

Abstract
Coenzyme Q10 (CoQ10) deficiency is an autosomal recessive disorder with heterogenous phenotypic manifestations and genetic background. We describe seven patients from five independent families with an isolated myopathic phenotype of CoQ10 deficiency. The clinical, histological and biochemical presentation of our patients was very homogenous. All patients presented with exercise intolerance, fatigue, proximal myopathy and high serum CK. Muscle histology showed lipid accumulation and subtle signs of mitochondrial myopathy. Biochemical measurement of muscle homogenates showed severely decreased activities of respiratory chain complexes I and II +III, while complex IV (COX) was moderately decreased. CoQ10 was significantly decreased in the skeletal muscle of all patients. Tandem mass spectrometry detected multiple acyl-CoA deficiency, leading to the analysis of the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene, previously shown to result in another metabolic disorder, glutaric aciduria type II (GAII). All of our patients carried autosomal recessive mutations in ETFDH, suggesting that ETFDH deficiency leads to a secondary CoQ10 deficiency. Our results indicate that the late-onset form of GAII and the myopathic form of CoQ10 deficiency are allelic diseases. Since this condition is treatable, correct diagnosis is of the utmost importance and should be considered both in children and in adults. We suggest to give patients both CoQ10 and riboflavin supplementation, especially for long-term treatment.
Keywords: coenzyme Q10 myopathy, ETFDH mutations, riboflavin and CoQ10 supplementation, late-onset glutaric aciduria type II
Introduction
Ubiquinone (coenzyme Q10 or CoQ10) is a lipid-soluble component of the cell membranes, where it functions as a mobile electron and proton carrier. Primary coenzyme Q10 (CoQ10) deficiency is thought to be an autosomal recessive disorder with five major phenotypes described: (1) an encephalomyopathic form with exercise intolerance, mitochondrial myopathy, myoglobinuria, seizures and ataxia (Ogasahara et al., 1989; Sobreira et al., 1997); (2) a multisystem infantile variant with encephalopathy, cardiomyopathy, ataxia, optic nerve atrophy, deafness and nephrotic syndrome resulting in renal failure (Rotig et al., 2000); (3) a predominantly cerebellar form with ataxia and cerebellar atrophy (Lamperti et al., 2003); (4) Leigh syndrome with growth retardation, ataxia and deafness (Maldergem et al., 2002) and (5) an isolated myopathic phenotype (Lalani et al., 2005; Horvath et al., 2006).
The concept that this clinical heterogeneity may reflect blocks at different levels in the complex biosynthetic pathway of CoQ10 is supported by the increasing number of molecular defects that are being identified in different clinical variants. Thus, two siblings of consanguineous parents with the multisystem infantile form of CoQ10 deficiency had autosomal recessive mutations in the COQ2 gene (Quinzii et al., 2006), which encodes para-hydroxybenzoate-polyprenyl transferase, whereas an infant with Leigh syndrome and nephrosis had autosomal recessive mutations in the PDSS2 gene, which encodes a subunit of COQ1, the first enzyme of CoQ10 biosynthesis (López et al., 2006). These defects were detected in single families, and the frequencies of mutations in COQ1 and COQ2 are still unknown. On the other hand, severe CoQ10 deficiency could be secondary to other genetic defects, as shown by a family with ataxia and CoQ10 deficiency harbouring mutations in the aprataxin gene (APTX) previously associated with ataxia oculomotor apraxia syndrome (AOA) [MIM606350] (Quinziii et al., 2005).
We have reported three patients with pure myopathy and CoQ10 deficiency in muscle manifesting as proximal myopathy with exercise intolerance and increased serum CK and lactate levels (Horvath et al., 2006). In two of the three patients, tandem mass spectrometry (TMS) showed increased levels of short-, medium-and long-chain acyl-carnitines, as described in multiple acyl-coenzyme-A deficiency (glutaric aciduria type II, GA II). These findings prompted us to search for mutations in the nuclear genes that cause multiple acyl-CoA deficiency.
Here, we describe pathogenic mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene in seven patients from five independent families fulfilling the clinical, histological and biochemical criteria of the myopathic form of coenzyme Q10 deficiency. Our results indicate that the late-onset form of GAII and the myopathic form of CoQ10 deficiency may be allelic diseases, which has important implications for therapy.
