

Abstract
Background
A number of completely sequenced eukaryotic genome data are available in the public domain. Eukaryotic genes are either 'intron containing' or 'intronless'. Eukaryotic 'intronless' genes are interesting datasets for comparative genomics and evolutionary studies. The SEGE database containing a collection of eukaryotic single exon genes is available. However, SEGE is derived using GenBank. The redundant, incomplete and heterogeneous qualities of GenBank data are a bottleneck for biological investigation in comparative genomics and evolutionary studies. Such studies often require representative gene sets from each genome and this is possible only by deriving specific datasets from completely sequenced genome data. Thus Genome SEGE, a database for 'intronless' genes in completely sequenced eukaryotic genomes, has been constructed.
Availability: http://sege.ntu.edu.sg/wester/intronless
Description
Eukaryotic 'intronless' genes are extracted from nine completely sequenced genomes (four of which are unicellular and five of which are multi-cellular). The complete dataset is available for download. Data subsets are also available for 'intronless' pseudo-genes. The database provides information on the distribution of 'intronless' genes in different genomes together with their length distributions in each genome. Additionally, the search tool provides pre-computed PROSITE motifs for each sequence in the database with appropriate hyperlinks to InterPro. A search facility is also available through the web server.
Conclusions
The unique features that distinguish Genome SEGE from SEGE is the service providing representative 'intronless' datasets for completely sequenced genomes. 'Intronless' gene sets available in this database will be of use for subsequent bio-computational analysis in comparative genomics and evolutionary studies. Such analysis may help to revisit the original genome data for re-examination and re-annotation.
Background
Eukaryotic genes are often interrupted by intragenic, non-coding sequences called introns [1]. However, prokaryotic genes lack introns. Therefore, 'Intronless' genes are characteristic features of prokaryotes. Interestingly, many eukaryotic histone [2,3] and GPCR [4] genes are predominantly 'intronless'. A number of vertebrate 'intronless' genes have been complied [5]. The human genome report identified 901 Otto predicted single exon genes (The Celera approach to gene prediction is called Otto) [6]. The presence of a sizeable amount of single exon genes (SEG) in eukaryotic genomes is intriguing. The SEGE database contains eukaryotic SEG derived from GenBank [7]. For most genomes, SEGE does not provide representative 'intronless' gene sets because GenBank often contains redundant sequences from the same species deposited by different authors. It should also be noted that all sequences obtained from genome projects are not available in GenBank. Representative sets of SEG from specific genomes will provide meaningful biological insights to subsequent bio-computational analysis for comparative and evolutionary studies. In order to facilitate such research we developed Genome SEGE, a database containing all putative SEG from completely sequenced eukaryotic genomes. Here, we describe the usefulness and construction of Genome SEGE.
Construction and content
Data source and methodology
The annotated eukaryotic genome sequence data was downloaded from NCBI [8]. 'Intronless' genes were identified using the 'CDS' annotation in the FEATURE as described elsewhere [7]. It should be noted that organellar sequences (annotated as 'chloroplast', 'plastid', 'mitochondrial', 'mitochondrion') were removed from further analysis. A flowchart describing the construction of the database is shown (Figure 1).
Figure 1.
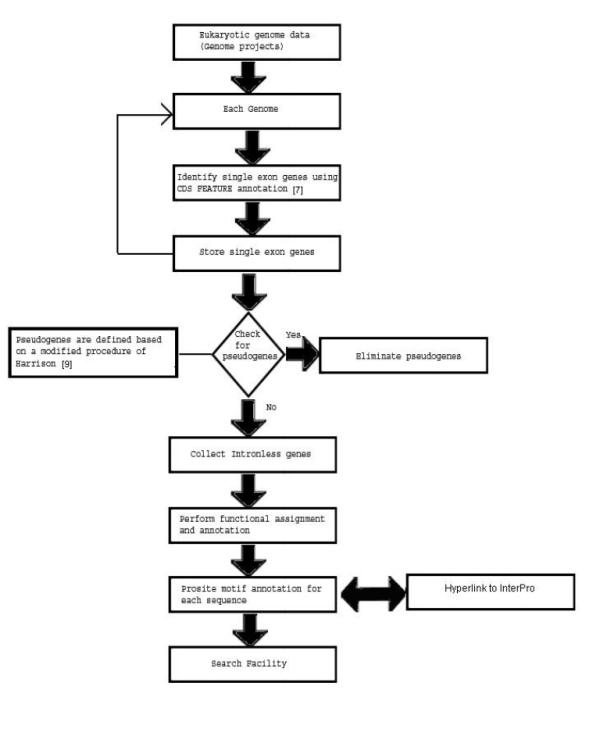
Database construction A flowchart describing the development of the database is shown. CDS = coding sequence.
