

Abstract
Since their description and identification in leukemias and solid tumors, cancer stem cells (CSC) have been the subject of intensive research in translational oncology. Indeed, recent advances have led to the identification of CSC markers, CSC targets, and the preclinical and clinical evaluation of the CSC-eradicating (curative) potential of various drugs. However, although diverse CSC markers and targets have been identified, several questions remain, such as the origin and evolution of CSC, mechanisms underlying resistance of CSC against various targeted drugs, and the biochemical basis and function of stroma cell-CSC interactions in the so-called ‘stem cell niche.’ Additional aspects that have to be taken into account when considering CSC elimination as primary treatment-goal are the genomic plasticity and extensive subclone formation of CSC. Notably, various cell fractions with different combinations of molecular aberrations and varying proliferative potential may display CSC function in a given neoplasm, and the related molecular complexity of the genome in CSC subsets is considered to contribute essentially to disease evolution and acquired drug resistance. In the current article, we discuss new developments in the field of CSC research and whether these new concepts can be exploited in clinical practice in the future.
Keywords: Cancer stem cells, Targeted therapy, Drug resistance
Introduction
The principle concept of cancer stem cells (CSC) has gained increasing acceptance in recent years [1-10]. By definition, CSC exhibit self-renewal activity and long-term cancer-propagating capacity [1-9]. By contrast, more mature clonal cells in the same neoplasm have limited proliferative potential. In leukemias, CSC are also known as leukemic stem cells (LSC) [5,8,11-18]. The concept of neoplastic stem cells may provide explanations for the failure of various cytoreductive agents to produce long-lasting responses in patients [1-9,11,15-18]. Notably, in many instances, anti-neoplastic drugs act on more mature neoplastic cells rather than CSC/LSC, a phenomenon that is explained in part by the fact that these cells exhibit intrinsic resistance [19-23]. Moreover, CSC often develop acquired drug resistance and thus produce more malignant subclones over time [11,24-26].
All these observations point to the need to develop new CSC-eliminating treatment strategies through which cure rates and survival can be improved [16,27-31]. In other words, CSC have been recognized as a major ‘target cell population’ in oncology in recent years, and considerable effort has been made to identify novel CSC markers and target expression profiles and to measure responses of these cells to various targeted drugs.
The present article provides a summary of our knowledge on CSC/LSC, with special focus on the possibility to translate CSC/LSC-targeting treatment concepts into clinical application. Unless otherwise stated, this article refers to CSC/LSC in primary human malignancies. With regard to cell line models and engineered CSC-like cells or other more ‘artificial’ models that may also support CSC research, we refer to the published literature.
Definition and function of cancer stem cells
In contrast to more mature cancer cells, CSC are self-renewing cells with long-term proliferative potential [1-9,11]. As a result, CSC can maintain a given neoplasm for prolonged time periods. In most cases, these cells can also produce a cancer (CSC) or leukemia (LSC) in immunodeficient mice (xenotransplantation model) which enables their detection and quantification [1-9,32,33]. Previous studies have used non-obese diabetic mice with severe combined immunodeficiency (NOD/SCID) [1-3,5,6,32,33]. In several tumor models, this mouse strain is a sufficient or even a preferable model to study CSC biology [34]. However, more recent data suggest that in several primary malignancies, NOD/SCID with loss-of-function-mutated IL-2Rgamma chain or IL-2Rgamma chain-knock out NOD/shi-SCID mice (NSG or NOG mice) provide superior engraftment rates [35-38]. Therefore, many current studies on primary CSC/LSC employ NSG mice. Depending on the type of disease, neoplastic cells are injected intravenously, subcutaneously, or directly into solid organs (orthotopic application) [27,39-44]. An important point is that ‘short-term engraftment’ (or just simple maintenance) of tumor/leukemic cells has to be differentiated from long-term engraftment, only the latter being indicative of the presence of functionally active (self-renewing) CSC. Long-term engraftment and growth of cancers/leukemias is best demonstrable by recovering engrafted cells from primary recipient mice and injecting these cells into secondary recipient animals [32,39,40,42,43,45-47].
Despite advanced technologies and novel mouse models, xenotransplantation assays for human CSC have several limitations. First, the microenvironment is often species-specific or tumor-specific. Second, in a neoplasm with low growth-rate (for example, indolent/low-grade/chronic tumor; premalignant neoplasms), the development phase of the neoplasm may exceed the lifetime of a mouse. Moreover, the CSC pool is composed of heterogeneous populations of tumor-initiating cells with subclone-specific molecular properties and varying growth characteristics in vivo [11,25,26,28,48]. Some of the CSC may be recognized (and eliminated) by the residual immune system of xeno-transplanted mice [37,38]. On the other hand, the lack of a natural immune system and thus tumor immune surveillance in highly immunodeficient mice may facilitate the uncontrolled expansion of clinically irrelevant sub-clones. Therefore, several attempts are currently made to establish NSG-mouse models harboring a human immune system.
A frequently discussed alternative to in vivo xenotransplantation studies are in vitro long-term culture experiments to study the growth and maintenance of CSC [47,49-53]. Although helpful as a screen approach, these assays are not sufficient for evaluating the in vivo self-renewal capacity of ‘true’ CSC. Several in vitro assays employ stromal cells which may provide some of the ‘niche-factors’ required for long-term growth CSC [47,49-53]. Solid tumor cells often grow in ‘spheres’ or clusters for prolonged time periods in such assays [47,49-53]. However, as mentioned above, the available in vitro assays cannot replace in vivo xenotransplantation models when long-term self renewal and tumor propagation should be examined.
Identification and enrichment of CSC/LSC
Several different approaches, through which CSC/LSC can be identified and enriched in primary cancer/leukemia samples, have been developed in the past [1-3,5-7,9,11-13,27,54-61]. A widely applied strategy is to use antibodies directed against certain cell surface antigens that are (or are not) expressed on CSC [1-3,5-7,9,11-13,27]. Expression of surface antigens is best determined by multicolor flow cytometry. Enrichment of CSC/LSC can be performed by fluorescence-activated cell sorting (FACS) or magnetic cell sorting [1-9,13,15-18,62-69]. Both techniques have certain limitations. One general problem is that the ‘so-called’ stem cell markers are often not specific for CSC or LSC. Likewise, the stem cell-related antigen CD34 is not only expressed on hematopoietic stem cells but also on myeloid progenitor cells and endothelial cells, and KIT is not only expressed on hematopoietic stem- and progenitor cells but also on mast cells, germ cells, and melanocytes [70,71]. Therefore, it is essential to apply combinations of antibodies when detecting and analyzing CSC/LSC in various tissues. Usually, one or two organ-specific markers are employed to confirm the primary origin of cells (Tables 1 and 2). The pan-hematopoietic marker CD45 is widely used to confirm the hematopoietic origin of cells or to exclude leukocytes in primary fractions obtained from solid tumors. Additional antibodies are applied to delineate CSC from more mature neoplastic cells [1-3,5-7,9,11-13,27,65-69,72,73]. In case of myeloid leukemias, the antigen profiles of more mature cells are well defined, and the approach to deplete these (Lin+) cells from LSC is well established. However, in certain leukemias, LSC may aberrantly express one or even several of the ‘lineage-related’ antigens. In such leukemias, application of the ‘Lin-cocktail’ may lead to a loss of LSC subsets. Another problem is that antibody-bound cells may be detected and eliminated by the residual immune system of NOD/SCID mice. This problem has been outlined in acute myeloid leukemia (AML) where CD38+ cells (CD38 antibody-laden) may be cleared by the residual immune system of NOD/SCID mice [38]. The problem has been addressed by switching from NOD/SCID mice to NSG (or NOG) mice that lack a functionally active cytokine receptor gamma chain [35-38]. As mentioned above, the lack of a natural immune system in these models is a remaining issue that will hopefully be solved by introducing a humanized immune system into these mice. Another caveat is that some of the antibody preparations used to define CSC may induce apoptosis in cancer cells [74].
Table 1.
Phenotype of neoplastic stem cells (NSC) in hematologic neoplasms
| Neoplasm | Defined cell subsets containing NSC | Cell surface antigens aberrantly expressed or overexpressed on neoplastic SC |
|---|---|---|
| AML | CD34+/CD38− [32] | CD25 [75], CD33 [76], CD52[77] |
| CD96 [68], CD123 [69] | ||
| CLL-1 [67] | ||
| AML | CD34+/CD38+ [38] | n.k. |
| AMLNPM1mutated | CD34− blast-like [78] | n.k. |
| MDS | CD34+ [79] | CD123 [80] |
| MDS with 5q- | CD34+/CD38− [81] | CD52 [77], CD123 |
| MPN | CD34+ [82] | n.k. |
| CML CD123 [86], IL-1RAP [87] | CD34+/CD38− [17] | CD25 [83], CD26 [84], CD33 [85] |
| Ph + ALL | CD34+/CD38−/CD19+ [88] | CD25, CD26a, CD52 |
| Ph − ALL | CD34+/CD19+ [89] | n.k. |
| CLL | CD34+/CD19+ [90] | CD5 |
| Myeloma | CD20+/CD27+/CD138− [91] | n.k. |
AML, acute myeloid leukemia; MDS, myelodysplastic syndrome(s); MPN, myeloproliferative neoplasm(s); CML, chronic myeloid leukemia; ALL, acute lymphoblastic leukemia; CLL; chronic lymphocytic leukemia; n.k., not known; NSC, neoplastic stem cells; SC, stem cell; Ph+, Philadelphia chromosome-positive; Ph−, Philadelphia chromosome-negative; IL-1RAP, interleukin-1 receptor accessory protein. aIn a subset of patients with Ph + ALL, LSC express CD26.
Table 2.
Phenotype of CSC-enriched fractions of neoplastic cells in solid tumors a
| Neoplasm | Phenotype of CSC-rich cell fraction | Reference |
|---|---|---|
| Breast cancer | CD326+/CD45−/CD44+/CD24− | [40] |
| CD44+/CD49f+/CD133+ | [92] | |
| CD326+/CD44+/CD47+/MET+ | [93] | |
| CD29f | ||
| Gastric cancer | CD326+/CD44+ | [94] |
| CD49f+ | [95] | |
| CD90+ | [96] | |
| LGR5b+ | [97] | |
| CD44+ | [98] | |
| Colon cancer | CD326+/CD44+/CD166+ | [99] |
| CD44+/CD49f+/CD133+ | [100] | |
| LGR5b+ | [101] | |
| CD133+ | [47] | |
| SCLC | CD133+ | [102] |
| NSCLC | CD133+ | [103] |
| Pancreatic cancer | CD44+/CD24+/CD326+ | [43] |
| CD133+/CXCR4+ | [39] | |
| HCC | CD326 | [104] |
| CD133+ | [105] | |
| CD44+/CD90+ | [44] | |
| Glioblastoma | CD133+ | [42] |
| CD15+/CD133+ | [106] | |
| CD15+/CD133+ | [106] | |
| CD133+/SSEA-1+ | [107] | |
| Ewing’s sarcoma | CD133+ | [108] |
| Osteosarcoma | CD133+ | [109] |
| CD117+/STRO-1+ | [110] | |
| CD271+ | [111] | |
| Ovarian cancer | CD24+/CD44+/CD326+ | [112] |
| CD44+/CD117+ | [113] | |
| CD133+ | [114] | |
| Prostate cancer | CD44+/CD49f/CD326+ | [115] |
| CD44+/CD24− | [116] | |
| CD44+/CD133+ | [117] | |
| Melanoma | CD271+ | [118] |
| ABCB5+ | [119] | |
| EPOR+ | [120] |
NSC, neoplastic stem cells; LGR5, Leucine-rich repeat-containing G-protein coupled receptor 5; SCLC, small cell lung cancer; n.k., not known; NSCL, non-small cell lung cancer; HCC, hepatocellular carcionoma; EPOR, erythropoietin receptor. aExpression of NSC markers refers to primary human cells tested in xenotransplantation assays and/or in a sphere-formation assay. bLGR5 is not detectable on human NSC by flow cytometry.
In solid tumors, a general problem is that for most neoplasms, robust markers discriminating between more mature and immature cells are not available. In colorectal cancer and some other solid tumors, the Wnt target gene LGR5 has been described as a potential CSC marker [121,122]. Other markers, such as CD44, are broadly expressed on tumor cells and also in other cell types (for example, leukocytes) present in the same organ sites. Another problem is that several CSC-homing receptors and their ligands are species specific which may prevent homing of CSC to their specific microenvironment (CSC niche) in mice. Such limitations can be overcame by direct (orthotopic) injection of CSC into target organs or into tissue scaffolds [39-44,46,123,124]. Other potential solutions may be to co-transplant ‘niche-relevant’ autologous (human) stroma cells together with CSC, to treat mice with cytokines promoting the growth of CSC/LSC or to employ NSG mice engineered to express human niche-associated cytokines such as stem cell factor (SCF) [125]. For the future, mouse models harboring a human immune system as well as human stromal cells might be desirable for studying CSC biology.
Probably the most important problem regarding CSC-recognition is stem cell plasticity and disease heterogeneity [9,11,25,28,54,126]. Likewise, depending on the subtype of myeloid leukemia, LSC may reside within the CD34+/CD38− fraction of the clone but also in the CD34+/CD38+ or even in CD34− cell populations [38,78,126]. It has also been described that LSC may be composed of CD133+ and CD133− subfractions [64,127]. Only a few markers, such as CLL-1 or interleukin-1 receptor accessory protein (IL-1RAP), may be more or less specific for LSC in certain human leukemia models [67,87]. These markers are interesting tools and may serve as diagnostic markers or/and therapeutic targets in the future. Tables 1 and 2 show a summary of markers expressed on CSC in hematopoietic neoplasms (Table 1) and non-hematologic malignancies (Table 2).
Regulation of growth and development of CSC/LSC
So far, little is known about the regulation of growth and survival of CSC/LSC in hematopoietic and non-hematopoietic malignancies. The development phase of CSC may often last for years if not decades [28,48,54,128]. In an early phase (pre-phase) of cancer or leukemia development, neoplastic stem cells may be slowly cycling cells that produce small-sized subclones [11,28,54,128]. At this early hypothetical stage of cancer evolution, it may be preferable to call these cells premalignant neoplastic stem cells (NSC) rather than CSC/LSC [24,28,128-131]. Later, when these premalignant cells have accumulated a sufficient number of molecular lesions (defects) and thereby have ‘learned’ how to escape all relevant surveillance mechanisms, their progeny can expand and form an overt malignancy within short time, so that the term malignant NSC (=CSC or LSC in leukemias) is appropriate [28,48,128,130,132] (Table 3). In early phases of NSC evolution (premalignant stage), the mechanisms and molecules regulating growth, survival, and asymmetrical cell division, may be similar if not the same compared to that in normal stem cells. These factors include cytokines and cytokine-receptors, niche-related factors, including stem cell-homing and chemotactic molecules, pro- and anti-apoptotic molecules, and signaling pathways involved in the regulation of self-renewal and proliferation [133-135]. Later, when NSC-derived neoplastic clones expand to an overt malignancy, several ‘physiologic’ mechanisms controlling growth and differentiation of normal (and premalignant neoplastic) stem cells may no longer work to prevent clonal expansion [28,48,128,130,136-138].
Table 3.
Classification of neoplastic stem cells (NSC)
| Defining properties | Premalignant NSC | Malignant NSC = CSC/LSC |
|---|---|---|
| Self-renewal | Yes | Yes |
| Cell cycle | Dormant or very slowly cycling | Slowly cycling or more rapidly cycling |
| Immediate tumor-initiating potential | Noa | Yes |
| Long-term tumor-initiating potential | Facultative potentiala | Yes |
| Numbers of somatic acquired molecular lesions/mutations | Relatively low | Relatively high |
| Drug response | Intrinsic resistance (based in part on quiescence) | Intrinsic and often also acquired resistance in malignant subclones |
aThe potential of a NSC to produce a neoplastic condition does not mean that this cell can form a tumor within a certain time period; however, after a certain latency period, when a sufficient number of molecular lesions have been accumulated, these premalignant NSC may transform to fully malignant NSC (=CSC/LSC) that have immediate tumor-initiating capacity in vivo in patients as well as in NSG mice. In a subset of patients, premalignant NSC will never convert into fully malignant NSC (= CSC/LSC). NSC, neoplastic stem cells; CSC/LSC, cancer stem cells/leukemic stem cells.