Patients and methods
Patients
Patients 1 and 2 were reported previously (Horvath et al., 2006). Briefly, patient 1 is a 34-year-old German woman with no family history of neuromuscular disease, in whom insulin-dependent diabetes mellitus was diagnosed at 14 years of age. Proximal muscle weakness started at age 32 during her first pregnancy and progressed rapidly. Serum CK was 2.000 U/l (normal <180) and increased further (up to 5000 U/l) after high doses of oral carnitine (3 g/day), lactate was 2.7 mmol/l (normal <2.0). Oral supplementation with high dose (500 mg/day) CoQ10 resulted in subjective improvement within a few days. After 1 month of supplementation, CK levels returned to normal and the patient reported a dramatic improvement of muscle strength, which was confirmed by manual and quantitative testing. After 6 months of supplementation, she was able to take care of her baby and all household chores. However, after 8 months of CoQ10 intake, the muscle symptoms reappeared, serum CK (2446 U/l) and lactate were again increased, and there was also an unexplained increase of the liver enzymes (GOT 259 U/l, normal < 32; GPT 226 U/l, normal <35; LDH 2175 U/l, normal <230). Serum TMS examinations showed multiple acyl-CoA derivatives and low carnitine. A repeated muscle biopsy was performed. Carnitine supplementation and a low-protein low-fat diet were ineffective, and associated with further worsening of symptoms. After sequencing the three genes responsible for GAII, we combined CoQ10 supplementation with riboflavin (100 mg/day), with marked and rapid improvement of her symptoms for the next 3 months. The elevation of LDH, CK and liver enzymes decreased to normal values and she was able to work and to take care of her 2-year-old daughter. CoQ10 supplementation was stopped and she was maintained on riboflavin therapy, which resulted in another deterioration of her symptoms within 3 weeks and prompted us to continue combined supplementation with CoQ10 and riboflavin (Table 1). Currently (12 months after the initiation of the combined CoQ10 and riboflavin treatment), she only has slight residual neck flexor weakness, and CK-levels are normal.
Table 1.
Follow-upTMS spectra of serum acylcarnitines in patient 1
| Follow-up | Before diagnosis | CoQ10 1000 mg/day | CoQ10 + low-protein, low-fat diet | CoQ10 + riboflavin without l-carnitine |
|---|---|---|---|---|
| Clinical symptoms | Subacute muscle Weakness, fatigue | Marked improvement | Weakness, ↑ liver Enzymes, re-biopsy | Complete recovery |
| Serum CK U/l normal< 180 | Up to 2000 | Normal | 2446 | Normal |
| C0 x-fold lower limit | Normal | Normal | 0.5 | 0.2 |
| C4 x-fold upper limit | 3.8 | Normal | 1.9 | Normal |
| C5 x-fold upper limit | 16.5 | 1.4 | 3.6 | Normal |
| C8 x-fold upper limit | 5.0 | 6.5 | 2.8 | Normal |
| C10 x-fold upper limit | 5.8 | 5.8 | 3.6 | Normal |
| C14 : 1x-fold upper limit | 8.9 | 1.8 | 6.4 | Normal |
Note: The combined elevation of short (C4, C5), medium (C8, C10) and long-chain (C14 : 1) acylcarnitines is characteristic of GA-II. Free carnitine (C0) was repeatedly diminished reflecting excretion of acylcarnitines in the urine. Reference ranges for free carnitine (C0) and acylcarnitines (μmol/l): C0 30–63; C4<0.6; C5<0.2; C8<0.3; C10<0.5; C14:1<0.3).
Patient 2 is a 29-year-old man of Turkish origin. His parents are consanguineous but there is no family history of similar disorders. Six months prior to examination, he developed premature fatigue, difficulty walking and weakness of shoulder, hip and neck muscles. Neurological examination showed bilateral scapular winging, waddling gait and proximal muscle weakness with decreased tendon reflexes. EMG showed myopathic alterations in proximal muscles. Nerve conduction velocities and brain MRI were normal. Serum CK was elevated up to 1000 U/l (normal <180), liver enzymes were normal and serum lactate was normal at rest but increased excessively after exercise (more than 10-fold). Oral supplementation with CoQ10 (150 mg/day) resulted in subjective improvement within 2 months. After documenting muscle CoQ10 deficiency, the dose of CoQ10 was increased to 500 mg/day. Within 3 months, CK levels normalized and the patient resumed work. After 1 year on CoQ10 alone, he is symptom-free, only his CK is mildly increased (320 U/l, normal <155). However, TMS examination showed accumulation of multiple acyl-CoA derivatives.
