

Abstract
Diabetes in humans and animals is accompanied by chronic low-grade inflammation, which could be a possible mediator of developing neuropathology and neurobehavioral deficits. The objective of the present study determined if decreasing inflammation could reverse diabetes-induced decreases in hippocampal cell proliferation, one aspect of hippocampal neurogenesis. C57BL/6J mice were made diabetic by administering streptozotocin (STZ; 195 mg/kg). STZ mice or vehicle controls received chronic treatment with the non-steroidal anti-inflammatory drug indomethacin (2 mg/kg for 14 days). Levels of glucose, corticosterone and, cytokines were measured from plasma, cell proliferation was measured using BrdU incorporation in the hippocampus and TNF-αR1 and TNF-αR2 mRNA was measured using real-time PCR. STZ-induced diabetes increased plasma levels of glucose and corticosterone and decreased body weight. Cell proliferation in the hippocampus was reduced in diabetic mice by 50%. The decreased level of cell proliferation was reversed by chronic treatment with indomethacin without changes to corticosterone and glucose levels. Plasma TNF-α levels increased in diabetic mice and were normalized by indomethacin treatment and IL-1 and IL-6 levels were unchanged by diabetes or indomethacin. In contrast, plasma levels of the cytokines IL-10 and IFN-gamma decreased in diabetic mice and were not affected by indomethacin treatment. STZ-induced diabetes decreased expression of TNF-αR2 but not TNF-αR1 mRNA. Indomethacin ameliorated the effects of STZ on hippocampal neurogenesis independent of corticosterone and glycemic control, possibly by mediating the proinflammatory cytokine TNF-α. Inflammation is a potential novel pharmacological target for alleviating neurobehavioral complications arising from diabetes.
Keywords: diabetes mellitus, streptozotocin, inflammation, indomethacin, corticosterone, neurogenesis
Introduction
Humans with diabetes frequently have comorbid neurobehavioral complications such as cognitive decline (Biessels et al. 2007; Brands et al. 2005) and depressed mood (Egede 2006; Holt et al. 2014; Lu et al. 2009). Changes in brain neuroplasticity, particularly in the hippocampus, produced by prolonged diabetes is likely a major contributor to these comorbid conditions (Ho et al. 2013). Numerous studies have shown that hippocampal cell proliferation is decreased in mice and rats rendered diabetic by streptozotocin (STZ), a compound that produces a model of type 1 diabetes by permanently destroying the insulin secreting pancreatic beta cells by uptake through the glucose transporter (Beauquis et al. 2006; Ho et al. 2011; Jackson-Guilford et al. 2000; Stranahan et al. 2008). In addition, decreased hippocampal neurogenesis has also been reported for rodent genetic models of type 2 diabetes associated with obesity (Stranahan et al. 2008; Yi et al. 2009). As decreased hippocampal neurogenesis has been associated with dysregulated cognition, affective behavior and endocrine regulation (Snyder and Cameron 2011; Snyder et al. 2011), these changes may contribute to the neurobehavioral complications associated with diabetes (Ho et al. 2013).
Understanding the physiological mediators responsible for decreasing hippocampal neurogenesis in diabetes can facilitate the development of novel therapeutic targets that could benefit patients by alleviating some of the associated neurobehavioral complications. Decreasing the hyperglycemia in STZ-treated diabetic mice directly by insulin treatment produced a reversal of the increased CORT levels and a corresponding normalization of hippocampal cell proliferation (Ho et al. 2011). However, this may not always be possible. For example, the increased secretion of adrenal steroids by the hypothalamic-pituitary-adrenal (HPA) axis is one of the principal regulators responsible for reducing hippocampal neurogenesis in diabetes. Thus, maintaining plasma CORT at normal levels with low-dose CORT replacement after adrenalectomy (Stranahan et al. 2008), or treatment with the glucocorticoid receptor antagonist mifepristone (Revsin et al. 2009), prevented the development of reduced hippocampal neurogenesis and stabilized cognitive deficits in STZ-induced diabetic rats independent of its effects on hyperglycemia.
Inflammation is another physiological mediator that may also regulate deficits in neuroplasticity and neurogenesis in diabetes independently of hyperglycemia or corticosterone secretion. Diabetes increases inflammation in humans and animals (Cho et al. 2006; Jain et al. 2007; Lukic et al. 1998; Manrique et al. 2008), which results in antineurogenic effects in the hippocampus through the activation of an orchestra of proinflammatory factors (Voloboueva and Giffard 2011; Zhao et al. 2008). However, the relationship between the HPA axis and inflammation is complex and bidirectional because stress hormones are responsible for changes in immunity and the immune system can affect the endocrine environment by regulating the secretion of stress hormones (Sternberg 2006). Although administration of indomethacin, a non-steroidal anti-inflammatory drug (NSAID), can restore hippocampal neurogenesis decreased by endotoxin challenge (Monje et al. 2003), it is unclear whether increased inflammation is sufficient to mediate decreased neurogenesis evoked by diabetes. The rodent experiments described in this paper determined if decreasing inflammation, using indomethacin, restored diabetes-induced decreases in hippocampal cell proliferation. Furthermore, the effects of indomethacin treatment on circulating levels of CORT, glucose and proinflammatory cytokines in diabetic animals were also examined to determine whether its effects on neurogenesis were mediated by regulating hyperglycemia, the secretion of stress hormones, or cytokine mediators of inflammation.