'Intronless' pseudogenes
Data processing and cleaning is an essential part of biological knowledge discovery. Hence, we eliminated all identifiable processed pseudogenes by scanning for polyadenylation signal (AATAAA) and polyadenylation tail using a modified procedure of Harrison and colleagues [9]. In this procedure, by definition, we consider a sequence to represent a pseudogene if it contains a polyadenylation tail (>15A) within 1000 nucleotides from the stop codon with a preceding polyadenylation signal.
Prosite motifs and InterPro
We characterized 'intronless' gene products using PROSITE, which is a method of identifying the functions of uncharacterized proteins translated from genomic sequences [10]. We chose PROSITE because it is complete, highly specific, fully documented and regularly updated. The search tool provides pre-computed PROSITE motifs for each sequence in the database with appropriate hyperlinks to InterPro [11].
Content
A Database is created to store all eukaryotic 'intronless' sequences derived from completely sequenced genomes. The database contains three sets of data for each genome: (1) 'intronless' sequence set, (2) 'intronless' pseudo-genes sequence set, and (3) 'intronless' sequence set without pseudo-genes.
Caveats
Genome annotation is an inherently dynamic process in which it is necessary to use many different sources of data, which are not updated in a rigorous fashion. It should also be noted that annotation is not generally uniform and consistent because various procedures are used by different groups for genome annotation. During genome annotation, a gene may have been annotated with a single exon CDS in the FEATURE for three main reasons: (1) the gene is truly 'intronless' and functional, (2) the gene is of retroposition origin [12,13], (3) false positive prediction by gene finding algorithms. False positives are not removed from the current dataset due to lack of a methodology. Nevertheless, the gene finding algorithms are reasonably optimized to find SEG.
Update
The database will be refreshed on a quarterly basis or as and when an update is noticed to genome files in the public domain.
Utility and discussion
Genome SEGE is an extension of SEGE [6] and these two databases complement each other in their biological utility and application. SEGE and Genome SEGE differ primarily in their content, as the datasets are created from different sources. The degree and quality of annotation also varies between them. SEGE could be used for general purpose studies involving 'intronless' genes from different genomes, while Genome SEGE is of particular interest for researchers interested in comparative genomics.
A wealth of information can be obtained by comparing 'intronless' gene sequences between two or more genomes to identify features conserved or diverged during evolution. Comparison of more closely related genomes can reveal similarities in gene order. Such analysis could also shed light on genome architecture and help understand why the genome is arranged the way it is and how its structure affects function. A systematic mapping between functional genes and their 'intronless' paralogs can provide a matrix for genomic rearrangement and gene duplication. Different 'intronless' gene sets available in the database will provide an opportunity to perform many-to-many comparison between genomes. Such analysis will provide information on paralogy and orthology at a molecular level. Analysis of the datasets using non-linear probabilistic models may provide acceptable evidence for retro-position events during evolution.
The search tool in the database provides options to scan through each dataset using gene name or protein name. The result page produces information on chromosomal location, organism name, gene name, product name, GenBank Index, nucleotide sequence and protein sequence. The result page also shows all PROSITE motifs in the sequence with specific hyperlinks to PROSITE documentation and InterPro (Figure 2).
Figure 2.
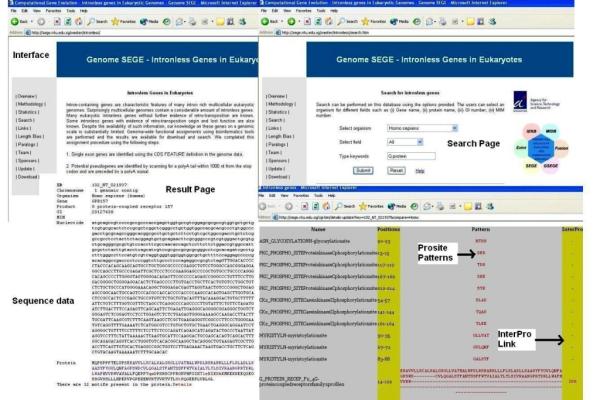
Illustration of an example search. This example illustrates a search for human 'G protein' in the database. The interface, search page and results (annotation, sequence, Prosite, InterPro links) are shown.