Cytokine regulation of NSC (CSC/LSC)
A number of recent data suggest that the cytokine network is involved in the regulation of self-renewal, growth, survival, and differentiation of NSC [64,69,125]. As mentioned above, the cytokines that regulate growth and function of premalignant NSC may be similar or the same as that regulating growth of normal stem cells. Likewise, in myeloid leukemias, NSC/LSC express receptors for various regulators of normal stem cells, including the IL-3 receptor (CD123/CD131), SCF receptor KIT (CD117), or G-CSF receptor (CD114) [64,69,139]. It has also been described that epidermal growth factor (EGF) receptor family members, including HER2, are expressed on epithelial NSC/CSC, such as mammary CSC [140,141]. There is also evidence that insulin-like growth factor (IGF) receptors and fibroblast growth factor (FGF) receptors play an important role in solid tumors and may be expressed on solid tumor CSC [142-144]. At least in leukemias, the cytokine ligands that bind to these receptors trigger proliferation of LSC-enriched cell fractions [139]. Depending on the type and phase of disease, these cytokines also promote differentiation and maturation of LSC. However, most of these cytokines may not cause self-renewal in LSC. Some of these cytokines, such as IL-3, are also produced in clonal cells and may thus act as autocrine growth regulators of LSC [17,87,145,146]. LSC are also considered to respond to various chemokines. In line with this assumption, LSC express chemokine receptors such as CXCR4 [39,147-150]. A clinically important question is whether premalignant NSC or CSC/LSC express receptors for erythropoietin (EPO), granulocyte colony-stimulating factor (G-CSF), or granulocyte/macrophage colony-stimulating factor (GM-CSF). These cytokines are often administered in tumor patients in order to correct disease-related anemia or to accelerate neutrophil production after chemotherapy. In AML as well as in the myelodysplastic syndromes (MDS), NSC/LSC indeed express receptors for G-CSF and sometimes also for GM-CSF [139]. By contrast, NSC/LSC usually do not express EPO receptors in these malignancies. However, the EPO receptor may be expressed on CSC in a few solid tumors as well as in melanoma-initiating cells [120,151-153]. Table 4 shows a summary of cytokine receptors expressed on CSC and LSC in various malignancies.
Table 4.
Cytokine/chemokine receptors detectable on neoplastic stem cells (NSC)
| Malignancy | Cytokine receptors expressed on NSC |
|---|---|
| AML | IL-2RA [154], IL-3RA [155], G-CSFR [156], FLT3 [157], SCFR/KIT [158], CXCR4 [159] |
| MDS | G-CSFR [160], SCFR/KIT, CXCR4 [161] |
| MPN | G-CSFR [160], SCFR/KIT [162], CXCR4 [163] |
| Ph + CML | IL-2RA [83], IL-3RA [17], G-CSFR [160], GM-CSFR [164], SCFR/KIT [165], IL-1RAP [87], CXCR4 [166] |
| Ph + ALL | IL-2RA [167], IL-3RA [168], CXCR4 [169] |
| Myeloma | CXCR4 [170] |
| Breast cancer | EGFR [171], ERBB2/Her2 [172], FGFR2 [173], TGFßR [174], MET [93] |
| Gastric cancer | EGFR [175], ERBB2/Her2 [176] |
| Colon cancer | EGFR [177], CXCR4 [178], IGF1R [179], TGFßR [180] |
| SCLC | EGFR [181] |
| Pancreatic cancer | EGFR [182], CXCR4 [39] |
| HCC | EGFR, IGF1R [183] |
| Glioblastoma | EGFR [184], PDGFRB [185], CXCR4 [186], TGFßR [187], MET [188] |
| Ovarian cancer | EGFR [189], ERBB2/Her2 [190], IGF1R, TGFßR [191] |
| Prostate cancer | CXCR4 [192] |
| Melanoma | CXCR1 [193], EPOR [120] |
AML, acute myeloid leukemia; IL, interleukin; G-CSFR, granulocyte colony-stimulating factor receptor; SCF, stem cell factor receptor; Ph+, Philadelphia chromosome-positive; CML, chronic myeloid leukemia; GM-CSF, granulocyte- macrophage colony-stimulating factor; ALL, acute lymphoblastic leukemia; CLL, chronic lymphocytic leukemia; EGFR, epidermal growth factor receptor; TGFßR, transforming growth factor ß receptor; IGF1R, insulin-like growth factor 1 receptor; SCLC, small cell lung cancer; NSCL, non-small cell lung cancer; n.k., not known; HCC, hepatocellular carcionoma; PDGFR, platelet-derived growth factor receptor; NGFR, nerve growth factor receptor; EPOR, erythropoietin receptor.
Oncogenic signaling pathways in NSC (CSC/LSC)
Growth and function of NSC, including self-renewal and malignant expansion, are considered to depend on a complex network of signaling cascades and molecules. Oncogenic signaling is considered to derive from three distinct classes of molecules, i) the driver lesions (primary oncogenic kinases) that are often disease-specific or at least disease-related, like BCR/ABL in chronic myeloid leukemia (CML), ii) broadly expressed mutated oncogenic kinases, and iii) cytokine-activated stem cell kinases that play a role in survival or/and growth of NSC (example: wt KIT in leukemias). The downstream signaling networks of ‘i,’ ‘ii,’ and ‘iii’ are in part overlapping, may often complement each other, and may sometimes even produce synergistic effects on downstream activation and thus oncogenesis [165,194-196]. In an early phase of cancer evolution, the driver mutation (‘i’) and otherwise physiologic mechanisms (‘iii’) may play a predominant role. However, with disease progression, more and more additional oncogenic signaling molecules (‘ii’) and pathways become activated [197-200]. Thus, in advanced phases of a malignancy, additional signaling cascades and networks may play a more and more decisive role in CSC/LSC expansion and resistance. All three classes of molecules may contribute to CSC/LSC resistance, and all three have been considered as potential targets of therapy in solid tumors and leukemias [7,14,16,28,198,201,202].
In the past 15 years, several of the driver kinases have been identified as major targets of therapy. The highlighting example is CML where BCR/ABL-targeting tyrosine kinase inhibitors (TKI) induce major and long-lasting responses [203]. Other similar treatment concepts are emerging in other types of cancers and leukemias as well as in lymphomas. However, it has also been described that in most tumor models, NSC (CSC/LSC) cannot be eradicated completely using these drugs [11,24,28,130,204] as CSC/LSC often grow and survive independent of the primary (major) driver lesion, such as BCR/ABL in CML [204,205].
During the past few years, several major attempts, supported by next-generation sequencing approaches, have been made to reveal additional molecular lesions and the resulting signaling cascades and to define additional target pathways in CSC/LSC [206,207]. Indeed, a number of different signaling pathways - often shared by normal and neoplastic stem cells - have been described to play a role in the evolution and maintenance of CSC/LSC. Several of these pathways have been implicated in stem cell self-renewal. One of these pathways is the Wnt/ß-catenin pathway. This pathway is involved in the maintenance of self-renewal of NSC in leukemias and melanoma as well as in breast, lung, and liver cancers [119,198,208-211]. The Notch signaling pathway has been implicated in self-renewal of CSC in breast cancer, colon cancer, and glioblastoma [201,212-214]. The hedgehog-signaling pathway is also considered to contribute to self-renewal of CSC in various malignancies, such as glioblastoma, breast cancer, colon cancer, pancreatic cancer, and also in leukemias [215-219]. Other signaling pathways may be involved in the regulation of proliferation, survival, and differentiation of CSC. These pathways include, among others, the PI3 kinase-mTOR pathway, the RAS-RAF-MEK-ERK pathway, or the JAK-STAT pathways [196,220-223].
Role of the microenvironment and cell-cell interactions
Depending on the stage and type of malignancy, growth and self-renewal of NSC (CSC/LSC) rely on a permissive microenvironment, the CSC niche [4,14,45,72,224,225]. In an early phase of cancer evolution, the CSC niche may regulate growth and self-renewal of premalignant NSC in a similar or in the same way as that of normal stem cells [54,211-216,226,227]. Relevant molecules contributing to stem cell niche interactions in healthy tissues and in ‘premalignant neoplastic states’ include adhesion molecules, chemotactic factors, cytokines, and growth factor receptors [99,225,228-235] (Figure 1). In addition, the local electrolyte milieu, the Ca2+ gradient as well as hypoxia may contribute to stem cell niche interactions and stem cell self-renewal in normal and (pre)malignant conditions [225].
Figure 1.
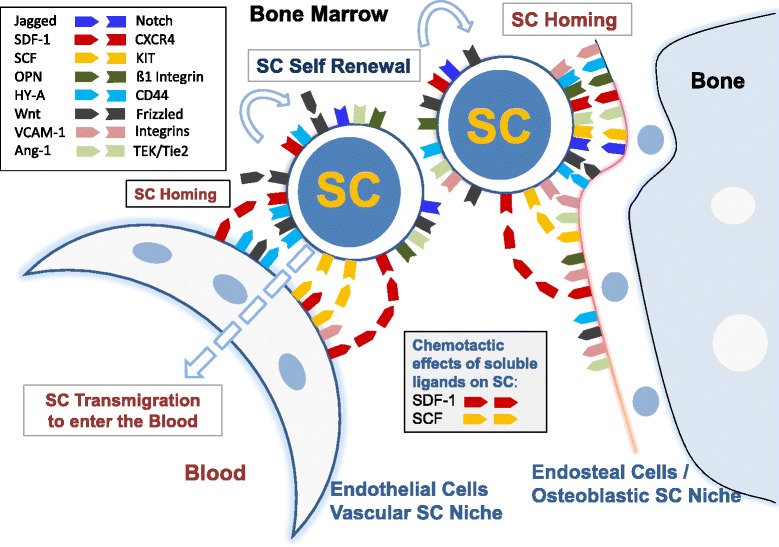
Cellular interactions in the bone marrow (BM) stem cell niches. Two types of BM stem cell (SC) niches have been postulated, the vascular SC niche and the endosteal (osteoblastic) SC niche. Both SC niches are considered to play a role in SC homing and SC self-renewal. A number of SC receptors and their ligands regulate qiuesence, self-renewal, proliferation, differentiation, and homing of SC. Relevant ligands are expressed in niche-related cells, including vascular endothelial cells, endosteal cells, and osteoblasts. Whereas several of these ligands are membrane-bound and act as homing receptors, some of them, such as stem cell factor (SCF) or stroma cell-derived factor-1 (SDF-1), can also be produced and released as soluble ligands and thus can act as chemotactic factors for SC. Abbreviations: OPN, osteopontin; HY-A, hyaluronic acid; Ang-1, Angiopoietin-1.
Stem cell homing and abnormal spread of NSC/CSC
Depending on the organ system, homing of stem cells is a physiologic process [225,236-239]. Likewise, normal hematopoietic stem cells are detectable in the peripheral blood and undergo homing in various organs. In most solid organs, however, stem cells do not undergo redistribution and homing, unless these cells transform to metastasizing CSC. Stem cell homing of normal hematopoietic stem cells and LSC is a multi-step process and the same holds true for the invasion-metastasis cascade of CSC [240,241]. Several different molecules are involved in the homing and invasion process, including selectins and selectin-ligands, integrins and their receptors, and other cell-cell and matrix-binding molecules [240,242]. However, ordered expansion and redistribution from and into the stem cell niches in various organs is usually deregulated in premalignant NSC and malignant CSC/LSC [84,99,225,228-235]. In the normal and leukemic bone marrow, several specific molecular interactions that may contribute to stem cell homing (to the niche) have been identified. These include, among others, SDF-1-CXCR4 interactions, SCF-KIT interactions, and Notch-Notch-ligand interactions (Figure 1) [64,139,225]. In solid tumors, interactions between CSC and the CSC niche are less well defined. One important type of molecules may be cytoadhesion receptors, including integrins, selectins, CD44, or members of the cadherin family. Most of these homing receptors, including chemokine receptors and ligands of matrix molecules such as L1 or CD44, have been detected on CSC [228,229,243]. Likewise, L1 is expressed on the edges of invasive colon cancers and its metastases [230,231] and the same holds true for CD44 and CD133, suggesting that these molecules play a role in tumor invasion and thus disease progression [99,230-232].
During progression of a tumor or leukemia, CSC/LSC may no longer depend on their interaction with the (original) organ-specific microenvironment (CSC niche). Rather, CSC/LSC often expand and redistribute from local sites to other organs to cause metastasis. In epithelial tumors, CSC redistribution is facilitated by the so-called epithelial-mesenchymal transition (EMT), a process that is associated with a loss of specific (adhesive) interactions between cancer cells and the surrounding microenvironment [233,244,245]. Several different molecules, including E-cadherin and L1, have been implicated in the process of EMT in solid tumors [230-233]. Since EMT may also involve CSC, metastasis formation is directly linked to EMT. In hematopoietic neoplasms, similar mechanisms may apply during disease evolution. However, so far, little is known about specific alterations in CSC niche interactions in these malignancies. In CML, LSC have been described to exhibit an adhesion defect that may explain the LSC escape from the bone marrow niche, and subsequent extramedullary spread of progenitors, which is a pathognomonic finding in this type of leukemia [234,235,246].
The endosteal and the vascular stem cell niche in the bone marrow
In the normal bone marrow (BM) and in hematopoietic neoplasms, two types of stem cell niches have been postulated, a vascular niche and an endosteal (osteoblastic) stem cell niche (Figure 1). Both niches are considered to act together and thereby trigger self-renewal, proliferation, migration, and redistribution of normal and neoplastic (leukemic) stem cells [225,247-249]. Whereas the endosteal niche is considered to regulate self-renewal and quiescence of normal and neoplastic stem cells, the vascular niche is considered to regulate self-renewal, redistribution, and the leukemic spread of these cells. The postulated vascular niche may primarily be composed of endothelial (arterial) cells and perivascular cells, whereas the endosteal stem cell niche is primarily represented by endosteal-lining cells and osteoblasts [225,247]. The endosteal niche is considered to provide a more hypoxic and hypercalcemic milieu than the vascular niche, which may also contribute to stem cell niche interactions [14,225,250-252] (Figure 1). Several different adhesion molecules, like hyaluronic acid, Jagged, N-cadherin, osteopontin, CAMs, VEGF, SCF, or SDF-1, are considered to contribute to stem cell homing in the niche [225,247-249]. Normal and neoplastic stem cells express receptors for these stromal ligand receptors (Figure 1).
Role of hypoxia
Hypoxia and hypoxia-inducible factors (HIF) may influence the fate and self-renewal capacity of stem cells in the micro-milieu of the stem cell niche in health and disease [14,225,250-254]. So far, little is known about the mechanisms through which hypoxia regulates self-renewal and proliferation of CSC. One important aspect may be that hypoxia upregulates not only HIF expression but also several angiogenic and growth-regulatory cytokines, such as SDF-1 (CXCR4) or VEGF [250,255,256]. These cytokines may promote tumor-associated angiogenesis. It has also been described that hypoxia maintains a more stem cell-like state of progenitor cells in the BM by regulating key signaling pathways responsible for stem cell growth and survival, such as Notch or Oct4 [253,254,257,258]. This may also hold true for CSC/LSC in hypoxic areas in the centers of solid tumors [259]. Another important aspect is that hypoxia can trigger the production of reactive oxygen species (ROS) in neoplastic (stem) cells, which in turn leads to DNA breaks and thereby increases mutagenesis and thus the generation of more malignant subclones [260,261]. Thus, hypoxia may be a trigger of oncogenesis and malignant progression as well as CSC/LSC resistance [262-264].