Methods
Animals
Mice were housed in a temperature- and humidity- controlled facility with a 12-hour light-dark cycle (lights on at 07:00) at the University of Pennsylvania, Translational Research Laboratories (Philadelphia, PA). Eight-week-old, male, C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME), weighing approximately 23–29 g at the beginning of experiments, were used. Mice were housed in groups of 4 in polycarbonate cages with standard laboratory pellet food and water freely available. Experimental procedures were conducted in accordance with the guidelines published in the NIH Guide for Care and Use of Laboratory Animals and protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Induction of diabetes and glucose monitoring
To induce diabetes, mice received a single intraperitoneal (i.p.) dose of 195 mg/kg STZ (Sigma-Aldrich, St. Louis, MO) prepared in 5 M sodium citrate, pH 4.5, or vehicle (Ho et al. 2011). Blood glucose levels were measured 7, 10 and 20 days after STZ or vehicle injection using a portable Freestyle glucometer (Abbott Laboratories, Abbott Park, IL). Blood was obtained via tail snip. Mice with blood glucose values > 300 mg/dl were included in the STZ groups. Glucose levels were then measured on a weekly basis, in the morning between 08:00 – 10:00, until study completion.
Indomethacin treatment
Indomethacin, a prototypic NSAID and non-specific cyclooxygenase (COX) inhibitor (Botting 2006), was given to decrease systemic inflammation. Starting at 7 days post STZ or vehicle injection, mice received 2 mg/kg i.p. of indomethacin in 1% carboxymethylcellulose, vehicle, or saline once daily for two weeks (Monje et al. 2003). A saline group was included to control for the potential effects of the carboxymethylcellulose vehicle on outcome measures. Each vehicle group had 12 mice per group, while each STZ group had 16 mice per group.
Plasma corticosterone (CORT) levels
Blood was collected from the trunk after decapitation between 08:00 – 10:00 a.m., and placed in EDTA tubes (BD, Franklin Lakes, NJ). Specimens were centrifuged at 14,000 x g for 15 min at 4°C. Plasma was separated and stored in −20°C until analysis. CORT levels were quantified using a commercially available enzyme immunoassay kit (Immunodiagnostic Systems Inc., Scottsdale, AZ). Each sample was analyzed in duplicate according to manufacturer’s instructions.
Hippocampal cell proliferation
To measure the effect of chronic diabetes and indomethacin treatment on hippocampal cell proliferation, all treated mice were injected with 5-bromo-2′-deoxyuridine (BrdU) (200 mg/kg, i.p.; Roche Applied Sciences, Indianapolis, IN), dissolved in 0.9% saline, 24 hours prior to sacrifice. Labeling of BrdU was measured in dissociated hippocampal cells displaying the nuclear marker 7-aminoactinomycin D (7-AAD) by flow cytometry as previously described and validated by immunohistochemistry (Balu et al. 2009). In brief, mice were decapitated and both hippocampal lobes were dissected on ice and then underwent mechanical trituration to make a single-cell suspension. After the cells were fixed and permeabilized, they were stained with the isothiocyanate (FITC) BrdU Flow Kit (BD Biosciences, San Jose, CA). The cells were analyzed on a BD FACS Canto system at the University of Pennsylvania Flow Cytometry Core Facility using BD FACSDiva software. Background signal was accounted for using stained tissue from untreated animals that did not receive BrdU injections. We have previously shown that experimental results obtained with this method are essentially similar to those obtained using immunohistochemistry (Balu et al. 2009).
Plasma levels of cytokines
Assessment of cytokine levels in plasma was performed using the Milliplex Mouse Cytokine/Chemokine Magnetic Bead Panel II, from Millipore (Billerica, MA) according to manufacturer’s instructions. Briefly, plasma samples were thawed and run in duplicate on a pre-wet 96 well plate with a pre-selected magnetic bead panel for IL-1β, IL-6, IL-10, IFN-γ, and TNF-α analytes. Following a 14-hour incubation, samples were removed by aspiration. After a triple plate wash, detection antibodies were added and incubated for 60 min. The plate was then stained with Streptamer PE and incubated for a final 30 min. Following a triple plate wash and addition of sheath fluid, the plate was read on a Bio-Plex 200 suspension array system (Bio-Rad, Hercules, CA). Protein concentrations were within expected ranges.