Patient 3, a 13-year-old boy, is the third of five children in a consanguineous Kurdish family. His parents and three of four siblings (age 19, 11 and 9 years) are healthy, while a 17-year-old sister is similarly affected (patient 4). Onset in the sister was at 14 years, with exercise intolerance, premature fatigue and proximal weakness. Serum CK was elevated at 1484 U/l (normal <180). There was no history of myoglobulinuria. Her proximal muscle strength was 3+/5 according to the MRC scale. There was no muscle hypertrophy. Our working diagnosis was either a limb-girdle dystrophy or a metabolic muscle disease. After reviewing the muscle biopsy from her affected brother (see later), supplementation with CoQ10 was initiated. This resulted in full recovery within 3 weeks. At the 4-month follow-up visit, her CK was 323 U/l. However, 9 months later her symptoms reappeared in the form of muscle cramps and weakness. At the time, her CK was significantly elevated (4824 U/l). No hepatopathy was observed. Interestingly, addition of riboflavin (100 mg/day) resulted in full recovery within a few days, with serum CK dropping to 141 U/l. Since then, several months have elapsed and she is totally symptom free. Her 13-year-old brother (patient 3) had just emerging symptoms. He recently began to experience muscle cramps and premature fatigue. No muscle weakness was detected. His CK was 2745 U/l. A muscle biopsy was performed from the vastus lateralis muscle. He was put on CoQ10, 200 mg/day, and his muscle complaints have ceased. However, 6 months after normalization of CK, a repeat test showed a CK of 373 U/l. In view of his sister’s response, he was also initiated on riboflavin 100 mg/day.
Patient 5 is a 12-year-old girl, the first child of a consanguineous Turkish family. Just 1 month prior to submission, she had subacute onset of muscle weakness and pain resembling polymyositis. She was weak and had a Gowers sign. Serum CK was 672 U/l at onset, and rose to 4188 U/l (normal <180), while liver enzymes were normal. EMG was normal. A metabolic myopathy was suspected and a muscle biopsy was performed. TMS examination showed accumulation of multiple acyl-CoA derivatives. Riboflavin, 100 mg/day, was introduced. This resulted in complete cessation of the muscle symptoms. One younger sister was found to be similarly affected (patient 6).
Patient 7 is a 13-year-old girl of a consanguineous Turkish family. She presented at age 12 with subacute onset of muscle weakness, myalgia and loss of ambulation within 20 days. Her serum CK was 538 U/l (normal <180), and liver enzymes were normal. A muscle biopsy was performed. TMS examination showed accumulation of multiple acyl-CoA derivatives. Riboflavin (100 mg/day) led to improvement over a few days and she started to walk again. At a 6-month follow-up visit she was doing fine. Several weeks later, her symptoms re-appeared when she incidentally stopped riboflavin. With re-initiation of riboflavin, she was back to normal. Two of her siblings had expired at ages 6 and 9 months with recurrent vomiting of unknown origin.
Muscle histology, biochemistry, CoQ10 measurement and tandem mass spectrometry (TMS)
Six micrometre thick serial cross-sections of muscle biopsy specimens were obtained for histochemical stains according to standard procedures. A frozen portion of the biopsy was used for biochemistry. Respiratory chain (RC) complexes I–IV activities were determined in skeletal muscle homogenate spectrophotometrically (Cary 50, VARIAN) as described (Fischer et al., 1986). In brief, frozen muscle specimen was first homogenized in SETH buffer (250 mM saccharose, 2 mM EDTA, 10 mM Tris–HCl pH 7.4, 50 U/ml heparin). For measuring NADH oxidation (complex I), we added 4 μg antimycin A to 480 μl test mix [28 mM KPO4, 2.825 mg/ml BSA, 0.56 mM MgCl2, 0.0225 mM NADH (Roche)] and after 10 min preincubation at 30°C, 15 μl decylubiquinon (1 mg/ml, Sigma) was added. Six minutes after, 20 μl muscle homogenate was added and the measurement was run for 2–5 min (till linearity). After adding 4 μg rotenone (Sigma), the estimated activity was corrected for rotenone-insensitive NADH activity. Succinate cytochrome c oxidoreductase (complexes II + III) activity was estimated spectrophotometrically after activation of the succinate dehydrogenase complex by preincubation for 10 min at 30°C of 460 μl of a test mix [20 mM KPO4 (pH 7.5), 2 mM EDTA, 2 mM NaN3, 0.7 μg/μl succinate, 0.004 μg/μl rotenon] and 8.4 mg cytochrome c (Sigma). The activity was estimated with and without 5 min preincubation with 125 μM CoQ1 on ice at 550 nm (cytochrome c).
In muscle homogenates, CoQ10 was measured by an HPLC method with UV detection (275 nm), using the Coenzym Q10 Kit (Chromsystems). The control range was determined on muscle biopsies of different age groups, without histological and biochemical evidence of a respiratory chain disease (n = 25). As disease controls, we measured CoQ10 in muscle of patients with primary respiratory chain disease and with fatty acid oxidation deficiency (n = 16). TMS was performed as described (Gempel et al., 2002).