Plasticity and subclone formation of NSC (CSC/LSC)
A remarkable aspect in the biology of neoplastic stem cells is plasticity and subclone formation during disease evolution which is relevant clinically as subclone formation is often associated with progression and drug resistance. Recent data suggest that in AML and CML, subclone formation is an early and frequent event in LSC development, and the same may hold true for other neoplasms, including solid tumors [26,54,128,129,131,206,220,265]. Plasticity is best explained by genetic instability. The excessive plasticity and subsequent formation of neoplastic subclones is somehow contradictory to the hypothesis that many (at least premalignant) NSC are quiescent cells. However, subclone formation is now considered to be a step-wise and long-lasting process, which may explain the formation of multiple CSC subclones with varying proliferative capacity (Figure 2) [28,48,54,128]. Subclone formation and plasticity of LSC in CML may also be associated with lineage commitment and differentiation or even a lineage switch. One good example is lymphoid or biphenotypic (mixed) blast crisis in Ph + CML [266-269]. In rare cases, subclone formation from LSC is excessive and may result in the development of two histologically unrelated but still monoclonal neoplasms [270-272]. Finally, it has also been reported that some of the hematopoietic neoplasms produce their own (clonal) microenvironment [273-275]. A related observation is ‘vasculogenic mimicry’ that involves the so-called ‘malignant stromal cells’ or ‘malignant endothelial cells.’ Such stromal cell progenitors have recently been detected in several malignancies, including AML [276]. All these observations suggest that the leukemia-associated microenvironment, including the LSC niche, is a new emerging target of therapy.
Figure 2.
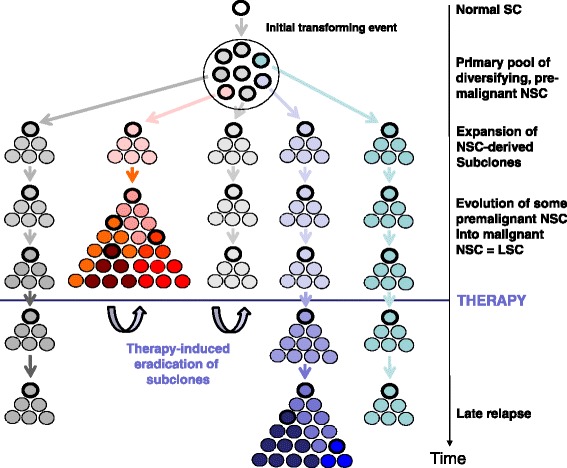
Subclone formation of CSC during evolution of a malignancy. During cancer/leukemia evolution, a large number of different subclones with varying combinations of mutational lesions develop. Each change in color is indicative of the acquisition of a relevant new molecular lesion. After a certain time, one or more malignant (dominant) subclones expand and develop into an overt malignancy. However, at the time of diagnosis of a cancer/leukemia, all the other premalignant subclones and their stem cells are also still present. Neoplastic stem cells are indicated by bold circles. After intensive therapy, many or most (sometimes all) of the cancer/leukemic stem cells may have been eradicated. However, the less malignant (pre-malignant) neoplastic stem cells may still survive (because of their quiescence and other resistance-related mechanisms) and may later expand and produce a relapse. Such late relapses may not necessarily express the same oncogenic lesions (driver mutations) compared to the original subclone but still are derived from the same initial stem cell clone. Today, the subclonal architecture is demonstrable by deep sequencing technologies in various malignancies.
Expression of molecular targets in NSC/CSC
An essential question in CSC research is whether certain therapeutic targets are expressed in or on CSC. Notably, targeting of CSC using drugs that can kill or permanently suppress these cells may be a pre-requisite for the development of new curative treatment approaches in cancers and leukemias [7,11,14,16,28]. However, unfortunately, in many instances, CSC and normal stem cells share the same target antigens [64]. As a result, CSC-targeting therapies often result in the occurrence of substantial adverse side effects such as prolonged cytopenia. In this regard, it is noteworthy that the only available curative drug-therapy in AML, which is polychemotherapy, is usually also associated with prolonged cytopenia. Therefore, current research is seeking novel markers and targets that are preferentially or even selectively expressed on CSC (LSC) but are not expressed (or less abundantly expressed) by normal stem cells [67,84,87].
Examples for surface markers/targets that have been described to be expressed primarily on LSC in myeloid leukemias, but less abundantly (or not at all) on normal stem cells, are CD25, CD26, CD33, CD47, CD52, CD96, CD123, IL-1RAP, and CLL-1 [65-69,72,73,85,277-280]. With regard to CD33 and CD52, clinically established targeting concepts are available [280-282]. Likewise, as assessed by in vitro and in vivo experiments, the CD52-targeting antibody alemtuzumab is able to kill LSC in AML and MDS [77]. Figure 3 shows the effect of alemtuzumab on AML LSC in vitro. However, unfortunately, normal stem cells also express low but detectable amounts of these surface antigens, and the respective drugs, gemtuzumab ozogamicin (GO, anti-CD33) and alemtuzumab (anti-CD52) have recently been removed from the oncologic market because of their toxicity profiles which may indeed result in part from their effects on normal stem cells [280-282]. There are also other antibody-based targeted drugs that are currently being developed, such as (among others) CLL-1, IL-1RAP, CD44, CD96, or CD123. The value of these agents is currently being tested preclinically and in clinical trials [283].
Figure 3.
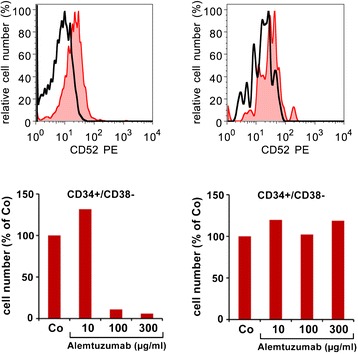
Leukemic stem cells express the cell surface target antigen CD52. Upper panels: bone marrow (BM) cells obtained from a patient with acute myeloid leukemia (AML; left panel) or control BM (right panel) cells were stained with antibodies against CD34, CD38, and CD52. The immature CD34+/CD38− stem cells were found to co-express CD52 (red histogram) in the patient with AML but did not express CD52 in the normal BM. The black open histogram represents the isotype-matched control antibody. Lower panels: BM cells were incubated in various concentrations of the CD52-targeted antibody alemtuzumab at 37°C for 1 h. Thereafter, the numbers of viable CD34+/CD38− stem cells were counted by flow cytometry using calibration beads. As visible, exposure to alemtuzumab resulted in a dose-dependent decrease in AML stem cells (left panel) but did not result in a decrease of normal BM stem cells (right panel).
A number of different signaling molecules and survival molecules have been identified as potential targets in LSC/CSC. Among these are the PI3K, mTOR, MEK, Smoothened, Notch, Wnt, heat shock proteins, and Bcl-2 family members. Table 5 provides an overview of molecular targets expressed in CSC and LSC in various malignancies. During the past few years, several potent targeted drugs directed against the primary dominant oncoproteins of various tumors and leukemias have been developed. An interesting example is CML, where BCR/ABL blockers are applied successfully to suppress the growth and expansion of LSC [203]. However, even BCR/ABL TKI may not be capable of suppressing all LSC for a prolonged time period, because of stem cell resistance [11,19-23,204]. Nevertheless, the effects of BCR/ABL TKI in CML are a highlighting example of LSC suppression. Notably, in many patients in whom TKI treatment has led to a complete continuous molecular response, treatment discontinuation can be performed, and only a subset of these patients relapse whereas others remain BCR/ABL-negative over years, suggesting that many (clinically relevant) LSC had been eradicated [284].
Table 5.
Molecular targets detectable in neoplastic stem cells (NSC)
| Target type | Molecular target example | Potentially relevant as NSC-target in | Targeted drug example |
|---|---|---|---|
| Surface antigens | CD20 | ALL, CLL | Rituximab [285,286] |
| CD33 | CML, AML | GO [85,282] | |
| CD44 | AML | mAb [72] | |
| CD52 | 5q-AML, CLL | Alemtuzumab [77,287] | |
| CD123 | AML | mAb [69] | |
| EGFR | Colon-Ca | Cetuximab [288] | |
| ERBB2 | Breast/Gastric/ | Trastuzumab [172] | |
| Ovarian-Ca | |||
| Cytokine receptors | KIT | GIST, CML | Imatinib [203,289] |
| PDGFRA | CEL, GIST | Imatinib | |
| EGFR | Pancreas-Ca | Erlotinib [182] | |
| ERBB2 | Breast-Ca | Lapatinib[290] | |
| Signaling molecules | Hedgehog | Basal cell carcinoma | Vismodegib [291] |
| BRAF | Melanoma | Vemurafenib [292] | |
| BTK | CLL | Ibrutinib [293] | |
| mTOR | Glioblastoma, | Temsirolimus [294,295] | |
| Renal cell carcinoma | |||
| Transcription factors | MYC | AML | JQ1 [296] |
| Niche-NSC-axis | CD26/DPPIV | CML | Gliptins [84] |
| CD184/CXCR4- | Plerixafor [159] | ||
| VEGF-VEGFR | - | Bevacizumab [297] | |
| SCF-KIT-axis- | Imatinib [203] |
Abbreviations: ALL, acute lymphoblastic leukemia; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; AML, acute myeloid leukemia; mAb, monoclonal antibodies, EGFR, epidermal growth factor receptor; Ca, carcinoma; GIST, gastrointestinal stroma cell tumor; CEL, chronic eosinophilic leukemia; PDGFR, platelet derived growth factor receptor; BTK, Bruton’s tyrosine kinase; mTOR, mammalian target of rapamycin; MDR-1, multidrug-resistance protein 1; CSA, cyclosporine A; DPPIV, dipeptidyl-peptidase IV; VEGF, vascular endothelial growth factor; SCF, stem cell factor.
Intrinsic and acquired resistance of NSC/CSC
Normal and neoplastic stem cells benefit from several repair mechanisms and defense systems through which these cells can escape or survive various stress reactions, toxin-exposure, or microbial attacks, and the same mechanisms are responsible for drug resistance [11,19-23,28,298,299]. In the context of neoplastic stem cells, intrinsic forms and acquired forms of resistance have been described. Intrinsic resistance is usually detectable in all CSC populations (subclones), including premalignant NSC and CSC/LSC, whereas acquired resistance is usually found in newly generated, more malignant, subclones and their (subclone-specific) CSC/LSC in advanced neoplasms [11,19-23,298,299].
The mechanisms underlying intrinsic resistance of LSC/CSC are poorly understood. In most neoplasms, multiple factors and mechanisms may act together to produce intrinsic resistance. One factor may be stem cell quiescence [11,19-23,298,299]. Another important factor are cytokine interactions and cell-cell interactions in the CSC niche [14,19-23,28,54]. Moreover, certain drug transporters are expressed differentially in CSC/LSC when compared to more mature neoplastic cells [300-304]. These transporters may mediate drug uptake (such as OCT-1, a drug transporter for Imatinib) but may also contribute to enhanced drug efflux from CSC/LSC. Likewise, in advanced leukemias, LSC often express MDR-1 and probably other drug efflux transporters [22,300-305]. Similar drug transporters have also been identified in solid tumors and in solid tumor CSC. Other mechanisms underlying intrinsic resistance of LSC/CSC may be an abnormal expression or upregulation of survival-related (stress) molecules (often after drug exposure), abnormal expression of signaling molecules or transcription factors, and the lack or loss of tumor suppressor genes or death regulators [11,19-23,28,41,306-312] (Table 6). In addition, the local organ-specific microenvironment, tissue hypoxia, and the interaction with the ‘CSC niche’ may contribute to the resistance of CSC/LSC [11,19-23,28,45,313].
Table 6.
Mechanisms of drug resistance in NSC and strategies to overcome resistance
| Mechanism | Proposed strategy to overcame resistance | Examples |
|---|---|---|
| NSC quiescence | Antibody-based killing of NSC | CD20, CD33, CD52, |
| Bi-specific mAb [77,280,286] | ||
| Mobilization of the immune system against NSC | Vaccination [314], | |
| IL-2 + histamine [315] | ||
| NSC mobilization into the cell cycle | Cytokine-priming [316] | |
| CTLA-4 inhibition | Ipilimumab [317] | |
| PD1 inhibition | Nivolumab [318] | |
| NSC-niche interactions | NSC mobilization out of the niche | Plerixafor [159] |
| Redirection of NSC into the niche | Gliptins (CML [84]) | |
| Targeting of niche cells | Revlimid [319] | |
| Targeting of Niche modulating-cytokines (for example, VEGF) or | Avastin [320] | |
| Cytokine (for example, VEGF) synthesis | Rapamycin [295] | |
| Enforced drug efflux Verapamil [321,322] | Blocking the efflux pumps | CSA |
| Expression of anti-apoptotic proteins | Blocking BCL-2 family members | Obatoclax [323] |
| Blocking heat shock proteins (Hsp) | Hsp70, Hsp90 [324] |
NSC, neoplastic stem cells; IL-2, interleukin-2; CSA, cyclosporine A; CML, chronic myeloid leukemia.
A number of different mechanisms may underlie acquired drug resistance in CSC/LSC. One is genetic instability and the ‘mutation capacity’ of the malignant genome, resulting in a plethora of mutations in critical target genes that can be detected in (more) malignant subclones in these patients [28,54,78,129,206,207,325]. These mutations may occur in an early phase (or even prephase) of the disease. They may develop in most, many, or only a few subclones and may either be detectable at diagnosis (prominent subclone/s) or they remain undetectable for a longer time period because they develop in slowly cycling NSC that are only be capable of generating small-sized subclones [26,28,54,128,325]. Nevertheless, as soon as these small-sized subclones acquire a sufficient number of additional hits (mutations), they can expand and develop into an overt disease in which neoplastic cells and CSC exhibit acquired resistance [26,28,54,128,325]. The use of targeted drugs must lead to a selection of these more malignant subclones over time. Mutations leading to drug resistance may occur in a number of different genes. Likewise, mutations in various tyrosine kinases may contribute to resistance against oncoprotein-targeting drugs [326-328]. The best studied model is CML, where multiple mutations in the BCR/ABL kinase domains have been identified in Imatinib-treated patients [326-328]. Such mutations have been detected in virtually all oncogenic kinases that play a key role in human leukemogenesis or myeloproliferation and also in most other tumor models [329].
Other mechanisms of acquired resistance include the amplifications of target genes (overexpressed targets) or activation of additional pro-oncogenic molecules (Table 5) [330-334]. These types of resistance are usually associated with a poor prognosis and are often accompanied by cytogenetic evidence of clonal evolution. Likewise, in CML and AML as well as in MDS, a complex karyotype usually indicates an unfavorable prognosis [334-336].
Can we translate the CSC concept into clinical practice?
Most of the conventional anti-cancer agents currently used in daily practice or in clinical trials are primarily acting on rapidly dividing cells that make up the bulk of the tumor, whereas most CSC (and premalignant NSC) are not affected. High-dose chemotherapy and novel targeted drugs may be able to eliminate the bulk of the neoplasm and to eradicate most CSC (or LSC) in a given tumor or leukemia. These debulking agents are still very useful and instrumental in anti-cancer therapy. However, relapses may develop from a few residual, drug-resistant, premalignant (quiescent) NSC that exhibit intrinsic stem cell resistance. Notably, even if all CSC/LSC can be eradicated by drug therapy, (late) relapses can develop from such residual, mostly quiescent premalignant NSC [26,28,54,128,131,220,325]. In other words, many new drug therapies can eliminate the mass of CSC/LSC that have generated the dominant clone but are unable to eradicate all quiescent premalignant NSC forming smaller subclones [26,54,128,284]. These drugs may even lead to operational cures without having the potential to eradicate the disease completely [128,284]. The question is how relevant the residual (often quiescent) NSC are in these patients. Notably, not all types of MRD and MRD-specific NSC may be relevant clinically, even if they may expand to another dominant clone [28,54,128]. Likewise, in hairy cell leukemia, cladribine (2CdA) may not be able to eradicate all LSC, and most premalignant NSC may survive. However, because of the relatively slow growth rate and low mutation rate of NSC, full blown relapses are relatively uncommon; and if they occur (typically after 3 to 5 years), leukemic cells are again responsive to the same drug. By contrast, in AML, the mutation rate is high and relapses are always indicative of a poor outcome and are often associated with multidrug resistance. The same holds true for most solid tumors. In CML, several novel TKI may induce complete continuous molecular remissions (CMR) [337,338]. Even imatinib can induce long-term CMR in a smaller fraction of patients [284]. When TKI are discontinued in these patients, some of them will relapse but may again respond to imatinib or other new TKI [284]. The exact curative potential of imatinib and of the new TKI in CML remains unknown. In solid tumor, novel TKI have also been applied in clinical trials and some of these agents are rather promising. However, long-term remissions are usually not induced with these agents even when combined with chemotherapy. Overall, with a few exceptions, in most advanced solid tumors, no drug-based CSC-eliminating treatment approach has been developed so far. However, there are several examples where targeted drugs as single agents may lead to long-term disease control. One example is the gastrointestinal stroma cell tumors (GIST), where TKI have shown encouraging results [339-341]. Another example is renal cell carcinoma, where inhibitors of the PI3K-mTOR pathway have shown to exert major anti-tumor effects [342,343].
