

Abstract
Background
Various methods for estimating protein expression levels are known. The level of correlation between these methods is only fair, and systematic biases in each of the methods cannot be ruled out. We here investigate systematic biases in the estimation of gene expression rates from microarray data and from abundance within the Expressed Sequence Tag (EST) database. We suggest that length is a significant factor in biases to measured gene expression rates.
As a specific example of the importance of the bias of expression rate with length, we address the following evolutionary question: Does the average C. elegans protein length increase or decrease with expression level? Two different answers to this question have been reported in the literature, one method using expression levels estimated by abundance within the EST database and another using microarrays. We have investigated this issue by constructing the full protein length versus expression curve for C. elegans, using both methods for estimating expression levels.
Results
The microarray data show a monotonic decrease of length with expression level, whereas the abundance within the EST database data show a non-monotonic behavior. Furthermore, the ratio of the expression level estimated by the EST database to that measured by microarrays is not constant, but rather systematically biased with gene length.
Conclusions
It is suggested that the length bias may lie primarily in the abundance within the EST database method, being not ameliorated by internal standards as it is in the microarray data, and that this bias should be removed before data interpretation. When this is done, both the microarray and the abundance within the EST database give a monotonic decrease of spliced length with expression level, and the correlation between the EST and microarray data becomes larger. We suggest that standard RNA controls be used to normalize for length bias in any method that measures expression.
Background
Estimation of protein expression levels is of significant interest in the genomics and proteomics fields. Protein expression levels provide one of the links between the genetic and functional properties of an organism. Average levels of protein expression determine the typical environments within cells. Changes in protein expression level determine developmental biology, response to stress or fluctuating environments, and progression of disease. The patterns of protein expression have recently become of great interest within evolutionary studies, in which relationships between expression level and various evolutionary rates have been examined [1-4].
While direct measurement of protein expression levels remains non-trivial, several methods to estimate mRNA or cDNA expression levels have been developed, including microarray [5-10], sequential analysis of gene expression (SAGE) [11] and its variants [12-14], enzymatic fragmentation fingerprints [15], polymerase chain reaction (PCR) amplification [16], RNAi library analysis [17], and EST abundance [18-21]. In this work, we focus on the microarray and the abundance within the EST database methods for measurement of mRNA expression levels. The microarray, or gene chip, method is perhaps the most popular approach in current use. The abundance within the EST database method, on the other hand, while growing in popularity, has only recently been proposed for estimation of expression levels [22].
One may hypothesize many possible biases in both the microarray and the abundance within the EST database methods. The microarray method, however, should generically be more reliable, as microarrays are explicitly intended to quantitatively measure expression levels. As such, a collection of gene standards is always included in microarray measurements, and these standards are used to normalize the experimental data, thus removing some of the systematic biases. It is important to note in this context that whereas expression data measured with the microarray method arise from a single, large experiment, the ESTs used in the abundance within the EST database method arise from the entire database, which is constructed from many experiments done under different conditions and often examining different subsets of genes of interest [21]. For these reasons, it would appear that the possible biases arising from the microarray method are better controlled and mitigated [9] in current experiments than are the biases that arise from using the abundance within the EST database to estimate expression levels [21]. Both the microarray and the abundance within the EST database methods are, of course, intrinsically noisy. The method of abundance within the EST database may even produce less noisy data that the method of microarrays. What we are pointing out is that there is a systematic bias in the ratio of the expression levels measured by these two methods. While noise is eliminated by averaging enough data, bias is not.
In the C. elegans genome, two different correlations between total exon length and expression level have been observed [22,23]. In one work, an estimation of expression level was made from abundance within the EST database for each gene [22]. In the other, gene expression was measured by microarrays [23-25]. Both approaches agreed about a negative correlation between length and expression for highly expressed genes, but they disagreed about the trends for moderately and lowly expressed genes. A negative relationship between protein length and expression is expected due to the increased metabolic cost to translate longer genes [3,26]. There are also evolutionary reasons to expect negative correlations between total protein length and expression rate [27].