Quantitative real-time PCR analysis
Mice were rapidly decapitated and brains were immediately removed and dissected on ice. The hippocampus was identified and macrodissected. RNA was subsequently isolated using the RNAqueous-4PCR kit for Isolation of DNA-free RNA (Ambion, Applied Biosystems, Austin, TX) following the manufacturers’ instructions. cDNA was synthesized using a Superscript Vilo cDNA synthesis kit (Invitrogen, Carlsbad, CA). All reactions were performed with a master mix of SYBR green (Applied Biosystems, Austin, TX) and 300 nM OligoDt primers (final concentration, Operon, Huntsville, AL). Reactions were run using the Stratagene MX3000 and MXPro QPCR software, with cycling parameters of 95°C (10 min) followed by 40 cycles of 95°C (30 sec), and 60°C (1 min). Reactions were performed in triplicate and the median cycle threshold was used for analysis and normalized to the housekeeping gene TATA binding protein (TBP). IL-6 Forward: 5′-AGTCCGGAGAGGAGACTTCAC-3′, Reverse: 5′-TCCAGTTTGGTAGCATCCATC-3′. IL-1beta Forward: 5′-GCAACTGTTCCTGAACTCAACT -3′ Il-1beta Reverse: 5′-ATCTTTTGGGGTCCGTCAACT -3′. TNFalpha Forward: 5′-GGTTCCTTTGTGGCACTTG-3′, TNFalpha Reverse: 5′-TTCTCTTGGTGACCGGGAG-3′. TNF alpha Receptor 1 Forward: 5′-CCGGGCCACCTGGTCCG-3′, Reverse: 5′-CAAGTAGGTTCCTTTGTG-3′. TNF alpha Receptor 2 Forward: 5′-CTCGCGCTGGTCTTCGAACTG-3′, Reverse: 5′-GGTATACATGCTTGCCTCACAGTC-3′
Data analysis
Data were analyzed using Prism GraphPad, Version 5.0b (La Jolla, CA). In all outcome measures, saline and vehicle treated groups did not differ from each other (p > 0.05) in the overall analysis of variance (ANOVA), which was run with two conditions (vehicle versus STZ) and three treatments (saline, vehicle, indomethacin). Therefore, the saline and vehicle groups were combined for presentation (labeled as vehicle) and analyzed by 2-way ANOVA, resulting in the following sample sizes: n = 24 for vehicle- vehicle; n = 12 for vehicle-indomethacin; n = 32 for STZ- vehicle; n = 16 for STZ-indomethacin. The Bonferroni test was used for post hoc comparisons. For the analysis of plasma cytokines, only a subset of 22 samples were analyzed from the STZ-vehicle group. For all tests, p < 0.05 was considered statistically significant and 0.05 < p < 0.10 was reported as a trend. Main effects for cell proliferation were analyzed as allowed for a trend between STZ and indomethacin.
Results
Glucose levels, CORT levels and body weight
All mice treated with STZ developed diabetes, defined as glucose > 300 mg/dl (Fig. 1A). STZ mice maintained an approximate 3.25-fold elevation in non-fasting glucose levels compared to controls. An overall ANOVA indicated significant effects of STZ on glucose levels (F(1, 80) = 1391, p < 0.001). There was no significant main effect or interaction between STZ and treatment with indomethacin on glucose levels (p > 0.05).
Fig 1.

Effect of diabetes and indomethacin treatments on plasma levels of glucose and CORT and changes in body weight. (A) Glucose levels were measured at 20 days post STZ or vehicle injection. Bars represent mean ± SEM. Asterisks denote significant difference compared to each of the corresponding vehicle control groups (***, p < 0.001). (B) Plasma CORT levels were measured at study completion, 20 days post STZ or vehicle injection. Bars represent mean values ± S.E.M. Asterisks denote significant difference compared to each of the corresponding vehicle control groups (*** p < 0.001). (C) Δ Weight is the change in body weight (g) from the beginning of the experiment and the end of the experiment. Bars represent mean ± SEM. Asterisks denote significant difference compared to each of the corresponding vehicle control groups (***, p < 0.001). Carrot (^) denotes significant difference compared to the treated groups (^, p < 0.05).
STZ-induced diabetes resulted in an approximate 2.5-fold elevation in CORT levels (Fig. 1B). The overall ANOVA indicated a significant effect of STZ on CORT levels (F(1, 77) = 22.24, p < 0.001), but there was no significant main effect of indomethacin or interaction between STZ and indomethacin (p > 0.05).
STZ mice showed a significant decrease in body weight compared to controls (F(1, 80) = 385, p < 0.001, Fig. 1C). Further, indomethacin treatment significantly increased body weight (F(2, 80) = 4.08, p < 0.05), but there was no significant interaction between STZ and indomethacin (p > 0.05).