A general problem in cancer evolution is that many CSC/LSC and most or all premalignant NSC may be dormant cells, and that dormancy is often associated with intrinsic resistance. One possible way to overcome this type of resistance may be to apply targeted antibodies, especially antibody-toxin conjugates which often act independent of the cell cycle and thus can destroy even dormant NSC. Likewise, in several types of lymphomas, the addition of pan-B-cell-targeting antibodies has substantially improved cure rates and the overall outcome (survival) in these patients [287,344,345]. An alternative strategy is to mobilize dormant cells into the cell cycle or out of the niche (where dormancy may be propagated) [159,346]. Finally, dormancy of NSC/CSC may be overcome by exposure to cytokines that promote cell cycle progression in NSC/CSC. Another principal strategy may be to promote CSC/LSC exhaustion by inducing differentiation and maturation in these cells or by mobilizing the immune system against CSC/LSC.
A major problem is that in advanced cancer lesions, CSC not only exhibit intrinsic (natural) stem cell resistance but often also acquired drug resistance in more resistant and thus more malignant subclones [28,54,128]. One strategy to address the multiple mechanisms of resistance accumulating in advanced tumor lesion is to apply drug combinations. Another strategy is to combine conventional or targeted drugs with response modifiers or agents that mobilize tumor cells into the cell cycle. An alternative approach is to select targeted drugs that can overcome acquired drug resistance resulting from point mutations in critical target genes. Likewise, in CML, second-generation BCR/ABL TKI can often overcome imatinib resistance associated with BCR/ABL mutations [338,347,348].
Another aspect in CSC/LSC evolution is that resistance of CSC/LSC is often associated with specific interactions between these cells and CSC niche. One strategy to overcome this form of resistance is to mobilize CSC/LSC from the niche where stem cells are considered to be protected and thus less accessible to targeted drugs. One example is the SDF-1/CXCR4 axis that can be disrupted by the CXCR4 blocking agent Plerixafor [349,350]. Recent data suggest that Plerixafor cannot only mobilize normal hematopoietic stem cells from the bone marrow stem cell niche but also LSC and that Plerixafor-mobilized LSC may be more sensitive against certain anti-leukemic drugs [159,346]. However, it remains unknown whether all LSC can be mobilized by Plerixafor, whether the mobilization is associated with a rebound of more rapidly growing LSC in the niche and whether addition of Plerixafor to conventional chemotherapy will indeed increase response and cure rates in patients with AML or other leukemias. In addition, more recent data suggest that in certain forms of leukemias (CML), LSC are already mobilized cells that can easily traffic between niches [84].
Recent data suggest that the vascular (arteriolar) stem cell niche is of particular importance for self-renewal of LSC [225,247-249]. Therefore, additional effects of targeted drugs on vascular cells or drug combinations employing anti-angiogenic agents are of considerable interest. One of these anti-angiogenic drugs is Lenalidomide, a major anti-angiogenic drug that produces major responses in various myeloid and lymphoid neoplasms, including 5q-MDS, multiple myeloma, and chronic lymphocytic leukemia. Several other anti-angiogenic drugs have been tested in hematopoietic and solid malignancies, with varying success. Encouraging results have been obtained when combining these agents with other anti-neoplastic agents. A remarkable observation is that the novel BCR/ABL TKI Nilotinib and Ponatinib (but not imatinib) are potent inhibitors of endothelial growth and angiogenesis [351]. Whether these additional effects of these TKI are responsible for their better efficacy in CML remains unknown. This is an attractive hypothesis, since their much stronger effects on BCR/ABL (when compared to imatinib) fail to explain their excellent clinical efficacy, as CML LSC are considered to survive independent of BCR/ABL [352,353]. Table 6 provides strategies aimed at overcoming LSC resistance in human malignancies.
Summary and future perspectives
During the past few years several CSC/LSC-targeting concepts have been developed with the aim to establish more effective treatment approaches in applied oncology. However, although such novel treatment concepts are straight-forward, several questions remain. First, the complexity of the somatic aberration networks and of the resulting signaling cascades that drive oncogenesis during CSC/LSC evolution and may lead to CSC/LSC resistance. To address this aspect, the use of drug combinations or broadly acting drugs has been suggested and may be required to eliminate or suppress all relevant CSC/LSC populations in a given neoplasm. Novel treatment concepts have to take additional aspects into account, including intrinsic resistance, the related issue of CSC/LSC quiescence, and the interaction of CSC/LSC with their organ- and disease-specific microenvironment (CSC niche). Additional local factors such as hypoxia may also play a role in CSC/LSC resistance. The hope for the future is that we will be able to exploit our increasing knowledge about CSC/LSC, in order to define new treatment concepts, with the ultimate aim to eradicate CSC/LSC in various cancer types as well as in leukemias. In advanced, drug-resistant neoplasms, such treatment concepts may need to be combined with high-dose chemotherapy and/or stem cell transplantation.
Funding
This study was supported by the Austrian Science Funds, Project SFB-F4704-B20#, a cancer stem cell grant of the Medical University of Vienna and a research grant of the Initiative Krebsforschung Vienna, Austria.
Abbreviations
- BM
bone marrow
- CD
cluster of differentiation
- CML
chronic myeloid leukemia
- CSC
cancer stem cell(s)
- CXCR-4
C-X-C chemokine receptor type 4
- EMT
epithelial-mesenchymal transition
- FGF
fibroblast growth factor
- G-CSF
granulocyte colony-stimulating factor
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- Her 2
human epidermal growth factor receptor 2
- IL
interleukin
- LSC
leukemic stem cell(s)
- NOD/SCID
non-obese diabetic mice with severe combined immunodeficiency
- NOG
IL-2Rgamma chain-knock out NOD/shi-SCID mice
- NSC
neoplastic stem cell(s)
- NSG
NOD/SCID with loss-of-function-mutated IL-2Rgamma chain
- PI3K
phosphatidylinositide-3 kinase
- SCF
stem cell factor
- SDF-1
stromal cell-derived factor 1
- TKI
tyrosine kinase inhibitor(s)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AS, BM, TWG, CCZ, and PV drafted the manuscript. KB, SC-R, IS, and HH helped to design the draft and provided figures and tables. All authors read and approved the final manuscript.
Contributor Information
Axel Schulenburg, Email: axel.schulenburg@meduniwien.ac.at.
Katharina Blatt, Email: katharina.blatt@meduniwien.ac.at.
Sabine Cerny-Reiterer, Email: sabine.cerny-reiterer@meduniwien.ac.at.
Irina Sadovnik, Email: irina.sadovnik@meduniwien.ac.at.
Harald Herrmann, Email: harald.herrmann@meduniwien.ac.at.
Brigitte Marian, Email: brigitte.marian@meduniwien.ac.at.
Thomas W Grunt, Email: thomas.grunt@meduniwien.ac.at.
Christoph C Zielinski, Email: christoph.zielinski@meduniwien.ac.at.
Peter Valent, Email: peter.valent@meduniwien.ac.at.
References
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007;18:460–6. doi: 10.1016/j.copbio.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–84. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 4.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 5.Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793–807. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]
- 6.Cho RW, Clarke MF. Recent advances in cancer stem cells. Curr Opin Genet Dev. 2008;18:48–53. doi: 10.1016/j.gde.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 7.Schatton T, Frank NY, Frank MH. Identification and targeting of cancer stem cells. Bioessays. 2009;31:1038–49. doi: 10.1002/bies.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulenburg A, Bramswig K, Herrmann H, Karlic H, Mirkina I, Hubmann R, et al. Neoplastic stem cells: current concepts and clinical perspectives. Crit Rev Oncol Hematol. 2010;76:79–98. doi: 10.1016/j.critrevonc.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Greaves M. Cancer stem cells: back to Darwin? Semin Cancer Biol. 2010;20(2):65–70. doi: 10.1016/j.semcancer.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells - perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 11.Valent P. Emerging stem cell concepts for imatinib-resistant chronic myeloid leukaemia: implications for the biology, management, and therapy of the disease. Br J Haematol. 2008;142(3):361–78. doi: 10.1111/j.1365-2141.2008.07197.x. [DOI] [PubMed] [Google Scholar]
- 12.Dick JE, Lapidot T. Biology of normal and acute myeloid leukemia stem cells. Int J Hematol. 2005;82:389–96. doi: 10.1532/IJH97.05144. [DOI] [PubMed] [Google Scholar]
- 13.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–43. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 14.Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol. 2011;29:591–9. doi: 10.1200/JCO.2010.31.0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sloma I, Jiang X, Eaves AC, Eaves CJ. Insights into the stem cells of chronic myeloid leukemia. Leukemia. 2010;24:1823–33. doi: 10.1038/leu.2010.159. [DOI] [PubMed] [Google Scholar]
- 16.Roboz GJ, Guzman M. Acute myeloid leukemia stem cells: seek and destroy. Expert Rev Hematol. 2009;2:663–72. doi: 10.1586/ehm.09.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holyoake TL, Jiang X, Jorgensen HG, Graham S, Alcorn MJ, Laird C, et al. Primitive quiescent leukemic cells from patients with chronic myeloid leukemia spontaneously initiate factor-independent growth in vitro in association with up-regulation of expression of interleukin-3. Blood. 2001;97:720–8. doi: 10.1182/blood.v97.3.720. [DOI] [PubMed] [Google Scholar]
- 18.Copland M. Chronic myelogenous leukemia stem cells: what’s new? Curr Hematol Malig Rep. 2009;4:66–73. doi: 10.1007/s11899-009-0010-9. [DOI] [PubMed] [Google Scholar]
- 19.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–51. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872–7. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- 21.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–84. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 22.Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21:926–35. doi: 10.1038/sj.leu.2404609. [DOI] [PubMed] [Google Scholar]
- 23.Barnes DJ, Melo JV. Primitive, quiescent and difficult to kill: the role of non-proliferating stem cells in chronic myeloid leukemia. Cell Cycle. 2006;5:2862–6. doi: 10.4161/cc.5.24.3573. [DOI] [PubMed] [Google Scholar]
- 24.Welch JS. Mutation position within evolutionary subclonal architecture in AML. Semin Hematol. 2014;51:273–83. doi: 10.1053/j.seminhematol.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Wang M, Wang Y, Zhong J. Side population cells and drug resistance in breast cancer. Mol Med Rep. 2015;doi:10.3892/mmr.2015.3291. [DOI] [PubMed]
- 26.Preuner S, Mitterbauer G, Mannhalter C, Herndlhofer S, Sperr WR, Valent P, et al. Quantitative monitoring of BCR/ABL1 mutants for surveillance of subclone-evolution, -expansion, and -depletion in chronic myeloid leukaemia. Eur J Cancer. 2012;48:233–6. doi: 10.1016/j.ejca.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 27.Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–9. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valent P. Targeting of leukemia-initiating cells to develop curative drug therapies: straightforward but nontrivial concept. Curr Cancer Drug Targets. 2011;11(1):56–71. doi: 10.2174/156800911793743655. [DOI] [PubMed] [Google Scholar]
- 29.Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. 2010;120:41–50. doi: 10.1172/JCI41004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Besancon R, Valsesia-Wittmann S, Puisieux A, de Fromentel CC, Maguer-Satta V. Cancer stem cells: the emerging challenge of drug targeting. Curr Med Chem. 2009;16:394–416. doi: 10.2174/092986709787315531. [DOI] [PubMed] [Google Scholar]
- 31.Korkaya H, Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. BioDrugs. 2007;21:299–310. doi: 10.2165/00063030-200721050-00002. [DOI] [PubMed] [Google Scholar]
- 32.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 33.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 34.Civenni G, Walter A, Kobert N, Mihic-Probst D, Zipser M, Belloni B, et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011;71:3098–109. doi: 10.1158/0008-5472.CAN-10-3997. [DOI] [PubMed] [Google Scholar]
- 35.Pearson T, Greiner DL, Shultz LD. Creation of “humanized” mice to study human immunity. Curr Protoc Immunol. 2008;15(15):21. doi: 10.1002/0471142735.im1521s81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishikawa F, Saito Y, Yoshida S, Harada M, Shultz LD. The differentiative and regenerative properties of human hematopoietic stem/progenitor cells in NOD-SCID/IL2rgamma(null) mice. Curr Top Microbiol Immunol. 2008;324:87–94. doi: 10.1007/978-3-540-75647-7_5. [DOI] [PubMed] [Google Scholar]
- 37.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–8. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–75. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- 39.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 40.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 42.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 43.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 44.Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–66. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 45.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–21. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 46.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 47.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–5. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 48.Bohm A, Herrmann H, Mitterbauer-Hohendanner G, Hauswirth AW, Rabitsch W, Mitterbauer M, et al. Stable non-transforming minimal residual disease in Philadelphia chromosome positive acute lymphoblastic leukemia after autologous transplantation: origin from neoplastic yet ‘pre-leukemic’ stem cells? Leuk Lymphoma. 2011;52:842–8. doi: 10.3109/10428194.2011.557168. [DOI] [PubMed] [Google Scholar]
- 49.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–70. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–37. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 51.Dexter TM, Allen TD, Lajtha LG. Conditions controlling the proliferation of haemopoietic stem cells in vitro. J Cell Physiol. 1977;91:335–44. doi: 10.1002/jcp.1040910303. [DOI] [PubMed] [Google Scholar]
- 52.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–83. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vermeulen L, Todaro M, de Sousa MF, Sprick MR, Kemper K, Perez Alea M, et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci U S A. 2008;105:13427–32. doi: 10.1073/pnas.0805706105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerashchenko BI, Huna A, Erenpreisa J. Characterization of breast cancer DNA content profiles as a prognostic tool. Exp Oncol. 2014;36:219–25. [PubMed] [Google Scholar]