We address these questions about the protein length versus expression curve. The full length versus expression curve is constructed using both the EST abundance and the microarray data. The difference between the two methods of estimating expression levels is displayed. Assuming the microarray data to be the more accurate measurement of expression levels, due to reliable internal standards, it is shown that the abundance within the EST database method is biased by coding sequence length, and an explicit form of the length bias is presented. By removing the length bias from the EST database estimation, we achieve agreement between the two sets of data, thus explaining the apparent contradiction. Our results confirm the negative correlation between protein length and expression level expected from the energetic costs associated with translation and from evolutionary theory.
Results
Figure 1 shows the plot for the spliced (exonic) gene length as a function of expression levels measured by microarrays in C. elegans [24,25]. A monotonic decrease in gene length with expression is observed. Figure 2 shows the plot for the spliced (exonic) gene length as a function of expression levels estimated by abundance within the EST database for C. elegans [22]. A non-monotonic variation of the length with expression is observed in Figure 2, which is strikingly dissimilar to Figure 1. The two curves differ most significantly in region of low to moderate expression levels. The abundance within the EST database data [22] are redrawn in Figure 3, after the expression levels have been normalized for the length bias:
Figure 1.

Spliced gene length as a function of microarray expression level Spliced gene length is plotted as a function of expression level (ppm) for C. elegans. These are microarray expression data [24,25]. The standard errors are indicated by the error bars.
Figure 2.

Spliced gene length as a function of EST expression level Spliced gene length is plotted as a function of expression level (arbitrary consistent units) for C. elegans. These are expression data estimated by abundance within the EST database [22]. The standard errors are indicated by the error bars.
Figure 3.

Spliced gene length as a function of normalized EST expression level Spliced gene length is plotted as a function of normalized expression level (arbitrary consistent units) for C. elegans. Expression level is estimated by abundance within the EST database [22] and normalized for the length bias of the abundance within the EST database method that is proportional to length, Eq. 1. The standard errors are indicated by the error bars.
![]()
The length versus normalized expression curve is shown in Figure 3 and is quite similar to the length versus expression curve from microarray data shown in Figure 1.
Discussion
The monotonically decreasing behavior in Figure 1 is in accord with previous observations of a negative correlation between exon length and expression level [28,29]. This negative correlation is expected on the grounds that longer genes are more energetically costly to translate and so should be underrepresented in highly expressed genes. This negative correlation may also be expected on the grounds that genes at high levels of expression must also be highly controlled, and it is easier to modulate shorter genes due to the shorter translation time, fewer epistatic interactions, and fewer associated transcription factors. The curve arising when expression levels are estimated from abundance within the EST database, Figure 2, shows an unexpected non-monotonic behavior, increasing in the region of lowly expressed genes and decreasing in the region of highly expressed genes, in evident conflict with the data stemming from microarray experiments. We suggest that this discrepancy is due to the indirect way in which the abundance within the EST database method estimates expression levels. The expression level for a gene is defined [22] as the number of different ESTs matching the gene, divided by the total number of ESTs in the library or collection of libraries. Thus, it would seem that the relative abundance of different ESTs matching with a given gene is proportional to the relative proportion of mRNAs corresponding to this gene in living tissue. This argument, however, is incomplete. There may be biases in the construction of the cDNA libraries that are used to populate the EST databases. In Figure 4, we show the ratio of the expression level estimated by the abundance within the EST database method [22] to that measured by the microarray method [24,25]. The bias in the abundance within the EST database estimation of the expression level is approximately proportional to the gene length, as shown by the linear regression in Figure 4. Also shown in Figure 4 is the same length bias when the abundance is calculated from the full WormBase EST database [30,31], rather than by the method that separates the data into two developmental stages [22]. Either method of estimating the expression levels contains the length bias, which is observed to be proportional to gene length. The microarray data used [24,25] are representative of C. elegans microarray data: bias curves very similar Figure 4 are observed when microarray data averaged over 553 microarray experiments [32] are compared to the limited EST data [22] or the entire C. elegans EST database [30,31].
Figure 4.

The length bias The length bias, calculated as the ratio of abundance within the EST database [22] to microarray expression data [24,25] for each gene present in both datasets, is shown (solid curve). A linear fit to the length bias is shown (solid line). Also shown is the length bias when all of the C. elegans EST data from WormBase [30,31] are used to calculate the EST abundance (dashed curve).
