

Abstract
Novel genes are now identified at a rapid pace for many Mendelian disorders, and increasingly, for genetically complex phenotypes. However, new challenges have also become evident: (1) effectively managing larger exome and/or genome datasets, especially for smaller labs; (2) direct hands-on analysis and contextual interpretation of variant data in large genomic datasets; and (3) many small and medium-sized clinical and research-based investigative teams around the world are generating data that, if combined and shared, will significantly increase the opportunities for the entire community to identify new genes. To address these challenges we have developed GEnomes Management Application (GEM.app), a software tool to annotate, manage, visualize, and analyze large genomic datasets (https://genomics.med.miami.edu/). GEM.app currently contains ~1,600 whole exomes from 50 different phenotypes studied by 40 principal investigators from 15 different countries. The focus of GEM.app is on user-friendly analysis for non-bioinformaticians to make NGS data directly accessible. Yet, GEM.app provides powerful and flexible filter options, including single family filtering, across family/phenotype queries, nested filtering, and evaluation of segregation in families. In addition, the system is fast, obtaining results within 4 seconds across ~1,200 exomes. We believe that this system will further enhance identification of genetic causes of human disease.
Keywords: Next-generation sequencing, Exome sequencing, Next-generation sequencing analysis
INTRODUCTION
The development of next-generation sequencing technologies has revolutionized human genetics research (Biesecker, 2010). Whole exome sequencing (WES), an early genomic application, is a rapid, high-throughput, and cost-effective approach and has been widely used to identify pathogenic variation especially in Mendelian disorders (Velinov et al., 2012; Ng et al., 2010). In addition, the recent validation of an excess of rare variation (<0.5% minor allele frequency) in the human species provides further support for the hypothesis that rare changes explain potentially a significant portion of inheritance in so-called complex human diseases (Tennessen et al., 2012). Such considerations have further stimulated the large-scale production of genomic datasets leading to significant challenges for efficient data management and analysis. The data-intensive nature and lengthy computational pipelines for genomic data have also increasingly removed clinically and molecular trained investigators from direct access to data analysis. This leads to missed opportunities in identifying novel causative gene variants and creates unnecessary bottlenecks in the discovery process. Finally, large disease-oriented research consortia and collaborative networks of investigators are seeking new ways for communal data analysis and the sharing of variant data.
To address these concerns, a number of tools have been developed to analyze and visualize genomic variant data. While these available tools are very useful, they are also limited in specific ways. VAAST (Yandell et al., 2011) is a powerful tool to identify genes likely involved in disease, but is not intended for the browsing of variant and annotation data. In order to visualize variant data, VARSIFTER (Teer et al., 2012) was developed, but is designed for a desktop computer and can presently only manage a modest amount of data. Lastly, the Sequence Variant Analyzer (Ge et al., 2011) includes many powerful analysis packages, but is rather complex and doesn’t easily facilitate collaborative efforts. To address these limitations, we have developed Genomes Management Application (GEM.app), which is an analysis toolset accessible via modern web browsers allowing for easy, quick, and collaborative analysis of genomic data. GEM.app is currently growing significantly in size and we are observing the identification of disease genes at the pace of >1 per month (Velinov et al., 2012; Montenegro et al., 2012; Martin et al., 2013; Tesson et al., 2012; Gonzalez et al., in press).
METHODS
Bioinformatics
The Illumina CASAVA v1.8 pipeline was used to produce 100bp sequence reads. BWA software (Li and Durbin, 2010) was used to align sequence reads to the human genome (hg19) and variants were called using the GATK v1.4 software package (DePristo et al., 2011; McKenna et al., 2010). Variants were submitted to SeattleSeq for annotation. Further annotation was obtained using data from dbSNP137, variant frequency data from the NHLBI Exome Sequencing Project (Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA Project (Exome Variant Server, 2012), the HGMD Human Gene Mutation Database (Stenson et al., 2012), and the OMIM Online Mendelian Inheritance in Man database (OMIM Online Mendelian Inheritance in Man, December, 2012).