- 55.Moserle L, Ghisi M, Amadori A, Indraccolo S. Side population and cancer stem cells: therapeutic implications. Cancer Lett. 2010;288:1–9. doi: 10.1016/j.canlet.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 56.Fukaya R, Ohta S, Yamaguchi M, Fujii H, Kawakami Y, Kawase T, et al. Isolation of cancer stem-like cells from a side population of a human glioblastoma cell line, SK-MG-1. Cancer Lett. 2010;291:150–7. doi: 10.1016/j.canlet.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 57.Murase M, Kano M, Tsukahara T, Takahashi A, Torigoe T, Kawaguchi S, et al. Side population cells have the characteristics of cancer stem-like cells/cancer-initiating cells in bone sarcomas. Br J Cancer. 2009;101:1425–32. doi: 10.1038/sj.bjc.6605330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li T, Su Y, Mei Y, Leng Q, Leng B, Liu Z, et al. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab Invest. 2010;90:234–44. doi: 10.1038/labinvest.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ran D, Schubert M, Pietsch L, Taubert I, Wuchter P, Eckstein V, et al. Aldehyde dehydrogenase activity among primary leukemia cells is associated with stem cell features and correlates with adverse clinical outcomes. Exp Hematol. 2009;37:1423–34. doi: 10.1016/j.exphem.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 60.Chen YC, Chen YW, Hsu HS, Tseng LM, Huang PI, Lu KH, et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem Biophys Res Commun. 2009;385:307–13. doi: 10.1016/j.bbrc.2009.05.048. [DOI] [PubMed] [Google Scholar]
- 61.Moreb JS. Aldehyde dehydrogenase as a marker for stem cells. Curr Stem Cell Res Ther. 2008;3:237–46. doi: 10.2174/157488808786734006. [DOI] [PubMed] [Google Scholar]
- 62.Polyak K, Hahn WC. Roots and stems: stem cells in cancer. Nat Med. 2006;12:296–300. doi: 10.1038/nm1379. [DOI] [PubMed] [Google Scholar]
- 63.Weissman IL, Shizuru JA. The origins of the identification and isolation of hematopoietic stem cells, and their capability to induce donor-specific transplantation tolerance and treat autoimmune diseases. Blood. 2008;112:3543–53. doi: 10.1182/blood-2008-08-078220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Florian S, Sonneck K, Hauswirth AW, Krauth MT, Schernthaner GH, Sperr WR, et al. Detection of molecular targets on the surface of CD34+/CD38– stem cells in various myeloid malignancies. Leuk Lymphoma. 2006;47:207–22. doi: 10.1080/10428190500272507. [DOI] [PubMed] [Google Scholar]
- 65.Moshaver B, van Rhenen A, Kelder A, van der Pol M, Terwijn M, Bachas C, et al. Identification of a small subpopulation of candidate leukemia initiating cells in the side population (SP) of patients with acute myeloid leukemia. Stem Cells. 2008;26(12):3059–67. doi: 10.1634/stemcells.2007-0861. [DOI] [PubMed] [Google Scholar]
- 66.Blair A, Hogge DE, Ailles LE, Lansdorp PM, Sutherland HJ. Lack of expression of Thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood. 1997;89:3104–12. [PubMed] [Google Scholar]
- 67.van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B, et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110:2659–66. doi: 10.1182/blood-2007-03-083048. [DOI] [PubMed] [Google Scholar]
- 68.Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A. 2007;104:11008–13. doi: 10.1073/pnas.0704271104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14:1777–84. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 70.Fina L, Molgaard HV, Robertson D, Bradley NJ, Monaghan P, Delia D, et al. Expression of the CD34 gene in vascular endothelial cells. Blood. 1990;75:2417–26. [PubMed] [Google Scholar]
- 71.Kitamura Y, Hirotab S. Kit as a human oncogenic tyrosine kinase. Cell Mol Life Sci. 2004;61:2924–31. doi: 10.1007/s00018-004-4273-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–74. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 73.Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, Jr, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–99. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim JB, Ko E, Han W, Lee JE, Lee KM, Shin I, et al. CD24 cross-linking induces apoptosis in, and inhibits migration of, MCF-7 breast cancer cells. BMC Cancer. 2008;8:118. doi: 10.1186/1471-2407-8-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med. 2010;2:17ra19. doi: 10.1126/scitranslmed.3000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106:4086–92. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blatt K, Herrmann H, Hoermann G, Willmann M, Cerny-Reiterer S, Sadovnik I, et al. Identification of campath-1 (CD52) as novel drug target in neoplastic stem cells in 5q-patients with MDS and AML. Clin Cancer Res. 2014;20(13):3589–602. doi: 10.1158/1078-0432.CCR-13-2811. [DOI] [PubMed] [Google Scholar]
- 78.Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(−) fraction. Blood. 2010;115:1976–84. doi: 10.1182/blood-2009-02-206565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nilsson L, Astrand-Grundstrom I, Anderson K, Arvidsson I, Hokland P, Bryder D, et al. Involvement and functional impairment of the CD34(+)CD38(−)Thy-1(+) hematopoietic stem cell pool in myelodysplastic syndromes with trisomy 8. Blood. 2002;100:259–67. doi: 10.1182/blood-2001-12-0188. [DOI] [PubMed] [Google Scholar]
- 80.De Smet D, Trullemans F, Jochmans K, Renmans W, Smet L, Heylen O, et al. Diagnostic potential of CD34+ cell antigen expression in myelodysplastic syndromes. Am J Clin Pathol. 2012;138:732–43. doi: 10.1309/AJCPAGVO27RPTOTV. [DOI] [PubMed] [Google Scholar]
- 81.Nilsson L, Astrand-Grundstrom I, Arvidsson I, Jacobsson B, Hellstrom-Lindberg E, Hast R, et al. Isolation and characterization of hematopoietic progenitor/stem cells in 5q-deleted myelodysplastic syndromes: evidence for involvement at the hematopoietic stem cell level. Blood. 2000;96:2012–21. [PubMed] [Google Scholar]
- 82.Jamieson CH, Gotlib J, Durocher JA, Chao MP, Mariappan MR, Lay M, et al. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci U S A. 2006;103:6224–9. doi: 10.1073/pnas.0601462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kobayashi CI, Takubo K, Kobayashi H, Nakamura-Ishizu A, Honda H, Kataoka K, et al. The IL-2/CD25 axis maintains distinct subsets of chronic myeloid leukemia-initiating cells. Blood. 2014;123:2540–9. doi: 10.1182/blood-2013-07-517847. [DOI] [PubMed] [Google Scholar]
- 84.Herrmann H, Sadovnik I, Cerny-Reiterer S, Rulicke T, Stefanzl G, Willmann M, et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood. 2014;123(25):3951–62. doi: 10.1182/blood-2013-10-536078. [DOI] [PubMed] [Google Scholar]
- 85.Herrmann H, Cerny-Reiterer S, Gleixner KV, Blatt K, Herndlhofer S, Rabitsch W, et al. CD34(+)/CD38(−) stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-targeting drug gemtuzumab/ozogamicin. Haematologica. 2012;97:219–26. doi: 10.3324/haematol.2010.035006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nievergall E, Ramshaw HS, Yong AS, Biondo M, Busfield SJ, Vairo G, et al. Monoclonal antibody targeting of IL-3 receptor alpha with CSL362 effectively depletes CML progenitor and stem cells. Blood. 2014;123:1218–28. doi: 10.1182/blood-2012-12-475194. [DOI] [PubMed] [Google Scholar]
- 87.Jaras M, Johnels P, Hansen N, Agerstam H, Tsapogas P, Rissler M, et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc Natl Acad Sci U S A. 2010;107:16280–5. doi: 10.1073/pnas.1004408107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cobaleda C, Gutierrez-Cianca N, Perez-Losada J, Flores T, Garcia-Sanz R, Gonzalez M, et al. A primitive hematopoietic cell is the target for the leukemic transformation in human philadelphia-positive acute lymphoblastic leukemia. Blood. 2000;95:1007–13. [PubMed] [Google Scholar]
- 89.Kong Y, Yoshida S, Saito Y, Doi T, Nagatoshi Y, Fukata M, et al. CD34 + CD38 + CD19+ as well as CD34 + CD38-CD19+ cells are leukemia-initiating cells with self-renewal capacity in human B-precursor ALL. Leukemia. 2008;22:1207–13. doi: 10.1038/leu.2008.83. [DOI] [PubMed] [Google Scholar]
- 90.Rossi FM, Zucchetto A, Tissino E, Dal Bo M, Bomben R, Caldana C, et al. CD49d expression identifies a chronic-lymphocytic leukemia subset with high levels of mobilized circulating CD34(+) hemopoietic progenitors cells. Leukemia. 2014;28:705–8. doi: 10.1038/leu.2013.331. [DOI] [PubMed] [Google Scholar]
- 91.Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008;68:190–7. doi: 10.1158/0008-5472.CAN-07-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meyer MJ, Fleming JM, Lin AF, Hussnain SA, Ginsburg E, Vonderhaar BK. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res. 2010;70:4624–33. doi: 10.1158/0008-5472.CAN-09-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013;31:539–44. doi: 10.1038/nbt.2576. [DOI] [PubMed] [Google Scholar]
- 94.Han ME, Jeon TY, Hwang SH, Lee YS, Kim HJ, Shim HE, et al. Cancer spheres from gastric cancer patients provide an ideal model system for cancer stem cell research. Cell Mol Life Sci. 2011;68:3589–605. doi: 10.1007/s00018-011-0672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fukamachi H, Seol HS, Shimada S, Funasaka C, Baba K, Kim JH, et al. CD49f(high) cells retain sphere-forming and tumor-initiating activities in human gastric tumors. PLoS One. 2013;8:e72438. doi: 10.1371/journal.pone.0072438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jiang J, Zhang Y, Chuai S, Wang Z, Zheng D, Xu F, et al. Trastuzumab (herceptin) targets gastric cancer stem cells characterized by CD90 phenotype. Oncogene. 2012;31:671–82. doi: 10.1038/onc.2011.282. [DOI] [PubMed] [Google Scholar]
- 97.Jang BG, Lee BL, Kim WH. Distribution of LGR5+ cells and associated implications during the early stage of gastric tumorigenesis. PLoS One. 2013;8:e82390. doi: 10.1371/journal.pone.0082390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Takaishi S, Okumura T, Tu S, Wang SS, Shibata W, Vigneshwaran R, et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells. 2009;27:1006–20. doi: 10.1002/stem.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104:10158–63. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haraguchi N, Ishii H, Mimori K, Ohta K, Uemura M, Nishimura J, et al. CD49f-positive cell population efficiently enriches colon cancer-initiating cells. Int J Oncol. 2013;43:425–30. doi: 10.3892/ijo.2013.1955. [DOI] [PubMed] [Google Scholar]
- 101.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 102.Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–14. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- 103.Tirino V, Camerlingo R, Franco R, Malanga D, La Rocca A, Viglietto G, et al. The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur J Cardiothorac Surg. 2009;36(3):446–53. doi: 10.1016/j.ejcts.2009.03.063. [DOI] [PubMed] [Google Scholar]
- 104.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–24. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–58. doi: 10.1038/sj.onc.1210811. [DOI] [PubMed] [Google Scholar]
- 106.Kahlert UD, Bender NO, Maciaczyk D, Bogiel T, Bar EE, Eberhart CG, et al. CD133/CD15 defines distinct cell subpopulations with differential in vitro clonogenic activity and stem cell-related gene expression profile in in vitro propagated glioblastoma multiforme-derived cell line with a PNET-like component. Folia Neuropathol. 2012;50:357–68. doi: 10.5114/fn.2012.32365. [DOI] [PubMed] [Google Scholar]
- 107.Son MJ, Woolard K, Nam DH, Lee J, Fine HA. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009;4:440–52. doi: 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Suva ML, Riggi N, Stehle JC, Baumer K, Tercier S, Joseph JM, et al. Identification of cancer stem cells in Ewing’s sarcoma. Cancer Res. 2009;69:1776–81. doi: 10.1158/0008-5472.CAN-08-2242. [DOI] [PubMed] [Google Scholar]
- 109.Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, Fazioli F, et al. Human primary bone sarcomas contain CD133+ cancer stem cells displaying high tumorigenicity in vivo. Faseb J. 2011;25:2022–30. doi: 10.1096/fj.10-179036. [DOI] [PubMed] [Google Scholar]
- 110.Adhikari AS, Agarwal N, Wood BM, Porretta C, Ruiz B, Pochampally RR, et al. CD117 and Stro-1 identify osteosarcoma tumor-initiating cells associated with metastasis and drug resistance. Cancer Res. 2010;70:4602–12. doi: 10.1158/0008-5472.CAN-09-3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tian J, Li X, Si M, Liu T, Li J. CD271+ osteosarcoma cells display stem-like properties. PLoS One. 2014;9:e98549. doi: 10.1371/journal.pone.0098549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Meirelles K, Benedict LA, Dombkowski D, Pepin D, Preffer FI, Teixeira J, et al. Human ovarian cancer stem/progenitor cells are stimulated by doxorubicin but inhibited by Mullerian inhibiting substance. Proc Natl Acad Sci U S A. 2012;109:2358–63. doi: 10.1073/pnas.1120733109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen J, Wang J, Chen D, Yang J, Yang C, Zhang Y, et al. Evaluation of characteristics of CD44 + CD117+ ovarian cancer stem cells in three dimensional basement membrane extract scaffold versus two dimensional monocultures. BMC Cell Biol. 2013;14:7. doi: 10.1186/1471-2121-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Curley MD, Therrien VA, Cummings CL, Sergent PA, Koulouris CR, Friel AM, et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells. 2009;27:2875–83. doi: 10.1002/stem.236. [DOI] [PubMed] [Google Scholar]
- 115.Guo C, Liu H, Zhang BH, Cadaneanu RM, Mayle AM, Garraway IP. Epcam, CD44, and CD49f distinguish sphere-forming human prostate basal cells from a subpopulation with predominant tubule initiation capability. PLoS One. 2012;7:e34219. doi: 10.1371/journal.pone.0034219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hurt EM, Kawasaki BT, Klarmann GJ, Thomas SB, Farrar WL. CD44+ CD24(−) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer. 2008;98:756–65. doi: 10.1038/sj.bjc.6604242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Maitland NJ, Collins AT. Prostate cancer stem cells: a new target for therapy. J Clin Oncol. 2008;26:2862–70. doi: 10.1200/JCO.2007.15.1472. [DOI] [PubMed] [Google Scholar]
- 118.Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466:133–7. doi: 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wilson BJ, Saab KR, Ma J, Schatton T, Putz P, Zhan Q, et al. ABCB5 maintains melanoma-initiating cells through a proinflammatory cytokine signaling circuit. Cancer Res. 2014;74:4196–207. doi: 10.1158/0008-5472.CAN-14-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mirkina I, Hadzijusufovic E, Krepler C, Mikula M, Mechtcheriakova D, Strommer S, et al. Phenotyping of human melanoma cells reveals a unique composition of receptor targets and a subpopulation co-expressing ErbB4, EPO-R and NGF-R. PLoS One. 2014;9:e84417. doi: 10.1371/journal.pone.0084417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yui S, Nakamura T, Sato T, Nemoto Y, Mizutani T, Zheng X, et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nat Med. 2012;18:618–23. doi: 10.1038/nm.2695. [DOI] [PubMed] [Google Scholar]
- 122.Kemper K, Prasetyanti PR, De Lau W, Rodermond H, Clevers H, Medema JP. Monoclonal antibodies against Lgr5 identify human colorectal cancer stem cells. Stem Cells. 2012;30:2378–86. doi: 10.1002/stem.1233. [DOI] [PubMed] [Google Scholar]