This bias factor may arise from several mechanisms. It has been observed that, generally, microarray data possess a greater degree of internal consistency and reproducibility than do EST data [33]. It has also been observed that, under some conditions, the decay rate of mRNA is slower for longer sequences, thus leading to a possible observed expression bias for longer coding sequences [34] (interestingly, in this experiment, length was found to correlate positively with decay rate within subclasses of RNA sequences, despite the overall negative correlation for the entire data set). The way in which sequences are selected to be deposited within the EST database by individual research groups is an additional, unknown source of bias. We cannot exclude, however, that the observed bias of expression rates with length may substantially arise from the microarray, rather than abundance within the EST database, method. If this is the case, then the expression versus length curve, Figure 2, would have a very interesting non-monotonic behavior.
Another common concern for source of biases that may be present in either the abundance within the EST database or the microarray measurements of protein expression is CG content. For example, in both cases, if DNA annealing was not driven to completion, there may be a bias toward genes with above average CG content, since the CG base paring is significantly stronger than the AT base pairing. We examined how the length versus expression curve changes as the datasets are enriched or depleted in genes with above average CG content. Little sensitivity of the curve to these biases was found.
Since normalization of the length bias brings the expression curves calculated from the microarray and the abundance within the EST database data into agreement, we expect that the correlation between the two datasets should increase as well after the normalization. Thus, we define the correlation coefficient between the two measurements as
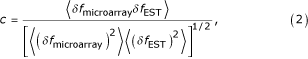
where the average is over all genes present in both datasets, δ fmicroarray = fmicroarray - 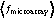 , δ fEST = fEST -
, δ fEST = fEST -  , fmicroarray is the expression rate measured by the microarray method, and fEST is the expression rate measured by the abundance within the EST database method. Since the most significant differences between Figures 1 and 2 occurs in the range of 0 to 200 ppm, we calculated the correlation coefficient Eq. 2 only for those genes with microarray estimated expression rates less than 200 ppm. For the raw data, Figures 1 and 2, the correlation is 0.29. For the data normalized for length bias, Figures 1 and 3, the correlation is 0.38. We thus conclude that the agreement between Figures 1 and 3 is due to the increased correlation between the normalized expression levels for each gene relative to that present in the raw data, Figures 1 and 2.
, fmicroarray is the expression rate measured by the microarray method, and fEST is the expression rate measured by the abundance within the EST database method. Since the most significant differences between Figures 1 and 2 occurs in the range of 0 to 200 ppm, we calculated the correlation coefficient Eq. 2 only for those genes with microarray estimated expression rates less than 200 ppm. For the raw data, Figures 1 and 2, the correlation is 0.29. For the data normalized for length bias, Figures 1 and 3, the correlation is 0.38. We thus conclude that the agreement between Figures 1 and 3 is due to the increased correlation between the normalized expression levels for each gene relative to that present in the raw data, Figures 1 and 2.
Conclusions
In summary, an explicit form of length bias between expression rates measured by microarray and abundance within the EST database methods has been found. Assuming the microarray data to be more reliable due to internal standards and protocols, this length bias stems from the increased representation of long genes within the EST databases, perhaps because longer genes are more likely to survive the enzymatic conditions within the homogenized samples that lead to the cDNA libraries represented in the EST databases. Normalizing for this bias, we find that both methods for measuring expression agree, and a monotonic decrease of gene length with expression is found, in accord with traditional expectations from genetics and evolutionary biology. We cannot completely rule out the presence of a length bias in the microarray data, for example due to decreased accessibility of long transcripts for the microarray surface, and we note that care must be taken to control for length bias in any method that measures expression. One means of control would be doping tissue and cell extracts with a standardized set of different length RNA samples. These standards would allow experimental calculation of the length dependent normalization factor for the expression rates.
Methods
To make use of the microarray expression data, we use the original microarray data [24,25]. A table is constructed that contains the names and corresponding experimental expression levels, measured at different times, for 18,588 different genes of C. elegans. As previously [23], these data are processed to remove data that are, according to the reference, not accurately measured in the experiments (the data marked with * or **). For each gene remaining in the table, the expression level averaged over all remaining data points in time was calculated. The spliced (exonic) sequence and length corresponding to each non-discarded gene was determined from WormBase [30,31]. Genes that were not within WormBase, or that have a zero reported length, were discarded. A total of 5,750 different genes remain after this processing of the original data set. From this data, the average gene length associated with each expression level was calculated. Expression levels were binned, with the bin width chosen to achieve an acceptably low level of error within the bins.