Molecular genetic analysis
Genomic sequence of the ETFDH genes (Goodman et al., 2002) was performed in all five index patients, ETFA and ETFB genes were sequenced in two additional patients as described elsewhere (Goodman et al., 2002). RFLP analysis for the codon 1130 T>C (L377P) mutation in families of patients 2, 3, 4, 5 and 6 was performed by amplifying exon 10 of ETFDH with a forward mismatch primer, 5′-TGT-TTC-CTC-AGT-CTA-TAC-CAA-GA-3′ and with the intronic exon 10 reverse primer (Goodman et al., 2002). The PCR product was digested with the MlyI restriction enzyme, which cuts the 227 bp wild-type product into two bands (204 and 23 bp) and mutant remains uncut.
Results
Muscle histology, biochemistry, CoQ10 measurement and TMS
Muscle histology in all five index cases revealed similar findings: moderate to severe myopathy with small vacuoles in most type 1 fibres (Fig. 1A, E, H and K). However, the vacuolar change was most prominent in patient 5 (Fig. 1K). Sudan black/oil-red-O stains showed excessive lipid droplets, predominantly in type 1 fibres (Fig. 1D, G, J and L). A few COX negative fibres were detected in all cases (Fig. 1C and F), but ragged red fibres were seen only in patients 1 and 2 (Fig. 1I). SDH stain was faint in two cases (patients 5 and 7, data not shown).
Fig 1.

Histological findings of patient 1 (A–G), patient 2 (H–J), patient 3 (L) and patient 5 (K). Muscle biopsies of all patients show moderate to severe vacuolar myopathy (H&E stain: A, E, K; semithin section, toluidin blue: H); the vacuolar change was most prominent in type 1 fibers (ATPase pH 9.4: B). There are few COX negative fibres (asterixis in C, F). Sudan black (D, G) or Oil-Red-O (J, L) stain shows lipid accumulation in type 1 fibres of all patients. Ragged red fibres were seen rarely (Gomori trichrome stain: I). There is no significant difference between the first and second biopsy specimens of patient 1 (A–D; E–G) Bars in A–L adjusted to 30μm.
Biochemically, the activities of RC complexes I and II + III and COX were decreased in all patients, especially when referred to the elevated mitochondrial marker enzyme citrate synthase (CS). Muscle CoQ10 was decreased in all five cases. The biochemical results are summarized in Table 2. Because of clinical progression on long-term CoQ10 monotherapy, patient 1 underwent a second muscle biopsy. Muscle histology was similar to the first biopsy; however, biochemical measurements of respiratory chain enzymes and CoQ10 were within normal range.
Table 2.
Summary of the clinical, histological, biochemical and genetic data of our five patients carrying mutations in ETFDH
| Patient | Family history | Disease onset (years) | Clinical signs | Muscle histology | RC I (normal 0.17–0.56) | RC II +III (normal 0.08–0.45) | RC IV (normal 1.1–5.0) | CS (normal 45–100) | CoQ10 nmol/UCS (normal 2.7–7.0) | ETFDH mutations |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 F | Negative | 32 | Proximal myopathy, exercise intolerance, hepatopathy, | Lipid-accumulation RRF |
0.05 | 0.03 | 0.7 | 173 | 0.6 | het P456L het K590E |
| 2 M | Consanguinous parents | 29 | Proximal muscle weakness, scapular winging, myalgia, CK up to 1000 U/L | Lipid-accumulation RRF |
0.09 | 0.04 | 0.9 | 172 | 0.8 | hom L377P |
| 3 M | Consanguinous parents, one affected sister | 12 | Exercise intolerance, fatigue, CK 2745 U/L | Lipid-accumulation COX-ve fibers SDH-ve fibers |
0.00 | 0.01 | 0.1 | 149 | 0.8 | hom L377P |
| 5 M | Consanguinous parents, one affected sister | 12 | Subacute muscle weakness, exercise intolerance, myalgia, CK up to 4188 U/L | Lipid-accumulation | 0.02 | 0.02 | 0.7 | 173 | 1.28 | hom L377P |
| 7 F | Consanguinous parents | 12 | Proximal muscle weakness, myalgia, CK 538 U/L | Lipid-accumulation | 0.00 | 0.01 | 0.4 | 193 | 0.87 | hom P483L |
Note: RC complex activities are normalized to citrate synthase (CS); for the RC complexes and for CS : 1 unit means 1 μmol/min; for the coenzyme Q10 from control measurements (n = 25) median value was 4.7. CK normal<180 U/l.
