

Abstract
Neuroblastoma is a fatal childhood cancer, but lack of knowledge about the underlying causative genes has hampered the development of effective therapies. The identification of anaplastic lymphoma kinase (ALK) mutations as drivers of neuroblastoma has indicated that targeted therapy with ALK inhibitors might be a valuable strategy in the fight against this lethal cancer.
In the era of personalized medicine, understanding the molecular drivers of oncogenesis will be likely to trump morphological characteristics with regard to diagnostics, prognostics and choice of therapies. Identifying single driver mutations from billions of possibilities used to require substantial insight. However, as the cost of deep genomic sequencing goes down each month, clever tricks such as identifying family cohorts may not be as necessary. Family cohorts have driven genetic discovery for decades, including the identification of most tumor suppressor genes and many oncogenes.
Neuroblastoma is a lethal cancer of early childhood that essentially comes in two forms: highly malignant and locally manageable, or, to paraphrase Audrey Evans, an early leader in the field, the ‘good’ies and the ‘bad’ies. The underlying genes responsible for neuroblastoma remain largely unknown, despite the discovery of a handful of genetic changes that have been implicated in neuroblastoma development. For example, Brodeur et al.1 and Look et al.2 correlated MYCN oncogene amplification with aggressive tumors that have a high propensity for metastasis and that cause mortality. Also, very small subsets of familial neuroblastoma were associated with PHOX2B (encoding paired-like homeobox-2) mutations, but these mutations accounted for only very few families (reviewed in ref. 3).
To identify additional oncogenes that cause familial neuroblastoma, Mossé et al.4 returned to old-fashioned family trees. They collected the pedigrees of 20 families that showed an autosomal dominant pattern of neuroblastoma inheritance. Using a genome-wide scan for linkage at 6,000 single nucleotide polymorphisms (SNPs), these investigators identified a previously unknown series of germline mutations in the ALK gene. Some but not all of these families had genomic amplicons on chromosome 2 that also included MYCN. Among the mutations identified, most were predicted to lead to amino acid substitutions in the ALK tyrosine kinase domain (Fig. 1). Five of the eight pedigrees with germline mutation led to proteins with the same R1275Q mutation. A neuroblastoma cell line containing the R1275Q mutation showed ALK activation, which was indicated by tyrosine phosphorylation at ALK Y1604.
Figure 1.
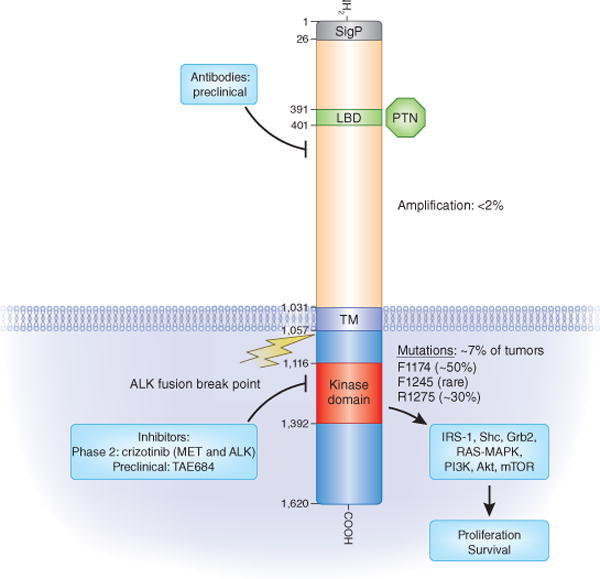
The ALK receptor kinase: its domains, pathways, mutations and inhibitors. The different domains of ALK are shown within their bordering amino-acid positions. The major mutations found by Mossé et al.4, Janoueix-Lerosey et al.5, Chen et al.6 and George et al.7 in the kinase domain and their relative frequencies (somatic or germline) are shown. The percentage of tumors that show ALK amplification or contain ALK mutations is indicated. Representative small-molecule kinase inhibitors that also inhibit ALK are listed according to their current status. The ALK breakpoint that leads to fusion proteins in different cancers is indicated. SigP, signal peptide; LBD, ligand binding domain; PTN, pleiotrophin; TM, transmembrane domain; IRS-1, insulin receptor substrate 1; Grb2, growth factor receptor–bound protein-2; mTOR, mammalian target of rapamycin.
Three additional publications by Janoueix-Lerosey et al.5, Chen et al.6 and George et al.7 used SNP- or amplicon-based sequencing methods to identify similar mutations in ALK (Fig. 1). Each of the three studies showed the effects of a dominant oncogene with a similar pattern of results. Not all mutations resulted in constitutive kinase activity; in fact, only the minority showed clear increases in ALK autophosphorylation and downstream target activation, such as phosphorylation of AKT. The R1275Q and F1174L(V) mutations were shown to have constitutive tyrosine phosphorylation in cell lines that endogenously expressed these mutant forms of ALK, or when expressed in either 3T3 or Ba/F3 cells6,7.