Conclusions
The biological role of 'intronless' genes in the genomes of higher organism is perplexing. 'Intronless' gene sets available in the database will be of use for subsequent bio-computational analysis in comparative genomics and evolutionary studies. Such analysis may help to revisit the original genome data for re-examination and re-annotation. Different eukaryotic genomes have varying proportions of 'intronless' genes and a sizeable fraction of them are found in many intron-rich multi-cellular genomes. We believe that these estimates will improve our understanding on the differential selection (as a process or force) of 'intronless' genes in different eukaryotic genomes. The different datasets made available in the database can serve as a data source for evolutionary and functional studies. They will also help to answer questions such as, (1) How many of 'intronless' genes are expressed in each genome? (2) How many of them are of prokaryotic origin? (3) How many of them have multi-exon correspondence within genome? (4) Do they evolve by retro-position? It is our hope that the database we make available will encourage molecular biologists and computational molecular evolutionist to address this problem. The unique features that distinguish Genome SEGE from SEGE is the service providing representative 'intronless' datasets for completely sequenced genomes. Such service will persuade researchers to use representative data sets for investigating a number of biologically significant evolutionary phenomena. We also hope to provide this service for other completely sequenced genomes as and when they are available in the public domain after appropriate examination and analysis. It is also our interest to compare the contents of SEGE and Genome SEGE on a genome by genome basis for the examination of data bias in SEGE.
Availability and requirements
Database is available freely at http://sege.ntu.edu.sg/wester/intronless.
List of abbreviations
GPCR G-protein coupled receptors
SEGE Single exon genes in eukaryotes
CDS Coding sequence
Acknowledgments
Acknowledgements
We thank Vincent Chow Tak Kwong, Iti Chaturvedi, Subbiah Subbramanian and Dmitri A. Petrov for their contribution and discussion. This research is supported by A*STAR-BMRC research grant #01/1/21/19/191. We thank the anonymous referees for their suggestions, which helped us make the manuscript contextual.
Contributor Information
Meena Kishore Sakharkar, Email: mmeena@ntu.edu.sg.
Pandjassarame Kangueane, Email: mpandjassarame@ntu.edu.sg.
References
- Gilbert W. Why genes in pieces? Nature. 1978;271:501. doi: 10.1038/271501a0. [DOI] [PubMed] [Google Scholar]
- Old RW, Woodland HR. Histone genes: not so simple after all. Cell. 1984;38:624–626. doi: 10.1016/0092-8674(84)90256-3. [DOI] [PubMed] [Google Scholar]
- Mollapour M, Piper P. Targeted gene deletion in zygo-saccharomyces bailii. Yeast. 2001;18:173–186. doi: 10.1002/1097-0061(20010130)18:2<173::AID-YEA663>3.3.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Gentles AJ, Karlin S. Why are human G-protein-coupled receptors predominantly 'intronless'? Trends Genet. 1999;15:47–49. doi: 10.1016/S0168-9525(98)01648-5. [DOI] [PubMed] [Google Scholar]
- Brosius J. Genomes were forged by massive bombardments with retro-elements and retrosequences. Genetica. 1999;107:209–238. doi: 10.1023/A:1004018519722. [DOI] [PubMed] [Google Scholar]
- Venter CJ, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Sakharkar MK, Kangueane P, Petrov DA, Kolaskar AS, Subbiah S. SEGE: A database on 'intronless/single exonic' genes from eukaryotes. Bioinformatics. 2002;18:1266–1267. doi: 10.1093/bioinformatics/18.9.1266. [DOI] [PubMed] [Google Scholar]
- ftp://ftp.ncbi.nih.gov/genomes
- Harrison MP, Hegyi H, Balasubramanian S, Luscombe NM, Bertone P, Echols N, Johnson T, Gerstein M. Molecular Fossils in the Human Genome: Identification and Analysis of the pseudogenes in Chromosomes 21 and 22. Genome Res. 2002;12:272–280. doi: 10.1101/gr.207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falquet L, Pagni M, Bucher P, Hulo N, Sigrist CJA, Hoffman K, Bairoch A. The PROSITE database, its status in 2002. Nucl Acids Res. 2002;30:235–238. doi: 10.1093/nar/30.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Barrell D, Bateman A, Binns D, Biswas M, Bradley P, Bork P, Bucher P, Copley RR, Courcelle E, Das U, Durbin R, Falquet L, Fleischmann W, Griffiths-Jones S, Haft D, Harte N, Hulo N, Kahn D, Kanapin A, Krestyaninova M, Lopez R, Letunic I, Lonsdale D, Silventoinen V, Orchard SE, Pagni M, Peyruc D, Ponting CP, Selengut JD, Servant F, Sigrist CJ, Vaughan R, Zdobnov EM. The InterPro Database, 2003 brings increased coverage and new features. Nucl Acids Res. 2003;31:315–318. doi: 10.1093/nar/gkg046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosius J. Many G-protein coupled receptors are encoded by retro-genes. Trends Genet. 1999;15:304–305. doi: 10.1016/S0168-9525(99)01783-7. [DOI] [PubMed] [Google Scholar]
- Fink GR. Pseudogenes in yeast? Cell. 1987;49:5–6. doi: 10.1016/0092-8674(87)90746-x. [DOI] [PubMed] [Google Scholar]