- 123.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–23. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 124.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–67. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Takagi S, Saito Y, Hijikata A, Tanaka S, Watanabe T, Hasegawa T, et al. Membrane-bound human SCF/KL promotes in vivo human hematopoietic engraftment and myeloid differentiation. Blood. 2012;119:2768–77. doi: 10.1182/blood-2011-05-353201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sarry JE, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac-deficient mice. J Clin Invest. 2011;121:384–95. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vercauteren SM, Sutherland HJ. CD133 (AC133) expression on AML cells and progenitors. Cytotherapy. 2001;3:449–59. doi: 10.1080/146532401317248054. [DOI] [PubMed] [Google Scholar]
- 128.Valent P, Bonnet D, Wohrer S, Andreeff M, Copland M, Chomienne C, et al. Heterogeneity of Neoplastic Stem Cells: Theoretical, Functional, and Clinical Implications. Cancer Res. 2013;73:1037–45. doi: 10.1158/0008-5472.CAN-12-3678. [DOI] [PubMed] [Google Scholar]
- 129.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108:7950–5. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. 2012;12:767–75. doi: 10.1038/nrc3368. [DOI] [PubMed] [Google Scholar]
- 131.Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Valent P, Lion T, Wolf D, Sillaber C, Agis H, Petzer A, et al. Diagnostic algorithms, monitoring, prognostication, and therapy in chronic myeloid leukemia (CML): a proposal of the Austrian CML platform. Wien Klin Wochenschr. 2008;120:697–709. doi: 10.1007/s00508-008-1100-8. [DOI] [PubMed] [Google Scholar]
- 133.Renström J, Kröger M, Peschel C, Oostendorp RAJ. How the niche regulates hematopoietic stem cells. Chem Biol Interact. 2010;184:7–15. doi: 10.1016/j.cbi.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 134.Wang P, Hou SX. Regulation of intestinal stem cells in mammals and Drosophila. J Cell Physiol. 2009;222:33–7. doi: 10.1002/jcp.21928. [DOI] [PubMed] [Google Scholar]
- 135.Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133–43. doi: 10.1038/nrc3184. [DOI] [PubMed] [Google Scholar]
- 136.Niebuhr B, Fischer M, Tager M, Cammenga J, Stocking C. Gatekeeper function of the RUNX1 transcription factor in acute leukemia. Blood Cells Mol Dis. 2008;40:211–8. doi: 10.1016/j.bcmd.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 137.Wang X, Tripodi J, Kremyanskaya M, Blouin A, Roda P, Hoffman R, et al. BCR-ABL1 is a secondary event after JAK2V617F in patients with polycythemia vera who develop chronic myeloid leukemia. Blood. 2013;121:1238–9. doi: 10.1182/blood-2012-11-467787. [DOI] [PubMed] [Google Scholar]
- 138.Itzykson R, Solary E. An evolutionary perspective on chronic myelomonocytic leukemia. Leukemia. 2013;27:1441–50. doi: 10.1038/leu.2013.100. [DOI] [PubMed] [Google Scholar]
- 139.Herrmann H. 50th ASH Annual Meeting and Exposition. 2008. Phenotypic and functional characterization of CD34+/CD38−/CD123+ leukemic progenitor (stem) cells in AML: a flow cytometric approach. [Google Scholar]
- 140.Morimoto K, Kim SJ, Tanei T, Shimazu K, Tanji Y, Taguchi T, et al. Stem cell marker aldehyde dehydrogenase 1-positive breast cancers are characterized by negative estrogen receptor, positive human epidermal growth factor receptor type 2, and high Ki67 expression. Cancer Sci. 2009;100:1062–8. doi: 10.1111/j.1349-7006.2009.01151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27:6120–30. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hewish M, Chau I, Cunningham D. Insulin-like growth factor 1 receptor targeted therapeutics: novel compounds and novel treatment strategies for cancer medicine. Recent Patents Anticancer Drug Discov. 2009;4:54–72. doi: 10.2174/157489209787002515. [DOI] [PubMed] [Google Scholar]
- 143.Gotoh N. Control of stemness by fibroblast growth factor signaling in stem cells and cancer stem cells. Curr Stem Cell Res Ther. 2009;4:9–15. doi: 10.2174/157488809787169048. [DOI] [PubMed] [Google Scholar]
- 144.Koneczny I, Schulenburg A, Hudec X, Knofler M, Holzmann K, Piazza G, et al. Autocrine fibroblast growth factor 18 signaling mediates Wnt-dependent stimulation of CD44-positive human colorectal adenoma cells. Mol Carcinog. 2014. doi:10.1002/mc.22146. [DOI] [PMC free article] [PubMed]
- 145.Jiang X, Lopez A, Holyoake T, Eaves A, Eaves C. Autocrine production and action of IL-3 and granulocyte colony-stimulating factor in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 1999;96:12804–9. doi: 10.1073/pnas.96.22.12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jiang X, Fujisaki T, Nicolini F, Berger M, Holyoake T, Eisterer W, et al. Autonomous multi-lineage differentiation in vitro of primitive CD34+ cells from patients with chronic myeloid leukemia. Leukemia. 2000;14:1112–21. doi: 10.1038/sj.leu.2401752. [DOI] [PubMed] [Google Scholar]
- 147.Zhang SS, Han ZP, Jing YY, Tao SF, Li TJ, Wang H, et al. CD133(+)CXCR4(+) colon cancer cells exhibit metastatic potential and predict poor prognosis of patients. BMC Med. 2012;10:85. doi: 10.1186/1741-7015-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Furusato B, Mohamed A, Uhlen M, Rhim JS. CXCR4 and cancer. Pathol Int. 2010;60:497–505. doi: 10.1111/j.1440-1827.2010.02548.x. [DOI] [PubMed] [Google Scholar]
- 149.Tavor S, Petit I. Can inhibition of the SDF-1/CXCR4 axis eradicate acute leukemia? Semin Cancer Biol. 2010;20:178–85. doi: 10.1016/j.semcancer.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 150.Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64:2817–24. doi: 10.1158/0008-5472.can-03-3693. [DOI] [PubMed] [Google Scholar]
- 151.Kumar SM, Zhang G, Bastian BC, Arcasoy MO, Karande P, Pushparajan A, et al. Erythropoietin receptor contributes to melanoma cell survival in vivo. Oncogene. 2012;31:1649–60. doi: 10.1038/onc.2011.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sinclair AM, Todd MD, Forsythe K, Knox SJ, Elliott S, Begley CG. Expression and function of erythropoietin receptors in tumors: implications for the use of erythropoiesis-stimulating agents in cancer patients. Cancer. 2007;110:477–88. doi: 10.1002/cncr.22832. [DOI] [PubMed] [Google Scholar]
- 153.Jelkmann W, Bohlius J, Hallek M, Sytkowski AJ. The erythropoietin receptor in normal and cancer tissues. Crit Rev Oncol Hematol. 2008;67:39–61. doi: 10.1016/j.critrevonc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 154.Gonen M, Sun Z, Figueroa ME, Patel JP, Abdel-Wahab O, Racevskis J, et al. CD25 expression status improves prognostic risk classification in AML independent of established biomarkers: ECOG phase 3 trial, E1900. Blood. 2012;120:2297–306. doi: 10.1182/blood-2012-02-414425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Testa U, Riccioni R, Diverio D, Rossini A, Lo Coco F, Peschle C. Interleukin-3 receptor in acute leukemia. Leukemia. 2004;18:219–26. doi: 10.1038/sj.leu.2403224. [DOI] [PubMed] [Google Scholar]
- 156.Lowenberg B, van Putten W, Theobald M, Gmur J, Verdonck L, Sonneveld P, et al. Effect of priming with granulocyte colony-stimulating factor on the outcome of chemotherapy for acute myeloid leukemia. N Engl J Med. 2003;349:743–52. doi: 10.1056/NEJMoa025406. [DOI] [PubMed] [Google Scholar]
- 157.Frohling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100:4372–80. doi: 10.1182/blood-2002-05-1440. [DOI] [PubMed] [Google Scholar]
- 158.Hassan HT, Zander A. Stem cell factor as a survival and growth factor in human normal and malignant hematopoiesis. Acta Haematol. 1996;95:257–62. doi: 10.1159/000203893. [DOI] [PubMed] [Google Scholar]
- 159.Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917–24. doi: 10.1182/blood-2011-10-383406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hanumanthu VS, Pirruccello SJ. GCSF-R expression in myelodysplastic and myeloproliferative disorders and blast dysmaturation in CML. Am J Clin Pathol. 2013;140:155–64. doi: 10.1309/AJCPCLHZR5KUHUBM. [DOI] [PubMed] [Google Scholar]
- 161.Zhang Y, Guo Q, Zhao H, Zhao D, Wu X, Zhao W, et al. Expression of CXCR4 is an independent prognostic factor for overall survival and progression-free survival in patients with myelodysplastic syndrome. Med Oncol. 2013;30:341. doi: 10.1007/s12032-012-0341-6. [DOI] [PubMed] [Google Scholar]
- 162.Nakata Y, Kimura A, Katoh O, Kawaishi K, Hyodo H, Abe K, et al. c-kit point mutation of extracellular domain in patients with myeloproliferative disorders. Br J Haematol. 1995;91:661–3. doi: 10.1111/j.1365-2141.1995.tb05364.x. [DOI] [PubMed] [Google Scholar]
- 163.Bogani C, Ponziani V, Guglielmelli P, Desterke C, Rosti V, Bosi A, et al. Hypermethylation of CXCR4 promoter in CD34+ cells from patients with primary myelofibrosis. Stem Cells. 2008;26:1920–30. doi: 10.1634/stemcells.2008-0377. [DOI] [PubMed] [Google Scholar]
- 164.Smith BD, Kasamon YL, Kowalski J, Gocke C, Murphy K, Miller CB, et al. K562/GM-CSF immunotherapy reduces tumor burden in chronic myeloid leukemia patients with residual disease on imatinib mesylate. Clin Cancer Res. 2010;16:338–47. doi: 10.1158/1078-0432.CCR-09-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Corbin AS, O’Hare T, Gu Z, Kraft IL, Eiring AM, Khorashad JS, et al. KIT signaling governs differential sensitivity of mature and primitive CML progenitors to tyrosine kinase inhibitors. Cancer Res. 2013;73:5775–86. doi: 10.1158/0008-5472.CAN-13-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Beider K, Darash-Yahana M, Blaier O, Koren-Michowitz M, Abraham M, Wald H, et al. Combination of imatinib with CXCR4 antagonist BKT140 overcomes the protective effect of stroma and targets CML in vitro and in vivo. Mol Cancer Ther. 2014;13:1155–69. doi: 10.1158/1535-7163.MCT-13-0410. [DOI] [PubMed] [Google Scholar]
- 167.Gaikwad AS, Donohue RE, Elghetany MT, Sheehan AM, Lu XY, Gramatges MM, et al. Expression of CD25 is a specific and relatively sensitive marker for the Philadelphia chromosome (BCR-ABL1) translocation in pediatric B acute lymphoblastic leukemia. Int J Clin Exp Pathol. 2014;7:6225–30. [PMC free article] [PubMed] [Google Scholar]
- 168.Kim HP, Frankel AE, Hogge DE. A diphtheria toxin interleukin-3 fusion protein synergizes with tyrosine kinase inhibitors in killing leukemic progenitors from BCR/ABL positive acute leukemia. Leuk Res. 2010;34:1035–42. doi: 10.1016/j.leukres.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 169.Parameswaran R, Yu M, Lim M, Groffen J, Heisterkamp N. Combination of drug therapy in acute lymphoblastic leukemia with a CXCR4 antagonist. Leukemia. 2011;25:1314–23. doi: 10.1038/leu.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Peled A, Abraham M, Avivi I, Rowe JM, Beider K, Wald H, et al. The high-affinity CXCR4 antagonist BKT140 is safe and induces a robust mobilization of human CD34+ cells in patients with multiple myeloma. Clin Cancer Res. 2014;20:469–79. doi: 10.1158/1078-0432.CCR-13-1302. [DOI] [PubMed] [Google Scholar]
- 171.LeBedis C, Chen K, Fallavollita L, Boutros T, Brodt P. Peripheral lymph node stromal cells can promote growth and tumorigenicity of breast carcinoma cells through the release of IGF-I and EGF. Int J Cancer. 2002;100:2–8. doi: 10.1002/ijc.10481. [DOI] [PubMed] [Google Scholar]
- 172.Korkaya H, Wicha MS. HER2 and breast cancer stem cells: more than meets the eye. Cancer Res. 2013;73:3489–93. doi: 10.1158/0008-5472.CAN-13-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U S A. 2010;107:21737–42. doi: 10.1073/pnas.1007863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sun L, Wu G, Willson JK, Zborowska E, Yang J, Rajkarunanayake I, et al. Expression of transforming growth factor beta type II receptor leads to reduced malignancy in human breast cancer MCF-7 cells. J Biol Chem. 1994;269:26449–55. [PubMed] [Google Scholar]
- 175.Pinto C, Di Fabio F, Siena S, Cascinu S, Rojas Llimpe FL, Ceccarelli C, et al. Phase II study of cetuximab in combination with FOLFIRI in patients with untreated advanced gastric or gastroesophageal junction adenocarcinoma (FOLCETUX study) Ann Oncol. 2007;18:510–7. doi: 10.1093/annonc/mdl459. [DOI] [PubMed] [Google Scholar]
- 176.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 177.Shaib W, Mahajan R, El-Rayes B. Markers of resistance to anti-EGFR therapy in colorectal cancer. J Gastrointest Oncol. 2013;4:308–18. doi: 10.3978/j.issn.2078-6891.2013.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kim J, Takeuchi H, Lam ST, Turner RR, Wang HJ, Kuo C, et al. Chemokine receptor CXCR4 expression in colorectal cancer patients increases the risk for recurrence and for poor survival. J Clin Oncol. 2005;23:2744–53. doi: 10.1200/JCO.2005.07.078. [DOI] [PubMed] [Google Scholar]
- 179.Wolpin BM, Meyerhardt JA, Chan AT, Ng K, Chan JA, Wu K, et al. Insulin, the insulin-like growth factor axis, and mortality in patients with nonmetastatic colorectal cancer. J Clin Oncol. 2009;27:176–85. doi: 10.1200/JCO.2008.17.9945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV, et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–84. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Okamoto I, Araki J, Suto R, Shimada M, Nakagawa K, Fukuoka M. EGFR mutation in gefitinib-responsive small-cell lung cancer. Ann Oncol. 2006;17:1028–9. doi: 10.1093/annonc/mdj114. [DOI] [PubMed] [Google Scholar]
- 182.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–6. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 183.Shan J, Shen J, Liu L, Xia F, Xu C, Duan G, et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology. 2012;56:1004–14. doi: 10.1002/hep.25745. [DOI] [PubMed] [Google Scholar]
- 184.Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313:144–7. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 185.Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B, et al. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992;52:3213–9. [PubMed] [Google Scholar]
- 186.Ehtesham M, Mapara KY, Stevenson CB, Thompson RC. CXCR4 mediates the proliferation of glioblastoma progenitor cells. Cancer Lett. 2009;274:305–12. doi: 10.1016/j.canlet.2008.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Penuelas S, Anido J, Prieto-Sanchez RM, Folch G, Barba I, Cuartas I, et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009;15:315–27. doi: 10.1016/j.ccr.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 188.Joo KM, Jin J, Kim E, Ho Kim K, Kim Y, Gu Kang B, et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012;72:3828–38. doi: 10.1158/0008-5472.CAN-11-3760. [DOI] [PubMed] [Google Scholar]
- 189.Glaysher S, Bolton LM, Johnson P, Atkey N, Dyson M, Torrance C, et al. Targeting EGFR and PI3K pathways in ovarian cancer. Br J Cancer. 2013;109:1786–94. doi: 10.1038/bjc.2013.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bookman MA, Darcy KM, Clarke-Pearson D, Boothby RA, Horowitz IR. Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: a phase II trial of the Gynecologic Oncology Group. J Clin Oncol. 2003;21:283–90. doi: 10.1200/JCO.2003.10.104. [DOI] [PubMed] [Google Scholar]
- 191.Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J, et al. TGF-beta modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013;73:5016–28. doi: 10.1158/0008-5472.CAN-13-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Dubrovska A, Elliott J, Salamone RJ, Telegeev GD, Stakhovsky AE, Schepotin IB, et al. CXCR4 expression in prostate cancer progenitor cells. PLoS One. 2012;7:e31226. doi: 10.1371/journal.pone.0031226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Singh S, Nannuru KC, Sadanandam A, Varney ML, Singh RK. CXCR1 and CXCR2 enhances human melanoma tumourigenesis, growth and invasion. Br J Cancer. 2009;100:1638–46. doi: 10.1038/sj.bjc.6605055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Karamboulas C, Ailles L. Developmental signaling pathways in cancer stem cells of solid tumors. Biochim Biophys Acta. 1830;2013:2481–95. doi: 10.1016/j.bbagen.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 195.Okuhashi Y, Itoh M, Nara N, Tohda S. Effects of combination of Notch inhibitor plus hedgehog inhibitor or Wnt inhibitor on growth of leukemia cells. Anticancer Res. 2011;31:893–6. [PubMed] [Google Scholar]