To document that CoQ10 deficiency was reasonably specific in our patients and not a general finding in mitochondrial myopathies, we measured CoQ10 in 16 patients with different primary respiratory chain or fatty acid oxidation defects (single mtDNA deletions, n = 3; multiple mtDNA deletions, n = 2; A3243G MELAS mutation, n = 2; novel mtDNA tRNAGlu mutation, n = 1; complex III deficiency caused by CYTB mutation, n = 1; complex IV deficiency caused by SURF1 mutation, n = 1; autosomal dominant POLG1 mutation, n = 1; isolated complex III deficiency, n = 1; CPTII deficiency, n = 1; lipid storage myopathy of unknown origin, n = 1; myoglobinuria of unknown origin, n = 1; ataxia of unknown cause, n = 1). Notably, most of these patients had severe isolated or combined respiratory chain defects. No significant decrease in CoQ10 levels was observed in any of them, CoQ10 was slightly below the normal range (70% of mean control) only in the patient with CPTII deficiency. TMS was performed in four of our five index patients and showed a combined elevation of short (C4, C5), medium (C8, C10) and long-chain (C14:1) acylcarnitines (Table 1). This TMS result is characteristic of GAII. Free carnitine (C0) was repeatedly diminished reflecting excretion of acylcarnitines in the urine (Table 1).
Molecular genetic analysis
Sequence analysis of ETFDH in patient 1 revealed two heterozygous missense mutations, c.1367 C>T (P456L) and c.1768 A>G (K590E), in compound heterozygosity. The mutation c.1367 C>T (P456L) had been previously described in late-onset GAII patients (Goodman et al., 2002; Beresford et al., 2006). The mutation c.1768 A>G (K590E) affects a highly conserved amino acid residue (Fig. 2) in the C-terminal part of the protein. Each parent was heterozygous for one of the mutations. Patients 2, 3, 4, 5 and 6 harboured the same homozygous missense mutation, c.1130 T>C (L377P). As all these patients belonged to three families of Turkish or Kurdish origin, this implies that L377P is more frequent in this region. This mutation was not detected in 50 Turkish normal control samples and it co-segregated with the disease phenotype within the families (Fig. 3). One healthy sibling of patient 3 also carried the mutation in homozygous form, but this child was below the age of onset in his siblings. This mutation had not been described previously, but it affects a conserved amino acid (Fig. 2) in the carboxyl-terminal domain of the protein. Patient 7 harboured a novel homozygous missense mutation, c.1448 C>T (P483L) in the carboxyl-terminal domain, which had not been detected, previously. This mutation affects a conserved amino acid (Fig. 2) and is in close proximity to a previously described mutation (P456L). Mutations in this part of the protein affect most likely the catalytic activity and the stability of the tetramer (Westover et al., 2003).
Fig 2.
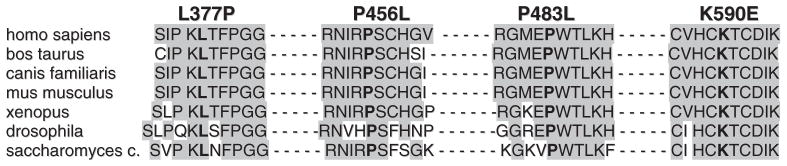
Conservation of the ETFDH mutations L377P, P456L, P483L and K590E.
Fig 3.

RFLP analysis for the mutation L377P in the family of patient 2. Presence of a homozygous state in muscle DNA (lane 2) and blood DNA (lane 7) of the patient.The mutation is heterozygous in both parents (motherçlane1; fatherçlane 3) and in one healthy sibling (lane 6), and absent in another healthy sibling (lane 4) and in the patients wife (lane 5) as well as in a normal control (lane 8).
Discussion
We present seven patients from five independent families with isolated myopathy, severe CoQ10 deficiency, and mutations in the ETFDH gene, previously associated with GAII. Our data indicate that ETFDH is the fourth gene associated with CoQ10 deficiency. Mutations in two genes involved in CoQ10 biosynthesis cause primary CoQ10 deficiency with infantile mitochondrial encephalomyopathy. Mutations in the aprataxin gene cause secondary CoQ10 deficiency associated with ataxia oculomotor apraxia (AOA1) and mutations in the ETFDH gene cause secondary CoQ10 deficiency associated with isolated myopathy.
