates from groups A2 and B have C-G-C-C configuration, except for GZ50, BJ04 being TGCC (Figure 2, Figure 9). The two sequence variants correspond to the epidemiological spread, so that those that originated in the Hotel M in Hong Kong have the T-T-T-T configuration – covering A1, C groups in our classification – Canada (Tor2), Singapore (all Sins), Frankfurt, Taiwan, Hong Kong (HKU39849), Hanoi (Urbani), Italy (HSR1), China (ZJ01), etc, and others having C-G-C-C configuration (A2, B in our classification) which originated in Guangdong, China (GD01 and GZ50), Hong Kong (CUHK-W1, SZ3 and SZ16), Beijing (BJ01-BJ04). The fact that the enlarged number of isolates exhibits the same properties relating to the four loci supports the assumption that the mutations could not have arisen by chance base substitution during propagation in cell culture and the sequencing procedure [8].
Figure 9.
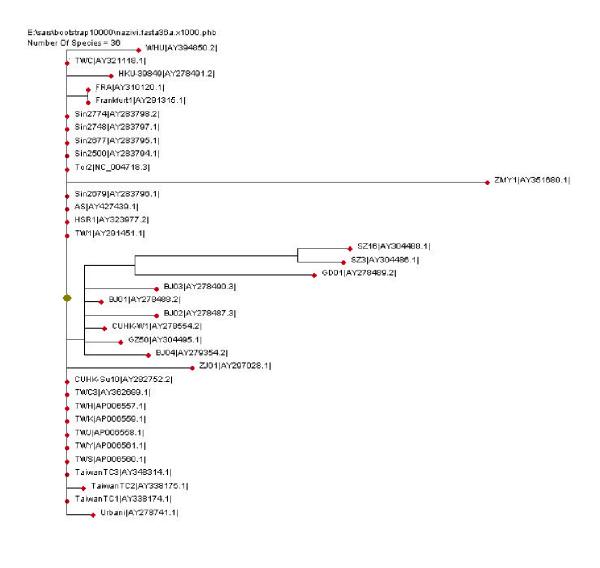
Phylogenetic tree of 36 SARS-CoV complete genome isolates. Distances represent degree of sequence variation. The largest distance is associated with ZMY 1, followed by ZJ01 isolate (groups C1, C2). Groups A1, A2 and B are clearly distinguished. The tree has been obtained using CLUSTALW and PhyloDraw programs.
Changes in amino acids
We analyzed amino acid changes in proteins for the annotated isolates (19 out of 36), and presumed proteins in non-annotated ones for multiple SNPs in all the isolates. Results of the analysis are represented in Figures 3 and 4. Figure 3 shows that silent mutations occurred in envelope protein E, while nucleotide changes resulted in amino acid changes in spike (S), membrane (M) and nucleocapside (N) proteins. All three SNPs in the spike protein are situated in the outer membrane region and not within the potential epitope region (amino acid position 469–882) as proposed by Ren Y. et al. [13]. Amino acid changes occurred in two multiple SNPs in M protein, one multiple SNPs in N protein and 7 (out of 13) multiple SNPs of the polyprotein 1ab, as well as in one multiple SNP of a hypothetical protein, while the silent mutations occurred in three hypothetical proteins. Figure 3 also represents properties of the corresponding amino acids resulted by SNPs. The only significant change in amino acid properties is in S protein Gly→Asp (A2, B groups, i.e., in CUHK-W1, GZ50, BJ01-BJ03, GD01, SZ3 and SZ16 isolates) and hypothetical protein Cys→Arg (the same isolates, BJ04 in addition). The only addition in non-annotated sequences is in hypothetical protein following S protein in TWH, exhibiting silent change, and in non-annotated BJ02 and BJ03, corresponding to the hypothetical protein, Gly→Glu. Similar analysis can be done for amino acid changes corresponding to SNPs at positions specific for B group isolates (Figure 4). Taking into account the only annotated isolate GD01, there are five amino acid changes in polyprotein 1ab, two amino acid properties changes in S protein (Ser→Leu and Tyr→Asp, the second being within the epitope region), one amino acid change in M protein and one amino acid property change (Cis→Arg) in BGI-PUP.