To analyze the abundance within the EST database method for expression analysis, we use the complete original EST data set [22]. These data are presented in a table that contains the name, total length, and estimated expression level for 17,082 different genes of C. elegans. This table was processed to remove entries that do not correspond to complete genes. From the genes remaining, the corresponding spliced (exonic) sequence and length were downloaded from WormBase. Genes that are not within WormBase, or that have a zero reported length, were discarded. A total of 12,707 different genes remain after this processing of the original data set. From this data, the average gene length associated with each expression level was calculated. Expression levels were binned, with the bin width chosen so that an acceptably low level of error was achieved within the bins. The expression estimated by the abundance within the EST database was also scaled by the gene length, Eq. 1. Protein length was then determined as a function of this normalized expression in an analogous fashion.
The ratio of the abundance within the EST database [22] to the microarray expression data [24,25] was calculated. This was done for each gene present in both the EST and microarray datasets, a total of 5334 genes. The ratio of the abundance within the entire C. elegans WormBase [30,31] EST database to that of the data from the microarray data [24,25] was also calculated. Finally, the abundance within the limited EST data set [22] or the entire WormBase [30,31] data set were compared to the data from a large analysis of 553 microarray experiments [32].
One simple mechanism for the bias factor admits that the homogenized sample which contains the mRNA also contains a large amount of Rnases. These Rnases degrade the mRNA from both the 5' and 3' ends. The 5' end may be the protected, terminal base or it may be an unprotected base generated by incomplete transcription or by an endonuclease. The 3' end is protected by the poly-A tail, to which the EST poly-dT primer binds. The 5' end, therefore, is degraded, and a shorter expressed mRNA might even be completely degraded. This enzymatic degradation is present in both the microarray and EST methods, but for the microarray method the established protocols and standards may ameliorate its impact by reducing enzymatic activity, concentration, and contact time. Making the assumption of a constant degradation rate
![]()
the probability that a given mRNA will survive the sample treatment is
![]()
where t is the time during which the mRNA is exposed to the Rnases in the homogenized sample. If we assume that this time is random in each experiment, uniform between 0 and tmax, then the average probability that a gene of length L will survive is given by
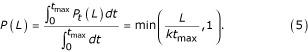
The expression level as estimated by abundance within the EST database, therefore, is biased by the above factor, Eq. 5. In other words, the estimated value is both proportional to the "true" expression level, as was argued [22], and also to this bias factor.
Authors' contributions
ETM carried out the bioinformatics studies. LDB participated in the data analysis. MWD conceived of the study and participated in its design and coordination. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgments
It is a pleasure to acknowledge stimulating discussions with Gabriel Marais. This research was supported by the U. S. National Institutes of Health.
Contributor Information
Enrique T Munoz, Email: munozt@king.rice.edu.
Leonard D Bogarad, Email: ldbogarad@king.rice.edu.
Michael W Deem, Email: mwdeem@rice.edu.