GEDI pipeline and GEM.app GUI
GEDI is an automated pipeline that uses different available software tools (VCFtools and ENSEMBL VEP) and PERL scripts to automate processing, annotation, backfilling of VCF files and data upload into our GEM.app database (mySQL 5.5). GEDI is optimized to extract relevant information for each variant and to transform and structure the genomic information to guarantee fast query execution using the GEM.app GUI. The GEM.app GUI was implemented in layers to allow enough flexibility to handle data efficiently in a fast growing environment. In order to execute queries, API’s were developed using PHP 5.3. GEM.app’s user friendly interface was built with JQUERY, HTML5, JSON, CCS3. Slickgrid was used to display query results.
RESULTS
Basic principles and description of GEM.app
Several design principles went into the development of GEM.app. We created a graphical user interface for this web application that makes genomic data accessible for physician scientists and molecular trained PhDs (Figure 1). The easy to use interface allows a user to design custom queries and results are typically returned in seconds. This is a key feature as it allows for an iterative working approach, instant refining of filters, and immediate testing of different Mendelian segregation patterns – even if hundreds of exomes are being queried. GEM.app is a web application developed using the latest Internet standards and languages – JQuery, JSON, HTML5, SlickGrid and is compatible with Safari 5.1.7, Chrome 22.0.1229.94, and Firefox 16.0.2 or later versions. We have developed a powerful access-control system that assigns an account to each user. This account system is context-aware and shows or hides phenotype-specific customized filter-options and variant annotation. Each user only sees their own detailed data; yet, visible to all users are anonymous variant counts derived from the entire database. Examples include global minor allele and genotype frequencies in GEM.app, number of familial segregation events of a specific variant under a selected Mendelian trait, or number of SNVs/indels per gene of interest. These latter features encourage collaboration for studies that are focused on rare Mendelian-type variants, as they give hints on the existence of a second family for a given new candidate gene. The access system further allows for sharing of access to specific exomes and a collaborative analysis. Examples of existing successful collaborations in GEM.app include the international Inherited Neuropathy Consortium with >250 exomes in GEM.app and a network of 15 collaborating groups in as many countries working on hereditary spastic paraplegia (>400 exomes).
Figure 1. GEM.app pipeline and Graphical User Interface.

A) Starting page with currently seven analysis modules. B) Example of “Variants within families” filter module. There are at least 13 context-specific different filter categories available. Preset filters auto-fill a number variables and allow for a “three click” search. C) Results screen of a GEM.app query. Different control options are detailed.
Data security
We only store de-identified data in GEM.app. This includes a numerical identifier for each sample and family, sex, and de-identified pedigrees. Access is password controlled and all data transfer between users and servers is encrypted by a VeriSign class 3 server certificate, which is comparable to online banking security. Further, GEM.app currently resides on servers of the University of Miami, which are behind a firewall and monitored 24/7 for cyber attacks. In the future, we envision moving GEM.app to a true cloud environment.
GEDI pipeline
Part of GEM.app is a processing pipeline, the GEnome Data Import module (GEDI), which handles processing of VCF files, annotation, “backfilling” of variant data, co-segregation analysis within families, and calculation of counts across all samples, including minor allele frequencies (Supplementary Figure 2). When data is processed and imported into GEM.app, information on each sample is required such as: affection status, pedigree individual ID, family ID, and possible Mendelian inheritance patterns. Using this information for each sample allows GEDI to automatically determine whether a specific variant follows a given segregation model (i.e. autosomal dominant). If a user selects a particular inheritance pattern to analyze, GEM.app will return variants that fit this model in a table format (Figure 1C). GEDI achieves annotation by utilizing the SeattleSeq annotation server (http://snp.gs.washington.edu/), which includes conservation and amino acid substitution scores (GERP, PhastCons, Grantham, PolyPhen2) (Davydov et al., 2010; Siepel et al., 2005; Adzhubei et al., 2010). In addition, GEDI obtains data from dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), NHLBI EVS (http://evs.gs.washington.edu/EVS/), OMIM (http://www.ncbi.nlm.nih.gov/omim), String-db (http://string-db.org/), and ENSEMBL (http://www.ensembl.org/). Backfilling is a process that retrieves sequence information from all previously added VCF files for each novel variant incorporated into GEM.app. Each addition of new exomes initiates a reanalysis of variant counts, recalculation of allele and genotype frequencies, etc. The GEDI process is fully automated and takes advantage of the 5,000-node computer cluster named Pegasus at the University of Miami.