Phylogenetic analysis
The SARS-CoV isolates have been multialigned using the CLUSTALW program [10] as the very first step in obtaining a phylogenetic tree. The aligned sequences have been submitted then to CLUSTALW for bootstrapping and phylogenetic tree production. Enlargement of the sequence set resulted in the refinement of the phylogenetic tree produced, as compared to previous results such as Ruan [8] and Zhang&Zheng [14], obtained for 14 and 16 isolates, respectively. The phylogenetic tree obtained, drawn using the PhyloDraw program [15], is represented in Figure 9. It is similar to our structural tree based on qualitative analysis of the isolates (Figure 2).
The results of the analysis of dissimilarities, described in previous paragraphs, are in accordance with the alignment obtained by CLUSTALW, but regrouped and formatted in a way that facilitates further interpretation and application.
Conclusion
Comparative analysis of genome sequence variations of 38 SARS-CoV isolates resulted in some conclusions that might be of interest in further investigation of the SARS-CoV genome:
1. All of the SARS-CoV isolates are highly homologous (more than 99% pairwise). Most of them have similar nucleotide structure, with the same 5' and 3' ends and poly-"a" at the 3' end of different length (0–24), some of them with a single short deletion close to the 3' end of the sequence; out of 312 SNPs in total, only two are in IGRs.
2. Three of the 38 isolates have long insertions within the sequence;
3. Two of the isolates have a large number of individual insertions / deletions, exhibiting different SNP positions;
4. All the isolates may be grouped according to sequence polymorphism into three groups (with up to two subgroups), reflecting their similarities / dissimilarities. Since the isolate sequences have a high degree of homology, different properties of groups are represented in a more transparent way in the classification tree obtained by such a qualitative analysis, than in a bootstrapped phylogenetic tree obtained from multialigned sequences using the CLUSTALW program [10].
5. The total number of amino acid changes caused by multiple SNPs is 15 (in isolates of A, C groups) and 34 in isolates of B group. The total number of silent mutations is 10 (for A, C groups) and 7 (for B group).
6. Since S protein is of special interest regarding its receptor affinity and antigenecity, it is interesting to notice that all amino acid properties' changes are located in its outer membrane region, one for A, C groups and two for B group.
7. The results obtained may be useful in further investigation aiming at identification of SARS-CoV genome regions responsible for its infectious nature.
Methods
Dataset
We investigated the complete genomes of 38 SARS-CoV isolates. Nucleotide sequences are taken from the PubMed NCBI Entrez database [9] in gbk and fasta formats (Table 1).
The coverage included all the isolates published by October 31st 2003 (with updated revisions). The identifiers, accession numbers, genomic size (in nucleotides), revision dates and country or source of the isolates considered are included in the table, together with labels as referred in this paper. The fully sequenced isolate TWH has been chosen as the referent isolate, since its average number of SNPs was the lowest as compared to all other isolates.
Methods for similarity analysis
For similarity analysis of isolates, the following procedure has been applied consisting of two steps:
1. identification of structurally identical parts of isolates, i.e., insertion and deletion sites
2. identification of SNPs in structurally identical parts.
Step 1 has been carried out by a function performing similarity analysis of subsequences of a given length (e.g., 100 bps), and identifying significantly non-matching strings as being inserted in the corresponding sequence (i.e. deleted from the other). Since significant number of isolates have the same length (29727 bases) and starting and ending subsequences (that seem to be the exact starts and ends of the complete SARS-CoV genome up to the poly-"a" at the 3' end), they may be considered as forming a representative group. The nucleotide structure of all other isolates was analyzed with respect to this representative group. For each pair of isolates (x,y) (x from the representative group), a file InsDelx-y has been produced containing positions and lengths of each of the insertions or deletions in the isolate y.
Step 2 has been carried out by comparing structurally identical parts (of the same length) of pairs of isolates. The starting and ending positions of those parts have been taken from the file InsDelx-y (for comparison of x and y), produced in step 1. The procedure returns results in a file with SNPs in the two sequences (files Mismx-y).