References
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA. The Evolution of Transcriptional Regulation in Eukaryotes. Mol Biol Evol. 2003;20:1377–1419. doi: 10.1093/molbev/msg140. [DOI] [PubMed] [Google Scholar]
- Kondrashov FA, Ogurtsov AY, Kondrashov AS. Bioinformatical Assay of Human Gene Mobidity. Nucleic Acids Res. 2004;32:1731–1737. doi: 10.1093/nar/gkh330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EPC, Danchin A. An Analysis of Determinants of Amino Acids Substitution Rates in Bacterial Proteins. Mol Biol Evol. 2004;21:108–116. doi: 10.1093/molbev/msh004. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li WH. Mammalian Housekeeping Genes Evolve More Slowly than Tissue-Specific Genes. Mol Biol Evol. 2004;21:236–239. doi: 10.1093/molbev/msh010. [DOI] [PubMed] [Google Scholar]
- Pease AC, Solas D, Sullivan EJ, Cronin MT, Holmes CP, Fodor SPA. Light-Generated Oligonucleotide Arrays for Rapid DNA-Sequence Analysis. Proc Natl Acad Sci USA. 1994;91:5022–5026. doi: 10.1073/pnas.91.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PO, Botstein D. Exploring the New World of the genome with DNA microarrays. Nat Genetics. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- Duggan DJ, Bitter M, Chen Y, Meltzer P, Trent JM. Expression Profiling using cDNA Microarrays. Nat Genetics. 1999;21:10–14. doi: 10.1038/4434. [DOI] [PubMed] [Google Scholar]
- Nagpal S, Karaman MW, Timmerman MM, Ho VV, Pike BL, Hacia JG. Improving the Sensitivity and Specificity of Gene Expression Analysis in Highly Related Organisms through the use of Electronic Masks. Nucleic Acids Res. 2004;32:e51. doi: 10.1093/nar/gnh048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romualdi C, Trevisan S, Celegato B, Costa G, Lanfranchi G. Improved Detection of Differentially Expressed Genes in Microarray Experiments through Multiple Scanning and Image Integration. Nucleic Acids Res. 2003;31:e149. doi: 10.1093/nar/gng149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asyali MH, Shoukri MM, Demirkaya O, Khabar KSA. Assessment of Reliability of Microarray Data and Estimation of Signal Thresholds using Mixture Modeling. Nucleic Acids Res. 2004;32:2323–2335. doi: 10.1093/nar/gkh544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velesculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial Analysis of Gene Expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Datson NA, van der Perk-de Jong J, van den Berg MP, de Kloet ER, Vreugdenhil E. MicroSAGE: A Modified Procedure for Serial Analysis of Gene Expression in Limited Amounts of Tissue. Nucleic Acids Res. 1999;27:1300–1307. doi: 10.1093/nar/27.5.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda M, Jantasuriyarat C, Dean RA, Wang GL. Robust-LongSAGE (RL-SAGE): A Substantially Improved LongSAGE Method for Gene Discovery and Transcriptome Analysis. Plant Physiology. 2004;134:890–897. doi: 10.1104/pp.103.034496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilain C, Libert F, Venet D, Costagliola S, Vassart G. Small Amplified RNA-SAGE: An Alternative Approach to Study Transcriptome from Limiting Amount of mRNA. Nucleic Acids Res. 2003;31:e24. doi: 10.1093/nar/gng024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimkets RA, Lowe DG, Tai JTN, Sehl P, Jin HK, Yang RH, Predki PF, Rothberg BEG, Murtha MT, Roth ME, Shenoy SG, Windemuth A, Simpson JW, Simons JF, Daley MP, Gold SA, McKenna MP, Hillan K, Went GT, Rothberg JM. Gene Expression Analysis by Transcript Profiling Coupled to a Gene Database Query. Nat Biotech. 1999;17:798–803. doi: 10.1038/11743. [DOI] [PubMed] [Google Scholar]
- Uematsu C, Nishida J, Okano K, Miura F, Ito T, Sakaki Y, Kambara H. Multiplex Polymerase Chain Reaction (PCR) with Color-Tagged Module-Shuffling Primers for Comparing Gene Expression Levels in Various Cells. Nucleic Acids Res. 2001;29::e84. doi: 10.1093/nar/29.16.e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirane D, Sugao K, Namiki S, Tanabe M, Iino M, Hirose K. Enzymatic Production of RNAi Libraries from cDNAs. Nat Genetics. 2001;36:190–196. doi: 10.1038/ng1290. [DOI] [PubMed] [Google Scholar]