Hippocampal cell proliferation
Administration of indomethacin for 14 days reversed the reduction of hippocampal cell proliferation produced by diabetes (Fig. 2). STZ treatment reduced hippocampal cell proliferation by 50% but only by 15% in mice treated with indomethacin. The overall ANOVA for hippocampal cell proliferation indicated a significant effect of the STZ condition (F(1, 80) = 13.15, p < 0.001) and a trend for an interaction between STZ condition and treatment (F(1, 80) = 3.27, p = 0.07). Post hoc tests showed that STZ-induced diabetes significantly decreased hippocampal cell proliferation levels compared to the vehicle control group (p < 0.001). On the other hand, the STZ group treated with indomethacin did not differ significantly from either control group treated with the SC vehicle (either the vehicle-indomethacin or vehicle-vehicle groups, p > 0.05).
Fig 2.
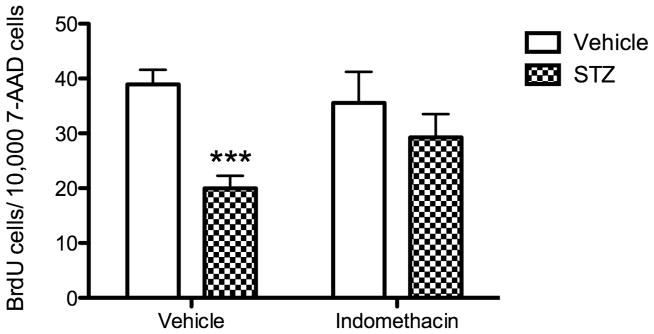
Effect of diabetes and indomethacin treatments on hippocampal cell proliferation. Measurement of hippocampal cell proliferation occurred 20 days post STZ or SC vehicle injection. To measure hippocampal cell proliferation, all mice were injected with BrdU, 200 mg/kg, i.p., 24 hours prior to sacrifice. Total n = 84; n = 24 for vehicle-vehicle; n = 12 for vehicle-indomethacin; n = 32 for STZ-vehicle; n = 16 for STZ-indomethacin. Data are expressed as number of positively labeled BrdU cells for every 10,000 7-AAD cells. Bars represent mean values ± S.E.M. Asterisks denotes significant difference compared to the corresponding vehicle control group (***, p < 0.001).
Plasma levels of cytokines
The effects of STZ-induced diabetes and treatment with indomethacin were examined on plasma levels of a panel of immunomodulatory cytokines. As shown in Fig. 3A and 3B, neither STZ or indomethacin significantly altered IL-1b and IL-6, respectively. STZ-induced diabetes significantly reduced levels of the anti-inflammatory cytokines IL-10 and IFN-γ (p < 0.01) and increased levels of the pro-inflammatory cytokine TNF-α (p < 0.001) (Figs. 3C–3E). Chronic treatment with indomethacin independently reduced levels of IL-10 and IFN-γ compared with controls, and chronic treatment with indomethacin did not further reduce levels of these cytokines in STZ-treated animals. In contrast, indomethacin administration reduced the levels of TNF-α that had been elevated by diabetes without producing any independent effect on TNF-α levels (Fig. 3E).
Fig 3.

Effects of STZ-induced diabetes and indomethacin treatments on plasma levels of cytokines. (A) IL-1 β; (B) IL-6; (C) IL-10; (D) IFN-γ; and (E) TNF-α. Bars represent mean values ± S.E.M. Asterisks indicate significant difference compared to each of the corresponding vehicle control groups (**p < 0.01 and *** p < 0.001).
TNF-α receptor gene expression in the hippocampus
In light of the changes observed in plasma TNF-α levels, we examined the effects of STZ-induced diabetes and indomethacin treatment on mRNA expression of TNF-α receptor types R1 and R2 in the hippocampus. STZ-induced diabetes alone and additional treatment with indomethacin had no significant effect on mRNA levels of TNF-αR1 (Fig. 4A). In contrast, STZ-induced diabetes alone significantly decreased expression of mRNA levels of TNF-αR2 (Fig. 4B) in the hippocampus (F(1,70) = 5.57, p < 0.05). Although treatment with indomethacin appeared to increase TNF-αR2 mRNA levels (Fig. 4B), there was no significant main effect for indomethacin treatment or interaction between STZ and indomethacin (p > 0.05).
Fig 4.

Effects of STZ-induced diabetes and indomethacin treatments on mRNA expression of TNF-α receptors in the hippocampus. mRNA levels were measured via qPCR at study completion, 20 days post STZ or SC vehicle injection. Fold change was quantified as compared to TATA-box binding protein (TBP) levels. Vertical bars represent mean values ± S.E.M. (A) TNF-αR1 gene expression. (B) TNF-αR2 gene expression. STZ treatment significantly decreased TNF-αR2 gene expression (see text). Asterisks indicate significant difference compared to the corresponding vehicle control groups (*p < 0.05).