We also used the CLUSTALW program [10] for multialignment as a control process, as well as for phylogenetic investigations.
Methods for phylogenetic investigation
In order to use similarity analysis results for drawing any phylogenetic conclusions about the SARS-CoV genome dataset, a CLUSTALW [10] multialigned output has been generated and a bootstrapped phylogenetic tree has been produced and drawn using the PhyloDraw program [15].
Authors' contributions
GMP-L performed the computational analysis and structural classification of SARS-CoV genome isolates, participated in drawing figures and drafted the manuscript.
NSM participated in sequence alignment, bootstrapping and phylogenetic tree production, in drawing figures and manuscript editing and formatting.
MVB participated in the design and overall coordination of the study.
All authors read and approved the final manuscript.
Supplementary Material
Positions of SNPs in A1 group. Positions are given on the relative and TWH scales. IDs of annotated isolates are in grey boxes; SNPs in ORFs (or corresponding to those in ORFs, for non-annotated isolate) are in red bold and SNPs in IGRs in blue bold. The total number of SNPs per isolate is given at the bottom, as well as number of SNPs in ORFs and IGRs for annotated isolates. A minus sign (-) denotes deletion.
Positions of SNPs in A2 group. Positions are given on the TWH scale. The same notation is applied as in the additional file 1.
Positions of SNPs and insertions in B group. The exact positions on all four scales (TWH, GD01, SZ3 and SZ16) are given. ID of the only annotated isolate (GD01) is in grey box; SNPs in ORFs (or corresponding to those in ORFs, for non-annotated isolates) are in red bold. The total number of SNPs per isolate is given at the bottom, as well as the number of SNPs in ORFs and IGRs for annotated isolate. Small letters denote insertion and a minus sign (-) denotes the corresponding deletion.
Positions of SNPs, insertions and deletions in C1 group. Positions of SNPs, insertions and deletions on both TWH and ZJ01 scales are given. The total number of SNPs is given. SNPs are in red bold. A minus sign (-) denotes deletion (insertion).
Positions of SNPs, insertions and deletions in C2 group. Positions of SNPs, insertions and deletions on both TWH and ZMY 1 scales are given. The total number of SNPs is given. SNPs are in red bold. A minus sign (-) denotes deletion (insertion).
Acknowledgments
Acknowledgements
The work presented has been financially supported by the Ministry of Science and Technology, Republic of Serbia, Project No. 1858.
Contributor Information
Gordana M Pavlović-Lažetić, Email: gordana@matf.bg.ac.yu.
Nenad S Mitić, Email: nenad@matf.bg.ac.yu.
Miloš V Beljanski, Email: mbel@matf.bg.ac.yu.
References
- Maskalyk J, Hoey J. SARS update. CMAJ. 2003;168:1294–1295. [PMC free article] [PubMed] [Google Scholar]
- Fouchier RA, Kuiken T, Schutten M, vanAmerongen G, vanDoornum GJ, vandenHoogen BG, Peiris M, Lim W, Stohr K, Osterhaus ADM. Aetiology: Koch's postulates fulfilled for SARS virus. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Peñaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TCT, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Günther S, Osterhaus ADME, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome, Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Marra MA, Jones SJM, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YSN, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. The Genome Sequence of the SARS-Associated Coronavirus, Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Thiel V, Ivanov KA, Putics A, Hertzig T, Schelle B, Bayer S, Weißbrich B, Snijder EJ, Rabenau H, Doerr HW, Gorbalenya AE, Ziebuhr J. Mechanisms and enzymes involved in SARS coronavirus genome expression, J Gen Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Qin E, Zhu Q, Yu M, Fan B, Chang G, Si B, Yang B, Peng W, Jiang