ALK mutations in neuroblastoma tissues account for small survival differences in the whole population of people with neuroblastoma; this is due to their very low incidence in the population. Still, individuals who have neuroblastomas with the ALK F1174 mutation show a significantly worse outcome relative to the overall cohort, suggesting that this mutation has an important impact on malignant progression, whereas the presence of the ALK R1275Q mutation did not make a difference8. Stratification according to ALK expression showed that individuals with high ALK expression in their tumors had a significantly worse overall survival and disease-free survival than those with low tumor ALK expression. Although this type of single-gene analysis can be useful to direct treatment with a targeted therapy, genomic instability in the tumors leads to complex mixtures of clonal cells; therefore, multigene analyses can provide a more robust predictor of disease course and may ultimately provide a more complex insight into pathways activated during tumorigenesis and even predict candidate drug sensitivity8.
The functional role of these ALK mutations was addressed in each of the four studies4–7; however, a deeper understanding will be necessary to optimize any ALK-targeted therapy. Each study used either ALK expression followed by inhibition in non-neuroblastoma cell lines, RNAi-mediated reduction of endogenous ALK expression in cell lines or small-molecule kinase inhibitors. Generally, the results from these studies supported a dominant role for mutant ALK. However, in some cases, RNAi-mediated reduction of wild-type ALK expression also reduced neuroblastoma cell growth, suggesting that the level of ALK expression might be as important as an activating mutation in driving tumorigenesis.
A recent paper by Sasaki et al.9 identified ALK F1174L as a mutation that occurred naturally in an individual with inflammatory myoblastic tumor undergoing treatment with the dual MET and ALK kinase inhibitor crizotinib. This tumor contained a somatic RANBP2-ALK translocation; however, when the person experienced recurrent disease, one ALK locus contained an additional F1174L mutation. An evaluation of the inflammatory myoblastic tumor cells harboring ALK F1174L showed that they had higher levels of phosphorylated ALK along with increased downstream phosphorylation of AKT. Thus, this study supports that F1174L is an activating mutation of ALK.
ALK was independently identified as a molecular target in neuroblastoma by screening >600 human cancer cell lines with pharmacological inhibitors of ALK kinase activity10. This work provided a platform from which to launch inhibitor development when a transforming EML4-ALK fusion gene was identified as a major driver in a subset of approximately 5% of non–small-cell lung cancers11. Indeed, a first clinical trial with crizotinib showed striking efficacy in individuals with non–small-cell lung cancer, providing evidence that a well-defined activated pathway may be targeted effectively12 (Fig. 1). A recent structural study of the ALK kinase domain revealed differences from the same domain in related receptor kinases in the insulin receptor family that will be helpful in designing new inhibitors as resistance to the initial ALK inhibitors emerges13.
As higher ALK receptor expression in neuroblastoma coincides with poorer disease outcome8, overexpressed ALK may thus be a potentially valuable therapeutic target. Indeed, two proteins—pleiotrophin and the related midkine—have been shown to activate mammalian ALK14 through extracellular interactions with it (Fig. 1). Therefore, antibody strategies to target overexpressed ALK in neuroblastoma may be an attractive additional approach. Antibody therapy may even act syn ergistically with small-molecule tyrosine kinase inhibitors.
It is clear that targeting ALK is now open season for the development of new therapies for neuroblastoma, as well as other cancers. The insights gained from ongoing trials are likely to directly benefit both subgroups of individuals whose tumors are driven by ALK and, in general, targeted approaches to cancer treatment. Obviously, genetic mutations can reveal new drivers and pathways activated in cancer and may present new therapeutic opportunities. However, fully validating any new target is a challenge at a new frontier of a deadly cancer.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturemedicine/.
References
- 1.Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Science. 1984;224:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- 2.Look AT, et al. J Clin Oncol. 1991;9:581–591. doi: 10.1200/JCO.1991.9.4.581. [DOI] [PubMed] [Google Scholar]
- 3.Maris JM. N Engl J Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mossé YP, et al. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janoueix-Lerosey I, et al. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, et al. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- 7.George RE, et al. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Brouwer S, et al. Clin Cancer Res. 2010;16:4353–4362. doi: 10.1158/1078-0432.CCR-09-2660. [DOI] [PubMed] [Google Scholar]
- 9.Sasaki T, et al. Cancer Res. 2010;70:10038–10043. doi: 10.1158/0008-5472.CAN-10-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDermott U, et al. Cancer Res. 2008;68:3389–3395. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 11.Soda M, et al. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 12.Kwak EL, et al. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee CC, et al. Biochem J. 2010;430:425–437. doi: 10.1042/BJ20100609. [DOI] [PubMed] [Google Scholar]
- 14.Stoica GE, et al. J Biol Chem. 2001;276:16772–16779. doi: 10.1074/jbc.M010660200. [DOI] [PubMed] [Google Scholar]