Discussion
Consistent with previous studies of rodents in a number of models of diabetes (Ho et al. 2013), the current experiment demonstrated that STZ-induced diabetes results in decreased hippocampal cell proliferation (Balu et al. 2009; Beauquis et al. 2006; Revsin et al. 2009; Saravia et al. 2004; Stranahan et al. 2008). The current study is the first to demonstrate the ability of an anti-inflammatory drug to restore deficits in hippocampal cell proliferation in diabetic mice. In non-diabetic animals, inflammation has been shown to exert detrimental effects on hippocampal neurogenesis and other forms of neuroplasticity (Song and Wang 2011), while treatment with an anti-inflammatory drug reversed the inhibitory effects of radiation on hippocampal cell proliferation (Monje et al. 2003).
The management of euglycemia is critically important in diabetic patients as a first defense against consequent medical problems. By directly reversing the hyperglycemia and hypercortisolemia associated with STZ-induced diabetes, treatment with insulin prevented the reduction of hippocampal cell proliferation and behavior (Ho et al. 2011). However, insulin is not always successful and controlling diabetes. Unlike insulin, indomethacin restored deficits in hippocampal cell proliferation in diabetic mice without lowering glucose levels suggesting a different mechanism of action. Other studies have shown that antidepressants, estrogen or exercise may also reverse diabetic brain complications in the absence of restoring euglycemia (Beauquis et al. 2006; Revsin et al. 2009; Saravia et al. 2004; Stranahan et al. 2008). Dysfunction of the hippocampus, particularly hippocampal neurogenesis, is associated with cognitive deficits, depression or augmented secretion of stress hormones (Becker and Wojtowicz 2007; Samuels and Hen 2011; Snyder et al. 2011). Because multiple mechanisms other than hyperglycemia are effective at regulating brain neuroplasticity and behavior in diabetes, integrative treatments targeting both neuroplasticity and glycemic control could be developed to prevent the risk of neurobehavioral complications from diabetes complications in a coordinated medical treatment plan.
Indomethacin treatment reversed deficits in hippocampal cell proliferation from diabetes in the present study without affecting hypercortisolemia. This is particularly important since previous studies have established CORT as a principal mediator of hippocampal neurogenesis in diabetic rodents (Stranahan et al. 2008). The glucocorticoid response to stress is thought to reduce hippocampal cell proliferation by downstream effects from the activation of NMDA receptors (Cameron et al. 1998). As a non-specific COX inhibitor, indomethacin decreases the synthesis of prostaglandins (Botting 2006) thus producing its effects by diminishing inflammation. Even though the immune system can potentially affect the endocrine system (Sternberg 2006), the present data indicate that hippocampal cell proliferation was likely not regulated by indomethacin in diabetes through endocrine mechanisms. Pro-inflammatory cytokines have been shown to alter neurotrophins, like BDNF, and its associated signaling pathways (Koo et al. 2010; Song and Wang 2011), and these mechanisms may be responsible for indomethacin’s effects on neurogenesis. Future studies may determine whether COX-1 or COX-2 enzymes are involved in mediating these changes and further explore the inflammatory cascade as a potential novel target for drug therapy in diabetes. In addition, these findings obtained with a model of type 1 diabetes in the mouse might also be applicable for managing the inflammatory consequences of type 2 diabetes.
There is ongoing discussion on the contribution of increased inflammation to the pathogenesis of type 1 diabetes, ranging from an active promotor of pancreatic β-cell death to a contributor to cardiovascular and neuropathic complications (Baumann et al. 2012; Korczak et al. 2011; Padgett et al. 2013). To investigate whether indomethacin regulated hippocampal cell proliferation in a model of type 1 diabetes by modulating inflammatory mechanisms, we examined the plasma levels of five common cytokines, IL-1β, IL-6, IL-10, IFN-γ, and TNF-α. STZ treatment alone increased inflammation in the diabetic animals as reflected by significantly increased plasma levels of the proinflammatory cytokine TNF-α and decreased levels of the anti-inflammatory cytokine IL-10 and IFN-γ, a cytokine with pleitrophic effects (Dinarello 2000). Increased plasma levels of TNF-α have been reported by other groups in experimental diabetes in mice and rats (Lukic et al. 1998; Jain et al. 2007) and in human type 1 diabetics (Padgett et al. 2013). Plasma IL-10 levels has been reported to fall in STZ rats (Sinzato et al. 2011) but an increase in plasma IL-6 (Jain et al. 2007) or IFN-γ levels (Lukic et al. 1998) reported previously in STZ-treated rats were not measured in the present study. Although plasma cytokine values may be important because they translate to human studies, there are relatively few studies and results may vary according to the animal model used.