- 196.Sinclair A, Latif AL, Holyoake TL. Targeting survival pathways in chronic myeloid leukaemia stem cells. Br J Pharmacol. 2013;169:1693–707. doi: 10.1111/bph.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Abetov D, Mustapova Z, Saliev T, Bulanin D. Biomarkers and signaling pathways of colorectal cancer stem cells. Tumour Biol. 2015;doi:10.1007/s13277-015-3198-4. [DOI] [PubMed]
- 198.Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–62. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- 199.Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012;337:1541–6. doi: 10.1126/science.1221711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kleppe M, Levine RL. Targeting beta-catenin in CML: leukemia stem cells beware! Cell Stem Cell. 2012;10:351–3. doi: 10.1016/j.stem.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 201.Pannuti A, Foreman K, Rizzo P, Osipo C, Golde T, Osborne B, et al. Targeting Notch to target cancer stem cells. Clin Cancer Res. 2010;16:3141–52. doi: 10.1158/1078-0432.CCR-09-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Merchant AA, Matsui W. Targeting hedgehog - a cancer stem cell pathway. Clin Cancer Res. 2010;16:3130–40. doi: 10.1158/1078-0432.CCR-09-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 204.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121:396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hamilton A, Elrick L, Myssina S, Copland M, Jorgensen H, Melo JV, et al. BCR-ABL activity and its response to drugs can be determined in CD34+ CML stem cells by CrkL phosphorylation status using flow cytometry. Leukemia. 2006;20:1035–9. doi: 10.1038/sj.leu.2404189. [DOI] [PubMed] [Google Scholar]
- 206.Wiseman DH, Greystoke BF, Somervaille TC. The variety of leukemic stem cells in myeloid malignancy. Oncogene. 2013;33(24):3091–8. doi: 10.1038/onc.2013.269. [DOI] [PubMed] [Google Scholar]
- 207.Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–61. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 208.Ysebaert L, Chicanne G, Demur C, De Toni F, Prade-Houdellier N, Ruidavets JB, et al. Expression of beta-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia. 2006;20:1211–6. doi: 10.1038/sj.leu.2404239. [DOI] [PubMed] [Google Scholar]
- 209.Li Y, Welm B, Podsypanina K, Huang SX, Chamorro M, Zhang XM, et al. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc Natl Acad Sci U S A. 2003;100:15853–8. doi: 10.1073/pnas.2136825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chien AJ, Moore EC, Lonsdorf AS, Kulikauskas RM, Rothberg BG, Berger AJ, et al. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci U S A. 2009;106:1193–8. doi: 10.1073/pnas.0811902106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yang W, Yan HX, Chen L, Liu Q, He YQ, Yu LX, et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008;68:4287–95. doi: 10.1158/0008-5472.CAN-07-6691. [DOI] [PubMed] [Google Scholar]
- 212.Gursel DB, Berry N, Boockvar JA. The contribution of Notch signaling to glioblastoma via activation of cancer stem cell self-renewal: the role of the endothelial network. Neurosurgery. 2012;70:N19–21. doi: 10.1227/01.neu.0000410937.38828.6f. [DOI] [PubMed] [Google Scholar]
- 213.Sikandar SS, Pate KT, Anderson S, Dizon D, Edwards RA, Waterman ML, et al. NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res. 2010;70:1469–78. doi: 10.1158/0008-5472.CAN-09-2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lee CW, Simin K, Liu Q, Plescia J, Guha M, Khan A, et al. A functional Notch-survivin gene signature in basal breast cancer. Breast Cancer Res. 2008;10:R97. doi: 10.1186/bcr2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Liu SL, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Huang FT, Zhuan-Sun YX, Zhuang YY, Wei SL, Tang J, Chen WB, et al. Inhibition of Hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. J Gastroen Hepatol. 2012;27:43. doi: 10.3892/ijo.2012.1597. [DOI] [PubMed] [Google Scholar]
- 217.Kawaguchi-Ihara N, Okuhashi Y, Itoh M, Murohashi I, Nara N, Tohda S. Promotion of the self-renewal capacity of human leukemia cells by sonic hedgehog protein. Anticancer Res. 2011;31:781–4. [PubMed] [Google Scholar]
- 218.Ferruzzi P, Mennillo F, De Rosa A, Giordano C, Rossi M, Benedetti G, et al. In vitro and in vivo characterization of a novel hedgehog signaling antagonist in human glioblastoma cell lines. Int J Cancer. 2012;131:E33–44. doi: 10.1002/ijc.27349. [DOI] [PubMed] [Google Scholar]
- 219.Mazumdar T, DeVecchio J, Shi T, Jones J, Agyeman A, Houghton JA. Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res. 2011;71:1092–102. doi: 10.1158/0008-5472.CAN-10-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, et al. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009;137:623–34. doi: 10.1016/j.cell.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009;7:e1000121. doi: 10.1371/journal.pbio.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kroon P, Berry PA, Stower MJ, Rodrigues G, Mann VM, Simms M, et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013;73:5288–98. doi: 10.1158/0008-5472.CAN-13-0874. [DOI] [PubMed] [Google Scholar]
- 223.Kanno H, Sato H, Yokoyama T-A, Yoshizumi T, Yamada S. The VHL tumor suppressor protein regulates tumorigenicity of U87-derived glioma stem-like cells by inhibiting the JAK/STAT signaling pathway. Int J Oncol. 2013;42:881–6. doi: 10.3892/ijo.2013.1773. [DOI] [PubMed] [Google Scholar]
- 224.Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. 2006;12:1175–80. doi: 10.1038/nm1489. [DOI] [PubMed] [Google Scholar]
- 225.Nair RR, Tolentino J, Hazlehurst LA. The bone marrow microenvironment as a sanctuary for minimal residual disease in CML. Biochem Pharmacol. 2010;80:602–12. doi: 10.1016/j.bcp.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Potten CS, Loeffler M. Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development. 1990;110:1001–20. doi: 10.1242/dev.110.4.1001. [DOI] [PubMed] [Google Scholar]
- 227.Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol. 2008;9:11–21. doi: 10.1038/nrm2319. [DOI] [PubMed] [Google Scholar]
- 228.Rosel M, Khaldoyanidi S, Zawadzki V, Zoller M. Involvement of CD44 variant isoform v10 in progenitor cell adhesion and maturation. Exp Hematol. 1999;27:698–711. doi: 10.1016/s0301-472x(98)00082-4. [DOI] [PubMed] [Google Scholar]
- 229.Alakel N, Jing D, Muller K, Bornhauser M, Ehninger G, Ordemann R. Direct contact with mesenchymal stromal cells affects migratory behavior and gene expression profile of CD133(+) hematopoietic stem cells during ex vivo expansion. Exp Hematol. 2009;37(4):504–13. doi: 10.1016/j.exphem.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 230.Raveh S, Gavert N, Ben-Ze'ev A. L1 cell adhesion molecule (L1CAM) in invasive tumors. Cancer Lett. 2009;282(2):137–45. doi: 10.1016/j.canlet.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 231.Gavert N, Ben-Shmuel A, Raveh S, Ben-Ze’ev A. L1-CAM in cancerous tissues. Expert Opin Biol Ther. 2008;8:1749–57. doi: 10.1517/14712598.8.11.1749. [DOI] [PubMed] [Google Scholar]
- 232.Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68:6043–8. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118:277–9. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 234.Peled A, Hardan I, Trakhtenbrot L, Gur E, Magid M, Darash-Yahana M, et al. Immature leukemic CD34(+)CXCR4(+) cells from CML patients have lower integrin-dependent migration and adhesion in response to the chemokine SDF-1. Stem Cells. 2002;20:259–66. doi: 10.1634/stemcells.20-3-259. [DOI] [PubMed] [Google Scholar]
- 235.Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee SU, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell. 2012;21:577–92. doi: 10.1016/j.ccr.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441:1075–9. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- 237.Greenberger JS. The hematopoietic microenvironment. Crit Rev Oncol Hematol. 1991;11:65–84. doi: 10.1016/1040-8428(91)90018-8. [DOI] [PubMed] [Google Scholar]
- 238.Torok-Storb B. Cellular interactions. Blood. 1988;72:373–85. [PubMed] [Google Scholar]
- 239.Mayani H, Guilbert LJ, Janowska-Wieczorek A. Biology of the hemopoietic microenvironment. Eur J Haematol. 1992;49:225–33. doi: 10.1111/j.1600-0609.1992.tb00053.x. [DOI] [PubMed] [Google Scholar]
- 240.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 241.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 242.Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood. 1996;88:146–57. [PubMed] [Google Scholar]
- 243.Schulenburg A, Cech P, Herbacek I, Marian B, Wrba F, Valent P, et al. CD44-positive colorectal adenoma cells express the potential stem cell markers musashi antigen (msi1) and ephrin B2 receptor (EphB2) J Pathol. 2007;213:152–60. doi: 10.1002/path.2220. [DOI] [PubMed] [Google Scholar]
- 244.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 245.Savagner P. The epithelial-mesenchymal transition (EMT) phenomenon. Ann Oncol. 2010;21(Suppl 7):vii89-92. doi: 10.1093/annonc/mdq292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Durig J, Rosenthal C, Elmaagacli A, Heyworth C, Halfmeyer K, Kasper C, et al. Biological effects of stroma-derived factor-1 alpha on normal and CML CD34+ haemopoietic cells. Leukemia. 2000;14:1652–60. doi: 10.1038/sj.leu.2401875. [DOI] [PubMed] [Google Scholar]
- 247.Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–43. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Scadden DT. Nice neighborhood: emerging concepts of the stem cell niche. Cell. 2014;157:41–50. doi: 10.1016/j.cell.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–34. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Konopleva M, Tabe Y, Zeng Z, Andreeff M. Therapeutic targeting of microenvironmental interactions in leukemia: mechanisms and approaches. Drug Resist Updat. 2009;12:103–13. doi: 10.1016/j.drup.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Sullivan WJ, Christofk HR. The metabolic milieu of metastases. Cell. 2015;160:363–4. doi: 10.1016/j.cell.2015.01.023. [DOI] [PubMed] [Google Scholar]
- 252.Xiang L, Gilkes DM, Hu H, Takano N, Luo W, Lu H, et al. Hypoxia-inducible factor 1 mediates TAZ expression and nuclear localization to induce the breast cancer stem cell phenotype. Oncotarget. 2014;5:12509–27. doi: 10.18632/oncotarget.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Clarke L, van der Kooy D. Low oxygen enhances primitive and definitive neural stem cell colony formation by inhibiting distinct cell death pathways. Stem Cells. 2009;27:1879–86. doi: 10.1002/stem.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Moreno-Manzano V, Rodriguez-Jimenez FJ, Acena-Bonilla JL, Fustero-Lardies S, Erceg S, Dopazo J, et al. FM19G11, a new hypoxia-inducible factor (HIF) modulator, affects stem cell differentiation status. J Biol Chem. 2010;285:1333–42. doi: 10.1074/jbc.M109.008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Levesque JP, Winkler IG, Hendy J, Williams B, Helwani F, Barbier V, et al. Hematopoietic progenitor cell mobilization results in hypoxia with increased hypoxia-inducible transcription factor-1 alpha and vascular endothelial growth factor A in bone marrow. Stem Cells. 2007;25:1954–65. doi: 10.1634/stemcells.2006-0688. [DOI] [PubMed] [Google Scholar]
- 256.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–64. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 257.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–28. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 258.Chen Y, De Marco MA, Graziani I, Gazdar AF, Strack PR, Miele L, et al. Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Res. 2007;67:7954–9. doi: 10.1158/0008-5472.CAN-07-1229. [DOI] [PubMed] [Google Scholar]
- 259.Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–6. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 260.Nieborowska-Skorska M, Kopinski PK, Ray R, Hoser G, Ngaba D, Flis S, et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood. 2012;119:4253–63. doi: 10.1182/blood-2011-10-385658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Kantner HP, Warsch W, Delogu A, Bauer E, Esterbauer H, Casanova E, et al. ETV6/RUNX1 induces reactive oxygen species and drives the accumulation of DNA damage in B cells. Neoplasia. 2013;15:1292–300. doi: 10.1593/neo.131310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Warsch W, Grundschober E, Berger A, Gille L, Cerny-Reiterer S, Tigan AS, et al. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukaemia. Oncotarget. 2012;3:1669–87. doi: 10.18632/oncotarget.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–70. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–72. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Horton SJ, Huntly BJ. Recent advances in acute myeloid leukemia stem cell biology. Haematologica. 2012;97:966–74. doi: 10.3324/haematol.2011.054734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Oh SH, Park TS, Kim HR, Lee JY, Kim JH, Shin JH, et al. Chronic myelogenous leukemia showing biphenotypic blast crisis followed by lineage switch to B lymphoblastic leukemia. Leuk Res. 2009;33:e195–8. doi: 10.1016/j.leukres.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 267.Schmidt CA, Przybylski GK. What can we learn from leukemia as for the process of lineage commitment in hematopoiesis? Int Rev Immunol. 2001;20:107–15. doi: 10.3109/08830180109056725. [DOI] [PubMed] [Google Scholar]
- 268.Reid AG, De Melo VA, Elderfield K, Clark I, Marin D, Apperley J, et al. Phenotype of blasts in chronic myeloid leukemia in blastic phase-analysis of bone marrow trephine biopsies and correlation with cytogenetics. Leuk Res. 2009;33:418–25. doi: 10.1016/j.leukres.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 269.Bettelheim P, Lutz D, Majdic O, Paietta E, Haas O, Linkesch W, et al. Cell lineage heterogeneity in blast crisis of chronic myeloid leukaemia. Br J Haematol. 1985;59:395–409. doi: 10.1111/j.1365-2141.1985.tb07326.x. [DOI] [PubMed] [Google Scholar]
- 270.Goradia A, Bayerl M, Cornfield D. The 8p11 myeloproliferative syndrome: review of literature and an illustrative case report. Int J Clin Exp Pathol. 2008;1:448–56. [PMC free article] [PubMed] [Google Scholar]
- 271.Tzankov A, Sotlar K, Muhlematter D, Theocharides A, Went P, Jotterand M, et al. Systemic mastocytosis with associated myeloproliferative disease and precursor B lymphoblastic leukaemia with t(13;13)(q12;q22) involving FLT3. J Clin Pathol. 2008;61:958–61. doi: 10.1136/jcp.2008.058073. [DOI] [PubMed] [Google Scholar]
- 272.Sperr WR, Horny HP, Valent P. Spectrum of associated clonal hematologic non-mast cell lineage disorders occurring in patients with systemic mastocytosis. Int Arch Allergy Immunol. 2002;127:140–2. doi: 10.1159/000048186. [DOI] [PubMed] [Google Scholar]
- 273.Streubel B, Chott A, Huber D, Exner M, Jager U, Wagner O, et al. Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. N Engl J Med. 2004;351:250–9. doi: 10.1056/NEJMoa033153. [DOI] [PubMed] [Google Scholar]
- 274.Gunsilius E, Duba HC, Petzer AL, Kahler CM, Grunewald K, Stockhammer G, et al. Evidence from a leukaemia model for maintenance of vascular endothelium by bone-marrow-derived endothelial cells. Lancet. 2000;355:1688–91. doi: 10.1016/S0140-6736(00)02241-8. [DOI] [PubMed] [Google Scholar]