Multiple acyl-CoA dehydrogenase deficiency or glutaric aciduria type II (GAII) is an autosomal recessive disorder of fatty acid and amino acid metabolism, due to a defect of electron transport from FAD-containing CoA dehydrogenases to CoQ in the mitochondrial electron transport chain (Przyrembel et al., 1976). The heterogeneous clinical features of patients with GAII fall into three main variants (Olsen et al., 2003). A neonatal-onset form (GAII type I) is characterized by congenital anomalies, severe non-ketotic hypoglycaemia, metabolic acidosis and excretion of large amounts of fatty acid- and amino acid-derived metabolites. A second neonatal-onset form (GAII type II) does not have congenital anomalies. A late-onset form (GAII type III) is characterized by hepatomegaly, lipid storage myopathy and recurrent episodes of lethargy, vomiting, hypoglycemia and metabolic acidosis often triggered by metabolic stress. Patients complain of muscle pain, exercise intolerance and proximal weakness, but glutaric aciduria is usually intermittent and coincides with worsening of the symptoms. All three clinical variants are caused by mutations in one of the three genes, two of which encode the alpha- and beta-subunits of the electron transfer flavoprotein (ETFA, OMIM : 608053; ETFB OMIM : 130410), while the third encodes ETF ubiquinone oxidoreductase (ETFDH OMIM 231675). All our patients harboured missense mutations in ETFDH and affected the C-terminal of the protein. Mutations in this protein domain most likely impair the catalytic activity and the folding/stability of the protein (Olsen et al., 2003; Westover et al., 2003). Nonsense mutations and missense mutations in the N-terminal part of the protein usually lead to the more severe neonatal GAII type I or type II. However, some cases of GAII are due to as yet unidentified disturbances of riboflavin metabolism. Riboflavin (vitamin B2) is the co-factor shared by ETF, ETFDH and all acyl-CoA dehydrogenases. Therapy with riboflavin, carnitine and low-fat, low-protein diet is beneficial, although the long-term treatment of patients with late-onset GAII is still challenging (Olsen et al., 2003).
The discovery of pathogenic ETFDH mutations in our patients with myopathic CoQ10 deficiency shows that late-onset GAII, usually caused by less severe mutations in the ETFDH gene, probably are one and the same disease, although the severe defect of CoQ10 in muscle was initially considered a primary aetiological event. This concept provides important clues to pathophysiology and useful tools for therapy.
How can ETFDH deficiency result in CoQ10 deficiency? As CoQ10 is the direct acceptor of electrons from ETF, it stands to reason that lack (or dysfunction) of the reducing enzyme could—via some feedback mechanism—downregulate the synthesis of CoQ10. Alternatively, faulty binding of the enzyme to CoQ10 could result in excessive degradation of the acceptor molecule (Olsen et al., 2003). CoQ10 had not been measured in previous patients with GAII, although it was repeatedly reported that respiratory chain complexes were decreased (Beresford et al., 2006). Respiratory chain dysfunction and clinical presentation suggest that the physiopathology of GAII may well be related to CoQ10 deficiency. Regarding the crucial point of ‘primary’ versus ‘secondary’ CoQ10 deficiency, our position is that the term ‘primary CoQ10 deficiency’ should be restricted to defects of CoQ10 biosynthesis, such as those due to COQ1 or COQ2 mutations, whereas defects of CoQ10 due to mutations in aprataxin or ETFDH are examples of secondary CoQ10 deficiency.
Not surprisingly, the clinical presentation of late-onset GAII patients (de Visser et al., 1986; Di Donato et al., 1986; Bell et al., 1990; Olsen et al., 2004) is very similar to that of our cases, with subacute (3–6 months) exercise intolerance and proximal weakness affecting predominantly hip and shoulder girdle muscles. Muscle weakness fluctuates and often worsens during intermittent infections, fasting, catabolic stress or pregnancy. Neck flexor weakness is relatively typical and was prominent in all patients described here. Episodes of hepatopathy, vomiting and somnolence or stupor (Reyés syndrome-like crises) were common in previously described patients with GAII (de Visser et al., 1986; Di Donato et al., 1986; Bell et al., 1990; Olsen et al., 2004), but muscle weakness and exercise intolerance usually preceded these events. Some patients also had respiratory failure requiring assisted ventilation. Of our patients, only one (patient 1) had an episode of weakness accompanied by LDH and liver transaminase elevation, which resolved over several weeks, whereas the other six had isolated myopathy, with no evidence of hepatopathy or encephalopathy, and none of our patients showed involvement of the respiratory muscles. The lack of extramuscular symptoms explains why initially we did not suspect GAII. Conversely, our cases show that GAII may present as a pure myopathy without clinical signs of a systemic metabolic disease.
In the late-onset form of GAII and in previous cases with the myopathic variant of CoQ10 deficiency, onset of symptoms was before age 15 years. Thus, it is noteworthy that two of our patients were 32 and 29 years old at presentation, implying that this diagnosis should be considered even in adult-onset cases.
Muscle histology in the five patients who were biopsied showed lipid storage myopathy with subtle signs of mitochondrial dysfunction, and biochemical measurement of respiratory chain enzymes showed reduced activities of complexes I and II + III and increased activity of citrate synthase. Thus, lipid storage myopathy and respiratory chain dysfunction are hallmarks of the disease. TMS suggested multiple acyl-CoA dehydrogenase deficiency in the four patients in whom it was performed.
In patient 1, the TMS profile and the low level of free carnitine in serum suggested a block in mitochondrial fatty acid oxidation and led to the genetic diagnosis of GAII.