Among the cytokines altered by STZ in the mouse, indomethacin moderated the levels of TNF-α alone without changing TNF-α levels in nondiabetic controls. This dual response pattern suggests that TNF-α could be a common mediator of both inflammation and hippocampal neurogenesis in diabetes, supporting this cytokine as a target for therapeutic intervention in type 1 diabetes (Baumann et al. 2012). TNF-α has been shown to affect insulin signaling by inhibiting kinase activity and directly involved in insulin resistance placing it as a direct mediator of diabetes (Baud and Karin 2001; Rui et al. 2001). Indomethacin also diminished the difference in IL-10 and IFN-γ levels between diabetic and control mice, but because indomethacin also reduced plasma levels of cytokines in controls, it is difficult to ascribe these particular changes to a disease-related process.
Although the results observed in plasma TNF-α levels are consistent with a proposed role for glial TNF-α in synaptic connectivity (Stellwagen and Malenka 2006), it is unclear from studying plasma cytokines whether central or peripheral mechanisms may have been involved. TNF–αR1 and R2 receptors are both expressed on hippocampal neural progenitors and have been proposed to mediate distinct effects on neuroplasticity, with TNF-αR1 contributing to neuronal death and TNF-αR2 being neuroprotective (Iosif et al. 2006). STZ decreased gene expression for TNF-αR2 receptors in the hippocampus, suggesting a potential role for TNF-αR2 as a compensatory mechanism in reducing cell proliferation. However, the lack of significant effect of indomethacin on TNF-αR2 gene expression suggests that its accommodating effects on cell proliferation were not mediated through this mechanism. It is not yet known whether decreased hippocampal expression of TNF-αR2 could be involved in other aspects of neuroplasticity deficits after STZ treatment. In future studies, microglia activation may be used to associate a direct marker of brain neuroinflammation with changes in hippocampal neurogenesis in diabetes (Rana et al. 2014).
In the current study, diabetes decreased body weight in mice. However, indomethacin treatment increased body weight in diabetic and non-diabetic mice, which may be attributable to increased fluid retention (Risser et al. 2009). Also, long-term use of NSAIDs results in gastric ulcers (Risser et al. 2009; Suleyman et al. 2010), while consumption of food relieves gastric ulcer pain. Thus, mice treated with indomethacin may have consumed more food to relieve this side effect, although food intake was not measured in this study. The potential negative effects of NSAIDs on bleeding risk and stomach irritation urge cautionary use of these drugs for primary prevention of complications with diabetes (Risser et al. 2009; Suleyman et al. 2010) and COX-2 inhibitors could potentially aggravate heart disease and hypertension (Barkin et al. 2010; Risser et al. 2009). However, aspirin was found not have similar bleeding risk in diabetes (Osorio 2012) and aspirin and statins, drugs with anti-inflammatory effects, have been given to diabetic patients to improve their quality of life and management of diabetes (Tricco et al. 2012). Future studies can explore the effects of other anti-inflammatory agents in animal studies of diabetes complications that could be used chronically in humans.
Neurogenesis is composed of multiple components, such as cell proliferation, maturation, and survival and confirmed to occur in two neurogenic regions in the brain, the hippocampus and the subventricular zone. The reduction of hippocampal cell proliferation by diabetes and inflammation may be a marker related to the deterioration of neurological processes that results in the loss of CNS neuroplasticity and behavioral disturbances (Ho et al. 2013; Stranahan et al. 2008). Greater than 80% of newborn cells in the hippocampus develop into neurons and neuronal cell fate is either unchanged or unchanged in experimental diabetes (Ho et al. 2013). Nevertheless, the cell fate of the newborn cells after indomethacin treatment in diabetes has not been identified. Hippocampal neurogenesis has been associated with maturation and aging, cognitive function, stress and the regulation of the HPA axis, and anxiety and depression (Balu and Lucki 2009; Samuels and Hen 2011; Zhao et al. 2008). Therefore, it is reasonable to suggest that the restoration of hippocampal cell proliferation by treatments that reduce inflammation, without the side effects of indomethacin, may prevent some of the long-term neurobehavioral consequences produced by long-term diabetes.
Acknowledgments
This research was supported by USPHS grants R01-MH86599, F31-NR010853 and T32-MH14654. Additional support was provided by the Eastern Nursing Research Society and the American Nurses Foundation Small Grants Program, grant #2009-033.
Footnotes
Conflict of interest
The authors have no conflict of interest related to this paper.