- 275.Wu J, Huang L, Huang M, Liu W, Zheng M, Cao Y, et al. Dominant contribution of malignant endothelial cells to endotheliopoiesis in chronic myeloid leukemia. Exp Hematol. 2009;37:87–91. doi: 10.1016/j.exphem.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 276.Mirshahi P, Rafii A, Vincent L, Berthaut A, Varin R, Kalantar G, et al. Vasculogenic mimicry of acute leukemic bone marrow stromal cells. Leukemia. 2009;23:1039–48. doi: 10.1038/leu.2009.10. [DOI] [PubMed] [Google Scholar]
- 277.Hauswirth AW, Florian S, Printz D, Sotlar K, Krauth MT, Fritsch G, et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur J Clin Invest. 2007;37:73–82. doi: 10.1111/j.1365-2362.2007.01746.x. [DOI] [PubMed] [Google Scholar]
- 278.Pearce DJ, Taussig DC, Bonnet D. Implications of the expression of myeloid markers on normal and leukemic stem cells. Cell Cycle. 2006;5:271–3. doi: 10.4161/cc.5.3.2393. [DOI] [PubMed] [Google Scholar]
- 279.Larson RA, Boogaerts M, Estey E, Karanes C, Stadtmauer EA, Sievers EL, et al. Antibody-targeted chemotherapy of older patients with acute myeloid leukemia in first relapse using Mylotarg (gemtuzumab ozogamicin) Leukemia. 2002;16:1627–36. doi: 10.1038/sj.leu.2402677. [DOI] [PubMed] [Google Scholar]
- 280.Sievers EL, Larson RA, Stadtmauer EA, Estey E, Lowenberg B, Dombret H, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19:3244–54. doi: 10.1200/JCO.2001.19.13.3244. [DOI] [PubMed] [Google Scholar]
- 281.Feldman E, Kalaycio M, Weiner G, Frankel S, Schulman P, Schwartzberg L, et al. Treatment of relapsed or refractory acute myeloid leukemia with humanized anti-CD33 monoclonal antibody HuM195. Leukemia. 2003;17:314–8. doi: 10.1038/sj.leu.2402803. [DOI] [PubMed] [Google Scholar]
- 282.Sievers EL, Appelbaum FR, Spielberger RT, Forman SJ, Flowers D, Smith FO, et al. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: a phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood. 1999;93:3678–84. [PubMed] [Google Scholar]
- 283.Majeti R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene. 2011;30:1009–19. doi: 10.1038/onc.2010.511. [DOI] [PubMed] [Google Scholar]
- 284.Mahon F-X, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029–35. doi: 10.1016/S1470-2045(10)70233-3. [DOI] [PubMed] [Google Scholar]
- 285.Pfreundschuh M, Schubert J, Ziepert M, Schmits R, Mohren M, Lengfelder E, et al. Six versus eight cycles of bi-weekly CHOP-14 with or without rituximab in elderly patients with aggressive CD20+ B-cell lymphomas: a randomised controlled trial (RICOVER-60) Lancet Oncol. 2008;9:105–16. doi: 10.1016/S1470-2045(08)70002-0. [DOI] [PubMed] [Google Scholar]
- 286.Patz M, Isaeva P, Forcob N, Muller B, Frenzel LP, Wendtner CM, et al. Comparison of the in vitro effects of the anti-CD20 antibodies rituximab and GA101 on chronic lymphocytic leukaemia cells. Br J Haematol. 2011;152:295–306. doi: 10.1111/j.1365-2141.2010.08428.x. [DOI] [PubMed] [Google Scholar]
- 287.Boyd K, Dearden CE. Alemtuzumab in the treatment of chronic lymphocytic lymphoma. Expert Rev Anticancer Ther. 2008;8:525–33. doi: 10.1586/14737140.8.4.525. [DOI] [PubMed] [Google Scholar]
- 288.Cunningham MP, Thomas H, Marks C, Green M, Fan Z, Modjtahedi H. Co-targeting the EGFR and IGF-IR with anti-EGFR monoclonal antibody ICR62 and the IGF-IR tyrosine kinase inhibitor NVP-AEW541 in colorectal cancer cells. Int J Oncol. 2008;33:1107–13. [PubMed] [Google Scholar]
- 289.Heinrich MC. Imatinib treatment of metastatic GIST: don’t stop (believing) Lancet Oncol. 2010;11:910–1. doi: 10.1016/S1470-2045(10)70225-4. [DOI] [PubMed] [Google Scholar]
- 290.Burris HA, 3rd, Hurwitz HI, Dees EC, Dowlati A, Blackwell KL, O’Neil B, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol. 2005;23:5305–13. doi: 10.1200/JCO.2005.16.584. [DOI] [PubMed] [Google Scholar]
- 291.Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171–9. doi: 10.1056/NEJMoa1113713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol. 2011;8:426–33. doi: 10.1038/nrclinonc.2011.69. [DOI] [PubMed] [Google Scholar]
- 293.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294–304. doi: 10.1200/JCO.2005.23.622. [DOI] [PubMed] [Google Scholar]
- 295.Hudes GR. mTOR as a target for therapy of renal cancer. Clin Adv Hematol Oncol. 2007;5:772–4. [PubMed] [Google Scholar]
- 296.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–8. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Fons P, Herault JP, Delesque N, Tuyaret J, Bono F, Herbert JM. VEGF-R2 and neuropilin-1 are involved in VEGF-A-induced differentiation of human bone marrow progenitor cells. J Cell Physiol. 2004;200:351–9. doi: 10.1002/jcp.20076. [DOI] [PubMed] [Google Scholar]
- 298.Shervington A, Lu C. Expression of multidrug resistance genes in normal and cancer stem cells. Cancer Invest. 2008;26:535–42. doi: 10.1080/07357900801904140. [DOI] [PubMed] [Google Scholar]
- 299.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–27. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 300.Engler JR, Frede A, Saunders V, Zannettino A, White DL, Hughes TP. The poor response to imatinib observed in CML patients with low OCT-1 activity is not attributable to lower uptake of imatinib into their CD34+ cells. Blood. 2010;116:2776–8. doi: 10.1182/blood-2010-01-267013. [DOI] [PubMed] [Google Scholar]
- 301.Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005;65:6207–19. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- 302.Nakai E, Park K, Yawata T, Chihara T, Kumazawa A, Nakabayashi H, et al. Enhanced MDR1 expression and chemoresistance of cancer stem cells derived from glioblastoma. Cancer Invest. 2009;27:901–8. doi: 10.3109/07357900801946679. [DOI] [PubMed] [Google Scholar]
- 303.Yamamoto A, Shofuda T, Islam MO, Nakamura Y, Yamasaki M, Okano H, et al. ABCB1 is predominantly expressed in human fetal neural stem/progenitor cells at an early development stage. J Neurosci Res. 2009;87:2615–23. doi: 10.1002/jnr.22094. [DOI] [PubMed] [Google Scholar]
- 304.Jin W, Liu Y, Xu SG, Yin WJ, Li JJ, Yang JM, et al. UHRF1 inhibits MDR1 gene transcription and sensitizes breast cancer cells to anticancer drugs. Breast Cancer Res Treat. 2010;124:39–48. doi: 10.1007/s10549-009-0683-8. [DOI] [PubMed] [Google Scholar]
- 305.Brendel C, Scharenberg C, Dohse M, Robey RW, Bates SE, Shukla S, et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia. 2007;21:1267–75. doi: 10.1038/sj.leu.2404638. [DOI] [PubMed] [Google Scholar]
- 306.Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, et al. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98:2301–7. doi: 10.1182/blood.v98.8.2301. [DOI] [PubMed] [Google Scholar]
- 307.Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–22. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 308.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 309.Hess CJ, Berkhof J, Denkers F, Ossenkoppele GJ, Schouten JP, Oudejans JJ, et al. Activated intrinsic apoptosis pathway is a key related prognostic parameter in acute myeloid leukemia. J Clin Oncol. 2007;25:1209–15. doi: 10.1200/JCO.2006.08.4061. [DOI] [PubMed] [Google Scholar]
- 310.Harper LJ, Costea DE, Gammon L, Fazil B, Biddle A, Mackenzie IC. Normal and malignant epithelial cells with stem-like properties have an extended G2 cell cycle phase that is associated with apoptotic resistance. BMC Cancer. 2010;10:166. doi: 10.1186/1471-2407-10-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 312.Skladanowski A, Bozko P, Sabisz M. DNA structure and integrity checkpoints during the cell cycle and their role in drug targeting and sensitivity of tumor cells to anticancer treatment. Chem Rev. 2009;109:2951–73. doi: 10.1021/cr900026u. [DOI] [PubMed] [Google Scholar]
- 313.Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood. 2009;114:1150–7. doi: 10.1182/blood-2009-01-202606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Teitz-Tennenbaum S, Wicha MS, Chang AE, Li Q. Targeting cancer stem cells via dendritic-cell vaccination. Oncoimmunology. 2012;1:1401–3. doi: 10.4161/onci.21026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Brune M, Castaigne S, Catalano J, Gehlsen K, Ho AD, Hofmann WK, et al. Improved leukemia-free survival after postconsolidation immunotherapy with histamine dihydrochloride and interleukin-2 in acute myeloid leukemia: results of a randomized phase 3 trial. Blood. 2006;108:88–96. doi: 10.1182/blood-2005-10-4073. [DOI] [PubMed] [Google Scholar]
- 316.Essers MA, Trumpp A. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol. 2010;4:443–50. doi: 10.1016/j.molonc.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207–12. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Weber DM, Chen C, Niesvizky R, Wang M, Belch A, Stadtmauer EA, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357:2133–42. doi: 10.1056/NEJMoa070596. [DOI] [PubMed] [Google Scholar]
- 320.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–8. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 321.Wulf GG, Wang RY, Kuehnle I, Weidner D, Marini F, Brenner MK, et al. A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood. 2001;98:1166–73. doi: 10.1182/blood.v98.4.1166. [DOI] [PubMed] [Google Scholar]
- 322.Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101:14228–33. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Peter B, Cerny-Reiterer S, Hadzijusufovic E, Schuch K, Stefanzl G, Eisenwort G, et al. The pan-Bcl-2 blocker obatoclax promotes the expression of Puma, Noxa, and Bim mRNA and induces apoptosis in neoplastic mast cells. J Leukoc Biol. 2014;95:95–104. doi: 10.1189/jlb.1112609. [DOI] [PubMed] [Google Scholar]
- 324.Moser C, Lang SA, Kainz S, Gaumann A, Fichtner-Feigl S, Koehl GE, et al. Blocking heat shock protein-90 inhibits the invasive properties and hepatic growth of human colon cancer cells and improves the efficacy of oxaliplatin in p53-deficient colon cancer tumors in vivo. Mol Cancer Ther. 2007;6:2868–78. doi: 10.1158/1535-7163.MCT-07-0410. [DOI] [PubMed] [Google Scholar]
- 325.Clappier E, Gerby B, Sigaux F, Delord M, Touzri F, Hernandez L, et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med. 2011;208:653–61. doi: 10.1084/jem.20110105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Hochhaus A. Chronic myelogenous leukemia (CML): resistance to tyrosine kinase inhibitors. Ann Oncol. 2006;17(Suppl 10):x274–9. doi: 10.1093/annonc/mdl273. [DOI] [PubMed] [Google Scholar]
- 327.Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808–17. doi: 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- 328.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 329.Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci U S A. 2005;102:11011–6. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Deininger M. Resistance and relapse with imatinib in CML: causes and consequences. J Natl Compr Canc Netw. 2008;6(Suppl 2):S11–21. [PubMed] [Google Scholar]
- 331.Quintas-Cardama A, Kantarjian HM, Cortes JE. Mechanisms of primary and secondary resistance to imatinib in chronic myeloid leukemia. Cancer Control. 2009;16:122–31. doi: 10.1177/107327480901600204. [DOI] [PubMed] [Google Scholar]
- 332.Andreeff M, Konopleva M. Mechanisms of drug resistance in AML. Cancer Treat Res. 2002;112:237–62. doi: 10.1007/978-1-4615-1173-1_12. [DOI] [PubMed] [Google Scholar]
- 333.Thomas X, Campos L, Le QH, Guyotat D. Heat shock proteins and acute leukemias. Hematology. 2005;10:225–35. doi: 10.1080/10245330500093120. [DOI] [PubMed] [Google Scholar]
- 334.Haase D, Germing U, Schanz J, Pfeilstocker M, Nosslinger T, Hildebrandt B, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110:4385–95. doi: 10.1182/blood-2007-03-082404. [DOI] [PubMed] [Google Scholar]
- 335.Mrozek K. Cytogenetic, molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin Oncol. 2008;35:365–77. doi: 10.1053/j.seminoncol.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Cortes JE, Talpaz M, Giles F, O’Brien S, Rios MB, Shan J, et al. Prognostic significance of cytogenetic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapy. Blood. 2003;101:3794–800. doi: 10.1182/blood-2002-09-2790. [DOI] [PubMed] [Google Scholar]
- 337.Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122:872–84. doi: 10.1182/blood-2013-05-501569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367:2075–88. doi: 10.1056/NEJMoa1205127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Blay JY. A decade of tyrosine kinase inhibitor therapy: historical and current perspectives on targeted therapy for GIST. Cancer Treat Rev. 2011;37:373–84. doi: 10.1016/j.ctrv.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 340.Kanda T, Ishikawa T, Takahashi T, Nishida T. Nilotinib for treatment of gastrointestinal stromal tumors: out of the equation? Expert Opin Pharmacother. 2013;14:1859–67. doi: 10.1517/14656566.2013.816676. [DOI] [PubMed] [Google Scholar]
- 341.Iorio N, Sawaya RA, Friedenberg FK. Review article: the biology, diagnosis and management of gastrointestinal stromal tumours. Aliment Pharmacol Ther. 2014;39(12):1376–86. doi: 10.1111/apt.12761. [DOI] [PubMed] [Google Scholar]
- 342.Shoji S, Nakano M, Sato H, Tang XY, Osamura YR, Terachi T, et al. The current status of tailor-made medicine with molecular biomarkers for patients with clear cell renal cell carcinoma. Clin Exp Metastasis. 2014;31:111–34. doi: 10.1007/s10585-013-9612-7. [DOI] [PubMed] [Google Scholar]
- 343.Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. 2013;369:722–31. doi: 10.1056/NEJMoa1303989. [DOI] [PubMed] [Google Scholar]
- 344.Plosker GL, Figgitt DP. Rituximab: a review of its use in non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Drugs. 2003;63:803–43. doi: 10.2165/00003495-200363080-00005. [DOI] [PubMed] [Google Scholar]
- 345.Cheson BD, Leonard JP. Monoclonal antibody therapy for B-cell non-Hodgkin’s lymphoma. N Engl J Med. 2008;359:613–26. doi: 10.1056/NEJMra0708875. [DOI] [PubMed] [Google Scholar]
- 346.Agarwal A, Fleischman AG, Petersen CL, MacKenzie R, Luty S, Loriaux M, et al. Effects of plerixafor in combination with BCR-ABL kinase inhibition in a murine model of CML. Blood. 2012;120:2658–68. doi: 10.1182/blood-2011-05-355396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251–9. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- 348.Khoury HJ, Cortes JE, Kantarjian HM, Gambacorti-Passerini C, Baccarani M, Kim DW, et al. Bosutinib is active in chronic phase chronic myeloid leukemia after imatinib and dasatinib and/or nilotinib therapy failure. Blood. 2012;119:3403–12. doi: 10.1182/blood-2011-11-390120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Damon LE. Mobilization of hematopoietic stem cells into the peripheral blood. Expert Rev Hematol. 2009;2:717–33. doi: 10.1586/ehm.09.54. [DOI] [PubMed] [Google Scholar]
- 350.Uy GL, Rettig MP, Cashen AF. Plerixafor, a CXCR4 antagonist for the mobilization of hematopoietic stem cells. Expert Opin Biol Ther. 2008;8:1797–804. doi: 10.1517/14712598.8.11.1797. [DOI] [PubMed] [Google Scholar]
- 351.Hadzijusufovic E, Herndlhofer S, Aichberger KJ, Ghanim V, Suppan V, Cerny-Reiterer S, et al. Nilotinib exerts direct effects on vascular endothelial cells and may act as a co-trigger of atherosclerosis in patients with Ph plus CML. Blood. 2011;118:1183–4. [Google Scholar]
- 352.Perl A, Carroll M. BCR-ABL kinase is dead; long live the CML stem cell. J Clin Invest. 2011;121:22–5. doi: 10.1172/JCI43605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;119:1501–10. doi: 10.1182/blood-2010-12-326843. [DOI] [PMC free article] [PubMed] [Google Scholar]