- Gitton Y, Dahmane N, Baik S, Ruiz A, Neidhardt L, Scholze M, Hermann BG, Kahlem P, Benkahla A, Schrinner S, Yildirimman R, Herwig R, Lehrach H, Yaspo ML. A Gene Expression Map of Human Chromosome 21 Orthologues in the Mouse. Nature. 2002;420:586–590. doi: 10.1038/nature01270. [DOI] [PubMed] [Google Scholar]
- Skrabanek L, Campagne F. TissueInfo: High-Throughput Identification of Tissue Expression Profiles and Specificity. Nucleic Acids Res. 2001;29:e102. doi: 10.1093/nar/29.21.e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Zhao S, Pershad R, Hsieh TF, Scarpa A, Wang SW, White RA, Beremand PD, Thomas TL, Gan L, Klein WH. Gene Expression in the Developing Mouse Retina by EST Sequencing and Microarray Analysis. Nucleic Acids Res. 2001;29:4983–4993. doi: 10.1093/nar/29.24.4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Safer HM. A Novel Algorithm for Computational Identification of Contaminated EST Libraries. Nucleic Acids Res. 2003;31:1067–1074. doi: 10.1093/nar/gkg170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G, Piganeau G. Hill-Robertson Interference is a Minor Determinant of Variations in Codon Bias Across Drosophila melanogaster and Caenorhabditis elegans Genomes. Mol Biol Evol. 2002;19:1399–1406. doi: 10.1093/oxfordjournals.molbev.a004203. [DOI] [PubMed] [Google Scholar]
- Castillo-Davis CI, Mekhedov SL, Hartl DL, Koonin EV, Kondrashov FA. Selection for Short Introns in Highly Expressed Genes. Nat Genet. 2002;31:415–418. doi: 10.1038/ng940. [DOI] [PubMed] [Google Scholar]
- Hill AA, Hunter CP, Tsung BT, Tucker-Kellogg G, Brown EL. Genomic Analysis of Gene Expression in C. elegans. Science. 2000;290:809–812. doi: 10.1126/science.290.5492.809. [DOI] [PubMed] [Google Scholar]
- Genomic Analysis of Gene Expression in C. elegans supplemental data files http://mcb.harvard.edu/hunter/Publications/1053496_supplemental.zip
- Duret L, Mouchiroud D. Expression Pattern and, Surprisingly, Gene Length Shape Codon Usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc Natl Acad Sci USA. 1999;96:4482–4487. doi: 10.1073/pnas.96.8.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T, Bogarad LD, Deem MW. Modulation of Base Specific Mutation and Recombination Rates Enables Functional Adaptation within the Context of the Genetic Code. J Mol Evol. 2004. [DOI] [PubMed]
- Coghlan A, Wolfe KH. Relationship of Codon Bias to mRNA Concentration and Protein Length in Saccharomyces cerevisiae. Yeast. 2000;16:1131–1145. doi: 10.1002/1097-0061(20000915)16:12<1131::AID-YEA609>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A. Synonymous Codon Bias is Related to Gene Length in Escherichia Coli: Selection for Translational Accuracy? Mol Biol Evol. 1996;13:864–872. doi: 10.1093/oxfordjournals.molbev.a025646. [DOI] [PubMed] [Google Scholar]
- Harris TW, Lee R, Schwarz E, Bradnam K, Lawson D, Chen W, Blasier D, Kenny E, Cunningham F, Kishore R, Chan J, Muller HM, Petcherski A, Thorisson G, Day A, Bieri T, Rogers A, Chen CK, Spieth J, Sternberg P, Durbin R, Stein LD. WormBase: A Cross-Species Database for Comparative Genomics. Nucleic Acids Res. 2003;31:133–137. doi: 10.1093/nar/gkg053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WormBase release WS114 http://www.wormbase.org
- Kim SK, Lund J, Kiraly M, Duke K, Jiang M, Stuart JM, Eizinger A, Wylie BN, Davidson GS. A Gene Expression Map for Caenorhabditis elegans. Science. 2001;293:2087–2092. doi: 10.1126/science.1061603. [DOI] [PubMed] [Google Scholar]
- Huminiecki L, Lloyd AT, Wolfe KH. Congruence of Tissue Expression Profiles from Gene Expression Atlas, SAGEmap and TissueInfo databases. BMC Genomics. 2003;4:31. doi: 10.1186/1471-2164-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago TC, Purvis IJ, Bettany AJE, Brown AJ. The Relationship between mRNA Stability and Length in Saccharomyces Cerevisiae. Nucleic Acids Res. 1986;14:8347–8360. doi: 10.1093/nar/14.21.8347. [DOI] [PMC free article] [PubMed] [Google Scholar]