Graphical User Interface
After the GEDI/GEM.app pipeline is completed, data is accessible to registered users through an online graphical user interface (https://genomics.med.miami.edu). Users interact with query modules, which are presented as tiles with descriptive names. Currently seven different modules are available (Figure 1a). These include simple modules (“Quick finds”) that search for genes (“Gene look-up”) or genomic positions (“Position look-up”). Further, all accessible exomes are individually listed (“My samples”) complete with basic phenotypes, external and internal numbering, quality measures from alignment and variant calling, possible traits, processing details, and other information. Most queries happen in extended filtering modules. “Variants within families” provides detailed options for variant filtering within single families, but can process as many families at once as requested. Fifteen different filter option fields are present, including selection of genomic positions, variant function class (synonymous, non-synonymous, etc.), conservations scores, quality scores, Mendelian traits, or lists of known genes for phenotypic groups (i.e. inherited peripheral neuropathy genes) (Figure 1b). The module “Genes across families” allows for identifying genes that have multiple hits in the same gene across multiple families and/or phenotypes. Advanced filter modules contain two-step or nested queries. This allows for filtering with a strict set of criteria resulting in few hits in a first step and then in a second step these gene are taken into account for a query with more relax criteria to find additional evidence for a gene in a larger set of exomes. All filter modules have the option of choosing from four pre-set filter criteria: user-defined, relaxed, moderate, or strict (Figure 1b).
The output is presented in a table that contains 28 different columns with annotation. Each row presents a variant. There are an additional 44 columns of annotation that can be added via a “column manager” (Supplementary Figure 1). Columns can also be sorted and rearranged via drag and drop. Several fields in the output table contain hyperlinks that directly link to pedigrees, to the UCSC genome browser (http://genome.ucsc.edu/), a gene-network viewer (http://string-db.org), NCBI (http://www.ncbi.nlm.nih.gov/), or OMIM (http://omim.org/) (Supplementary Figure 1). GEM.app connects several publicly available databases directly to data being analyzed within the same webpage, thus making follow-up analysis less tedious (Figure 1c).
Performance
Queries of individual families are typically finished in less than one second. Benchmarking of more than 10,000 queries of over 60 different users demonstrate that all the different query modules achieve an average query time of <8 seconds, with the exception of the two-step module (Figure 3). The most popular query module, “Variants within families”, produces output in ~4 seconds. Factors that negatively influence query times include: 1) querying across multiple phenotypes, 2) using fewer filter criteria, and 3) using nested filter options. Generally, the speed and simplicity of GEM.app allows an investigator to iteratively test different filtering strategies (such as multiple possible traits) within a few minutes without the need of programming. Searching across a large number of samples/families is fast: a query for conserved and rare mutations in 124 known genes for related neurodegenerative diseases (Charcot-Marie-Tooth, Hereditary Spastic Paraplegia, distal Hereditary Motor Neuron, etc.) across 481 exomes obtained results within 10 seconds. Querying these same 124 genes across 1,200 samples finished within 20 seconds.
Figure 3. Performance of GEM.app.