References
- Balu DT, Hodes GE, Hill TE, Ho N, Rahman Z, Bender CN, Ring RH, Dwyer JM, Rosenzweig-Lipson S, Hughes ZA, Schechter LE, Lucki I. Flow cytometric analysis of BrdU incorporation as a high-throughput method for measuring adult neurogenesis in the mouse. J Pharmacol Toxicol Methods. 2009;59:100–7. doi: 10.1016/j.vascn.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Lucki I. Adult hippocampal neurogenesis: regulation, functional implications, and contribution to disease pathology. Neuroscience and biobehavioral reviews. 2009;33:232–52. doi: 10.1016/j.neubiorev.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkin RL, Beckerman M, Blum SL, Clark FM, Koh EK, Wu DS. Should nonsteroidal anti-inflammatory drugs (NSAIDs) be prescribed to the older adult? Drugs Aging. 2010;27:775–89. doi: 10.2165/11539430-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends in cell biology. 2001;11:372–7. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- Baumann B, Salem HH, Boehm BO. Anti-inflammatory therapy in type 1 diabetes. Current diabetes reports. 2012;12:499–509. doi: 10.1007/s11892-012-0299-y. [DOI] [PubMed] [Google Scholar]
- Beauquis J, Roig P, Homo-Delarche F, De Nicola A, Saravia F. Reduced hippocampal neurogenesis and number of hilar neurones in streptozotocin-induced diabetic mice: reversion by antidepressant treatment. The European journal of neuroscience. 2006;23:1539–46. doi: 10.1111/j.1460-9568.2006.04691.x. [DOI] [PubMed] [Google Scholar]
- Becker S, Wojtowicz JM. A model of hippocampal neurogenesis in memory and mood disorders. Trends in cognitive sciences. 2007;11:70–6. doi: 10.1016/j.tics.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Kerssen A, de Haan EH, Kappelle LJ. Cognitive dysfunction and diabetes: implications for primary care. Prim Care Diabetes. 2007;1:187–93. doi: 10.1016/j.pcd.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Botting RM. Inhibitors of cyclooxygenases: mechanisms, selectivity and uses. J Physiol Pharmacol. 2006;57(Suppl 5):113–24. [PubMed] [Google Scholar]
- Brands AM, Biessels GJ, de Haan EH, Kappelle LJ, Kessels RP. The effects of type 1 diabetes on cognitive performance: a meta-analysis. Diabetes Care. 2005;28:726–35. doi: 10.2337/diacare.28.3.726. [DOI] [PubMed] [Google Scholar]
- Cameron HA, Tanapat P, Gould E. Adrenal steroids and N-methyl-D-aspartate receptor activation regulate neurogenesis in the dentate gyrus of adult rats through a common pathway. Neuroscience. 1998;82:349–54. doi: 10.1016/s0306-4522(97)00303-5. [DOI] [PubMed] [Google Scholar]
- Cho WC, Yip TT, Chung WS, Leung AW, Cheng CH, Yue KK. Potential biomarkers found by protein profiling may provide insight for the macrovascular pathogenesis of diabetes mellitus. Dis Markers. 2006;22:153–66. doi: 10.1155/2006/450762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–8. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- Egede LE. Disease-focused or integrated treatment: diabetes and depression. Med Clin North Am. 2006;90:627–46. doi: 10.1016/j.mcna.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Ho N, Balu DT, Hilario MR, Blendy JA, Lucki I. Depressive phenotypes evoked by experimental diabetes are reversed by insulin. Physiology & behavior. 2011;105:702–708. doi: 10.1016/j.physbeh.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho N, Sommers MS, Lucki I. Effects of diabetes on hippocampal neurogenesis: Links to cognition and depression. Neuroscience and biobehavioral reviews. 2013;37:1346–62. doi: 10.1016/j.neubiorev.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt RI, de Groot M, Lucki I, Hunter C, Sartorius N, Golden S. NIDDK International Conference Report on Diabetes and Depression: Current Understanding and Future Directions. Diabet Care. 2014;37:2067–2077. doi: 10.2337/dc13-2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z, Jacobsen SE, Lindvall O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 2006;26:9703–12. doi: 10.1523/JNEUROSCI.2723-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Guilford J, Leander JD, Nisenbaum LK. The effect of streptozotocin-induced diabetes on cell proliferation in the rat dentate gyrus. Neuroscience letters. 2000;293:91–4. doi: 10.1016/s0304-3940(00)01502-0. [DOI] [PubMed] [Google Scholar]
- Jain SK, Rains JL, Croad JL. Effect of chromium niacinate and chromium picolinate supplementation on lipid peroxidation, TNF-alpha, IL-6, CRP, glycated hemoglobin, triglycerides, and cholesterol levels in blood of streptozotocin-treated diabetic rats. Free Radic Biol Med. 2007;43:1124–31. doi: 10.1016/j.freeradbiomed.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A. 2010;107:2669–74. doi: 10.1073/pnas.0910658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korczak DJ, Pereira S, Koulajian K, Matejcek A, Giacca A. Type 1 diabetes mellitus and major depressive disorder: evidence for a biological link. Diabetologia. 2011;54:2483–93. doi: 10.1007/s00125-011-2240-3. [DOI] [PubMed] [Google Scholar]