The therapy and follow-up of our patients led us to important conclusions. After 3–6 months of CoQ10 supplementation, all patients showed dramatic clinical improvement and normalization of serum CK and lactate levels. This was also true in all other reported cases with the myopathic phenotype (Lalani et al., 2005; Horvath et al., 2006). As our initial diagnosis was primary myopathic CoQ10 deficiency, in four of our patients we initiated high-dose CoQ10, which resulted in prominent clinical and biochemical improvement. After diagnosing GAII, we added to the therapy of patient 1 a low-protein low-fat diet: far from improving, after 3 weeks on the diet, she became weaker, was unable to walk more than 100 m, had frequent vomiting and liver enzymes increased. After stopping the diet and introducing riboflavin supplementation (100 mg/day) she recovered within a few days. After 3 months of combined CoQ10 and riboflavin therapy, she was completely normal. Because of the good condition and cooperation of the patient, we stopped CoQ10 supplementation and continued with riboflavin monotherapy, but after 3 weeks the reappearance of proximal muscle weakness prompted us to continue with combined riboflavin and CoQ10 supplementation. It seems that patients with ETFDH deficiency in long-term need both CoQ10 and riboflavin to maintain a good muscle function. Because of the additional carnitine deficiency, carnitine supplementation was repeatedly tried, but never resulted in improvement, rather worsening of symptoms.
For cases 5 and 7, riboflavin was given alone as a single agent just based on the pattern of TMS screening which originally denoted a GAII pattern. This scheme really worked well, and they currently are not in need of CoQ10. Of course, longer follow-up is required.
We sequenced ETFDH in 10 other patients with CoQ10 deficiency. Eight of these patients presented with ataxia and epilepsy and only two showed myopathy. One of them was a 7-year-old boy with normal TMS result (patient 3 in Horvath et al., 2006), and the other was a 35-year-old woman with delayed motor milestones, partial complex seizures, exercise intolerance and recurrent myoglobinuria (Sobreira et al., 1997). Mutations of the ETFDH gene were not detected in any of these cases, suggesting further genetic heterogeneity.
Since CoQ10 deficiency/late-onset GAII is treatable, correct diagnosis is of utmost importance and should be considered both in children and in adults. The association of high-serum CK, proximal myopathy (with or without hepatopathy or encephalopathy), multiple acyl-CoA deficiency on TMS, lipid storage myopathy and decreased activity of respiratory chain complexes I and II + III (and IV) attributable to lack of CoQ10 in muscle homogenate are key tell–tale signs of the disease. We would suggest that patients should be kept on both CoQ10 and riboflavin supplementation, especially on the protracted course.
Acknowledgments
The authors thank Ira Kaus, Manja Thorwirt, Andrea Zöllner and Eva Schmidtmeyer for technical assistance. KG is supported by a grant from the Stiftung Pathobiochemie der Deutschen Gesellschaft für Klinische Chemie und Labormedizin (DGKL). BGS, PS and HL are members of the German network on muscular dystrophies (MD-NET, 01GM0302) funded by the German ministry of education and research (BMBF, Bonn, Germany). MD–NET is a partner of TREAT–NMD (EC, 6th FP, proposal # 036825; www.treat-nmd.eu). SDM is supported by a grant from the Muscular Dystrophy Association. HP is supported by the German National Genome Network (BMBF O1GR0411). RH is supported by a grant from the Deutsche Forschungsgemeinschaft (HO 2505/2-1).
Abbreviations
- TMS
tandem mass spectrometry
References
- Bell RB, Brownell AKW, Roe CR, Engel AG, Goodman SI, Frerman FE, et al. Electron transfer flavoprotein; ubiquinone oxidoreductase (ETF;QO) deficiency in an adult. Neurology. 1990;40:1779–82. doi: 10.1212/wnl.40.11.1779. [DOI] [PubMed] [Google Scholar]
- Beresford MW, Pourfarzam M, Turnbull DM, Davidson JE. So doctor, what exactly is wrong with my muscles? Glutaric aciduria type II presenting in a teenager. Neuromuscul Disord. 2006;16:269–73. doi: 10.1016/j.nmd.2006.01.001. [DOI] [PubMed] [Google Scholar]
- de Visser M, Scholte HR, Schutgens RBH, Bolhuis PA, Luyt-Houwen IEM, Vaandrager-Verduin MHM, et al. Riboflavin-responsive lipid-storage myopathy and glutaric aciduria type II of early adult onset. Neurology. 1986;36:367–72. doi: 10.1212/wnl.36.3.367. [DOI] [PubMed] [Google Scholar]