T, Liu B, Deng Y, Liu H, Zhang Y, Wang C, Li Y, Gan Y, Li X, Lu F, Tan G, Cao W, Yang R, Wang J, Li W, Xu Z, Li Y, Wu Q, Lin W, Cheng W, Tang L, Deng Y, Han Y, Li C, Lei M, Li G, Li W, Lu H, Shi J, Tong Z, Zhang F, Li S, Liu B, Liu S, Dong W, Wang J, Gane KSW, Yu J, Yang H. A complete sequence and comparative analysis of a SARS-associated virus (Isolate BJ01) Chin Sci Bull. 2003;48:941–948. doi: 10.1360/03wc0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng FY, Chan CW, Chan MN, Chen JD, Chow KY, Hon CC, Hui Li J, Li VY, Wang CY, Wang PY, Guan Y, Zheng B, Poon LL, Cha KH, Yuen KY, Peiris JS, Leung FC. The complete genome sequence of severe acute respiratory syndrome coronavirus strain HKU-39849 (HK-39) Exp Biol Med (Maywood) 2003;228:866–873. doi: 10.1177/15353702-0322807-13. [DOI] [PubMed] [Google Scholar]
- Ruan YJ, Wei CL, Ee LA, Vega VB, Thoreau H, Yun STS, Chia JM, Ng P, Chiu KP, Lim L, Tao Z, Peng CK, Ean LOL, Lee NM, Sin LY, Ng LFP, Chee RE, Stanton LW, Long PM, Liu ET. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection, The Lancet. 2003;361:1779–1785. doi: 10.1016/S0140-6736(03)13414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PubMed NCBI Entrez http://www.ncbi.nlm.nih.gov/entrez
- CLUSTALW ftp://ftp.ebi.ac.uk/software/dos/clustalw
- Wood L. Questions about comparative genomics of SARS coronavirus isolates, Lancet. 2003;362:578. doi: 10.1016/S0140-6736(03)14130-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh PR, Hsiao CH, Yeh SH, Wang WK, Chen SH, Wang JT, Chang SC, Kao CL, Yang PC. Microbiologic characteristics, serologic responses, and clinical manifestations in Severe Acute Respiratory Syndrome, Taiwan, Emerging Infectious Diseases. 2003;9:1163–1167. doi: 10.3201/eid0909.030367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Ren Y, Zhou Z, Liu J, Lin L, Li S, Wang H, Xia J, Zhao Z, Wn J, Zhou C, Wang J, Yin J, Xu N, Liu S. A strategy for searching antigenic regions in the SARS-CoV spike protein, Geno, Prot & Bioinfo. 2003;1:207–215. doi: 10.1016/S1672-0229(03)01026-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zheng N. Genomic phylogeny of SARS coronavirus suggested that Guangdong province is the origin area (personal communication)
- PhyloDraw V0.82 http://pearl.cs.pusan.ac.kr/phylodraw
- Russel RB, Betts MJ, Barnes MR. Amino acid properties http://www.russell.embl.de/aas/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Positions of SNPs in A1 group. Positions are given on the relative and TWH scales. IDs of annotated isolates are in grey boxes; SNPs in ORFs (or corresponding to those in ORFs, for non-annotated isolate) are in red bold and SNPs in IGRs in blue bold. The total number of SNPs per isolate is given at the bottom, as well as number of SNPs in ORFs and IGRs for annotated isolates. A minus sign (-) denotes deletion.
Positions of SNPs in A2 group. Positions are given on the TWH scale. The same notation is applied as in the additional file 1.
Positions of SNPs and insertions in B group. The exact positions on all four scales (TWH, GD01, SZ3 and SZ16) are given. ID of the only annotated isolate (GD01) is in grey box; SNPs in ORFs (or corresponding to those in ORFs, for non-annotated isolates) are in red bold. The total number of SNPs per isolate is given at the bottom, as well as the number of SNPs in ORFs and IGRs for annotated isolate. Small letters denote insertion and a minus sign (-) denotes the corresponding deletion.
Positions of SNPs, insertions and deletions in C1 group. Positions of SNPs, insertions and deletions on both TWH and ZJ01 scales are given. The total number of SNPs is given. SNPs are in red bold. A minus sign (-) denotes deletion (insertion).
Positions of SNPs, insertions and deletions in C2 group. Positions of SNPs, insertions and deletions on both TWH and ZMY 1 scales are given. The total number of SNPs is given. SNPs are in red bold. A minus sign (-) denotes deletion (insertion).