- Lu FP, Lin KP, Kuo HK. Diabetes and the risk of multi-system aging phenotypes: a systematic review and meta-analysis. PLoS One. 2009;4:e4144. doi: 10.1371/journal.pone.0004144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukic ML, Stosic-Grujicic S, Shahin A. Effector mechanisms in low-dose streptozotocin-induced diabetes. Developmental immunology. 1998;6:119–28. doi: 10.1155/1998/92198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrique C, Lastra G, Palmer J, Gardner M, Sowers JR. Aspirin and Diabetes Mellitus: revisiting an old player. Ther Adv Cardiovasc Dis. 2008;2:37–42. doi: 10.1177/1753944707088185. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–5. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Osorio J. Diabetes: Aspirin and diabetes mellitus-no increase in bleeding risk? Nature reviews Endocrinology. 2012 doi: 10.1038/nrendo.2012.108. [DOI] [PubMed] [Google Scholar]
- Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Annals of the New York Academy of Sciences. 2013;1281:16–35. doi: 10.1111/j.1749-6632.2012.06826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana I, Badoer E, Alahmadi E, Leo CH, Woodman OL, Stebbing MJ. Microglia are selectively activated in endocrine and cardiovascular control centres in streptozotocin-induced diabetic rats. Journal of neuroendocrinology. 2014;26:413–25. doi: 10.1111/jne.12161. [DOI] [PubMed] [Google Scholar]
- Revsin Y, Rekers NV, Louwe MC, Saravia FE, De Nicola AF, de Kloet ER, Oitzl MS. Glucocorticoid receptor blockade normalizes hippocampal alterations and cognitive impairment in streptozotocin-induced type 1 diabetes mice. Neuropsychopharmacology. 2009;34:747–58. doi: 10.1038/npp.2008.136. [DOI] [PubMed] [Google Scholar]
- Risser A, Donovan D, Heintzman J, Page T. NSAID prescribing precautions. Am Fam Physician. 2009;80:1371–8. [PubMed] [Google Scholar]
- Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, Dunaif A, White MF. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. The Journal of clinical investigation. 2001;107:181–9. doi: 10.1172/JCI10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels BA, Hen R. Neurogenesis and affective disorders. Eur J Neurosci. 2011;33:1152–9. doi: 10.1111/j.1460-9568.2011.07614.x. [DOI] [PubMed] [Google Scholar]
- Saravia F, Revsin Y, Lux-Lantos V, Beauquis J, Homo-Delarche F, De Nicola AF. Oestradiol restores cell proliferation in dentate gyrus and subventricular zone of streptozotocin-diabetic mice. J Neuroendocrinol. 2004;16:704–10. doi: 10.1111/j.1365-2826.2004.01223.x. [DOI] [PubMed] [Google Scholar]
- Sinzato YK, Damasceno DC, Laufer-Amorim R, Rodrigues MM, Oshiiwa M, Taylor KN, Rudge MV. Plasma concentrations and placental immunostaining of interleukin-10 and tumor necrosis factor-alpha as predictors of alterations in the embryo-fetal organism and the placental development of diabetic rats. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas/Sociedade Brasileira de Biofisica [et al] 2011;44:206–11. doi: 10.1590/s0100-879x2011007500015. [DOI] [PubMed] [Google Scholar]
- Snyder JS, Cameron HA. Could adult hippocampal neurogenesis be relevant for human behavior? Behavioural Brain Research. 2011 doi: 10.1016/j.bbr.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature. 2011;476:458–61. doi: 10.1038/nature10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Wang H. Cytokines mediated inflammation and decreased neurogenesis in animal models of depression. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:760–8. doi: 10.1016/j.pnpbp.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–9. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6:318–28. doi: 10.1038/nri1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci. 2008;11:309–17. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suleyman H, Albayrak A, Bilici M, Cadirci E, Halici Z. Different mechanisms in formation and prevention of indomethacin-induced gastric ulcers. Inflammation. 2010;33:224–34. doi: 10.1007/s10753-009-9176-5. [DOI] [PubMed] [Google Scholar]
- Tricco AC, Ivers NM, Grimshaw JM, Moher D, Turner L, Galipeau J, Halperin I, Vachon B, Ramsay T, Manns B, Tonelli M, Shojania K. Effectiveness of quality improvement strategies on the management of diabetes: a systematic review and meta-analysis. Lancet. 2012;379:2252–61. doi: 10.1016/S0140-6736(12)60480-2. [DOI] [PubMed] [Google Scholar]
- Voloboueva LA, Giffard RG. Inflammation, mitochondria, and the inhibition of adult neurogenesis. J Neurosci Res. 2011;89:1989–96. doi: 10.1002/jnr.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi SS, Hwang IK, Yoo KY, Park OK, Yu J, Yan B, Kim IY, Kim YN, Pai T, Song W, Lee IS, Won MH, Seong JK, Yoon YS. Effects of treadmill exercise on cell proliferation and differentiation in the subgranular zone of the dentate gyrus in a rat model of type II diabetes. Neurochem Res. 2009;34:1039–46. doi: 10.1007/s11064-008-9870-y. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–60. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]