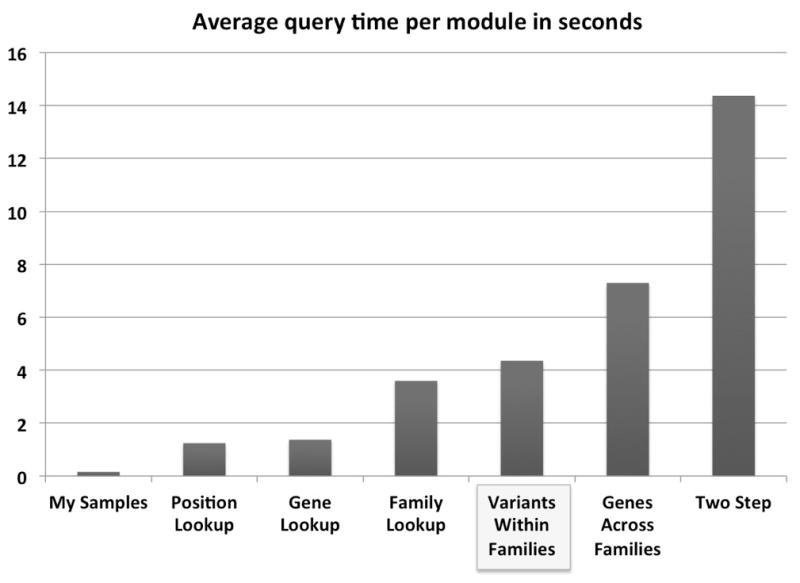
Average search times over 10,000 queries from over 60 different users. By far the most popular module is “Variants within families” which returns results in ~4 second across 1,200 exomes. Individual families are typically instantly returned.
Currently, 103 users are registered to use GEM.app, which includes 40 principle investigators from 15 different countries studying over 50 different phenotypes (Figure 2a). Since the release of GEM.app in April 2012, we have experienced a significant increase in the number of users and queries (Figure 2b and 2c).
Figure 2. Current usage of GEM.app.
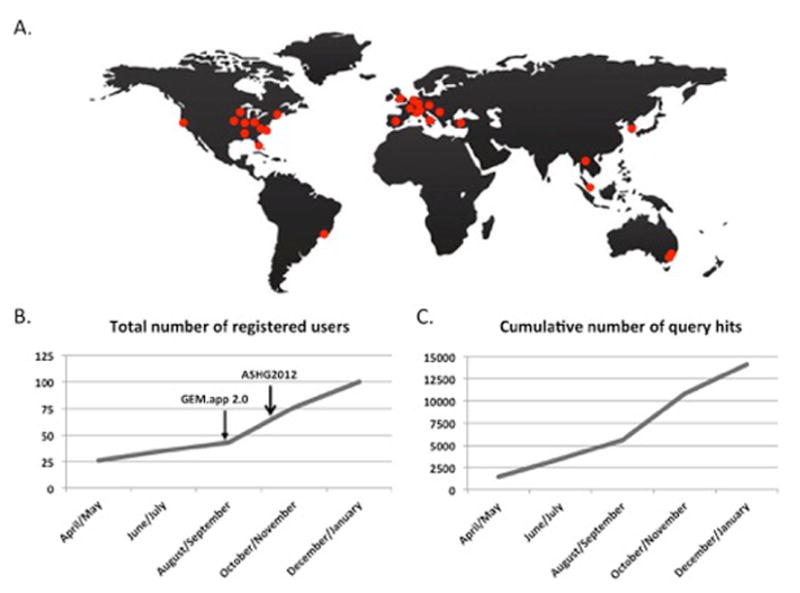
A) Geographical scheme of principle investigators with data in GEM.app. B) The number of registered users has grown to >103 in the past 6 months. C) The usage of GEM.app has increased significantly since its release. ASHG – American Society for Human Genetics annual meeting.