- Di Donato S, Frerman FE, Rimoldi M, Rinaldo P, Taroni F, Wiesmann UN. Systemic carnitine deficiency due to lack of electron transfer flavoprotein; ubiquinone oxidoreductase. Neurology. 1986;36:957–63. doi: 10.1212/wnl.36.7.957. [DOI] [PubMed] [Google Scholar]
- Fischer JC, Ruitenbeek W, Gabreels FJ, Janssen AJ, Renier WO, Sengers RC, et al. A mitochondrial encephalomyopathy: the first case with an established defect at the level of coenzyme Q. Eur J Pediatr. 1986;144:441–4. doi: 10.1007/BF00441735. [DOI] [PubMed] [Google Scholar]
- Gempel K, Kiechl S, Hofmann S, Lochmuller H, Kiechl-Kohlendorfer U, Willeit J, et al. Screening for carnitine palmitoyltransferase II deficiency by tandem mass spectrometry. J Inherit Metab Dis. 2002;25:17–27. doi: 10.1023/a:1015109127986. [DOI] [PubMed] [Google Scholar]
- Goodman SI, Binard RJ, Woontner MR, Frerman FE. Glutaric acidemia type II: gene structure and mutations of the electron transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO) gene. Mol Genet Metab. 2002;77:86–90. doi: 10.1016/s1096-7192(02)00138-5. [DOI] [PubMed] [Google Scholar]
- Horvath R, Schneiderat P, Schoser BGH, Gempel K, Neuen-Jacob E, Plöger H, et al. Coenzyme Q10 deficiency may cause isolated myopathy. Neurology. 2006;66:253–5. doi: 10.1212/01.wnl.0000194241.35115.7c. [DOI] [PubMed] [Google Scholar]
- Lalani SR, Vladutiu GD, Plunkett K, Lotze TE, Adesina AM, Scaglia F. Isolated mitochondrial myopathy associated with muscle coenzyme Q10 deficiency. Arch Neurol. 2005;62:317–20. doi: 10.1001/archneur.62.2.317. [DOI] [PubMed] [Google Scholar]
- Lamperti C, Naini A, Hirano M, De Vivo DC, Bertini E, Servidei S, et al. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology. 2003;60:1206–8. doi: 10.1212/01.wnl.0000055089.39373.fc. [DOI] [PubMed] [Google Scholar]
- López LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJT, Naini A, et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79:1125–30. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldergem LV, Trijbels F, DiMauro S, Sindelar PJ, Musumeci O, Janssen A, et al. Coenzyme Q-responsive Leigh’s encephalopathy in two sisters. Ann Neurol. 2002;52:750–4. doi: 10.1002/ana.10371. [DOI] [PubMed] [Google Scholar]
- Ogasahara S, Engel AG, Frens D, Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci. 1989;86:2379–82. doi: 10.1073/pnas.86.7.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RKJ, Andresen BS, Christensen E, Bross P, Skovby F, Gregersen N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl CoA dehydrogenation deficiency. Human Mutat. 2003;22:12–23. doi: 10.1002/humu.10226. [DOI] [PubMed] [Google Scholar]
- Olsen RKJ, Pourfarzam M, Morris AAM, Dias RC, Knudsen I, Andresen BS, et al. Lipid-storage myopathy and respiratory insufficiency due to ETFQO mutations in a patient with late-onset multiple acyl-CoA dehydrogenation deficiency. J Inher Met Dis. 2004;27:671–8. doi: 10.1023/b:boli.0000042986.10291.e9. [DOI] [PubMed] [Google Scholar]
- Przyrembel H, Wendel U, Becker K, Bremer HJ, Bruinvis L, Ketting D, et al. Glutaric aciduria type II: report on a previously undescribed metabolic disorder. Clin Chim Acta. 1976;66:227–39. doi: 10.1016/0009-8981(76)90060-7. [DOI] [PubMed] [Google Scholar]
- Quinzii CM, Kattah AG, Naini A, Akman HO, Mootha VK, DiMauro S, et al. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology. 2005;64:539–41. doi: 10.1212/01.WNL.0000150588.75281.58. [DOI] [PubMed] [Google Scholar]
- Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, Dimauro S, et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. 2006;78:345–9. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotig A, Appelkvist EL, Geromel V, Chretien D, Kadhom N, Edery P, et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356:391–5. doi: 10.1016/S0140-6736(00)02531-9. [DOI] [PubMed] [Google Scholar]
- Sobreira C, Hirano M, Shanske S, Keller RK, Haller RG, Davidson E, et al. Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology. 1997;48:1238–43. doi: 10.1212/wnl.48.5.1238. [DOI] [PubMed] [Google Scholar]
- Westover JB, Goodman SI, Frerman FE. Pathogenic mutations in the carboxyl-terminal domain of glutaryl-CoA dehydrogenase: effects on catalytic activity and the stability of the tetramer. Mol Genet Metab. 2003;79:245–56. doi: 10.1016/s1096-7192(03)00109-4. [DOI] [PubMed] [Google Scholar]