DISCUSSION
The development of GEM.app was motivated by the need of a next-generation sequencing analysis tools for physician scientists and biomedical investigators with limited computational experience. Importantly, we needed a tool to manage and organize large sets of exome data as they become available from large-scale sequencing projects. Increasing sample sizes are required to identify genes for rare, highly heterogeneous Mendelian disorders and rare familial forms of phenotypes with a complex genetic architecture. The modular structure of GEM.app allows for future implementation of new computational strategies, such as multi-core processing and multi-threading, to address the increasing size of data and scaling to whole genomes. Since investigators worldwide are producing small to large exome/genome datasets the exchange of variant information and uniform analysis provides an often untapped opportunity for increasing genetic power via collaborations. A variety of collaborative models from loose networks to tightly integrated consortia are feasible within the flexible access system in GEM.app. Existing projects range from the ability of direct shared access to full exomes at different sites to using the anonymous variant counts across all datasets, which are available to every registered user. Further, consortia can decide to limit data analysis to specific sites, or, potentially more advantageous, data analysis can be spread over a larger number of institutions. The intuitive interface allows for the participation of investigators with different skill sets, including expert geneticists, physician scientists, or basic scientists.
The GEM.app framework has recently been utilized to identify clinically relevant variants in a number of disorders, such as inherited deafness, Charcot-Marie-Tooth disease (CMT), Hereditary-Spastic Paraplegia (HSP), and dilated-cardiomyopathies (DCM) (Diaz-Horta et al, 2012; Montenegro et al., 2011; McCorquodale et al., 2011; Montenegro et al., 2012; Norton et al., 2012). GEM.app has also been applied to identify novel genes (Velinov et al., 2012; Montenegro et al., 2012; Martin et al., 2013; Tesson et al., 2012; Osterloh et al., 2012; Sirmaci et al., 2011). The majority of these studies are led by physician scientists interested in the application of WES for the elucidation of genetic variation involved in their disorders of interest, thus demonstrating the power of GEM.app to connect investigators to their NGS datasets. Some of the newer findings have only been able to accomplish by connecting relatively small datasets from around the world in this centralized resource and the ability of individual investigators to share their results on a trusted platform.
In summary, GEM.app will enable researchers of all computational backgrounds to visualize and analyze genomic variant data. Using an automated pipeline, GEM.app organizes thoroughly annotated variant data to be directly connected to various genomics resources and allows investigators to directly analyze and interpret mutations from large sets of samples. GEM.app offers the ability for biomedical researchers to share data and perform joint analysis simultaneously. These features promote collaborations leading to the identification of novel disease associated genes.
Supplementary Material
(A) The column manager allows for adding up to an additional 44 columns, deleting columns or resorting of columns. (B) An example of a public resource link-out in GEM.app is the STRING database.
{kind=link}
1) Raw sequence reads are aligned to reference and variants are called with available tools resulting in VCF files (ie BWA/GATK).; 2) From VCF files GEDI extracts variants to annotate and backfill all previous samples in GEM.app. GEDI also performs standard calculations, such as genotype counts across GEM.app samples and segregation analysis.; 3) After the GEDI pipeline is completed, analysis can be performed using the GEM.app GUI.
{kind=link}
Acknowledgments
We are thankful to Yamil Velez for helpful discussions about details of data mining. The study was supported by NINDS (1R01NS075764, 5R01NS052767). Michael Gonzalez is supported by a NIH supplement for promoting diversity in health related fields (1R01NS072248-02S2).
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker LG. Exome sequencing makes medical genomics a reality. Nat Genet. 2010;42:13–14. doi: 10.1038/ng0110-13. [DOI] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Horta O, Duman D, Foster J, 2nd, Sirmaci A, Gonzalez M, Mahdieh N, Fotouhi N, Bonyadi M, Cengiz FB, Menendez I, Ulloa RH, Edwards YJ, Züchner S, Blanton S, Tekin M. Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS One. 2012;7:e50628. doi: 10.1371/journal.pone.0050628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exome Variant Server. NHLBI exome sequencing project (ESP) 2012 [Google Scholar]
- Ge D, Ruzzo EK, Shianna KV, He M, Pelak K, Heinzen EL, Need AC, Cirulli ET, Maia JM, Dickson SP, Zhu M, Singh A, Allen AS, Goldstein DB. SVA: Software for annotating and visualizing sequenced human genomes. Bioinformatics. 2011;27:1998–2000. doi: 10.1093/bioinformatics/btr317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M, Nampoothiri S, Kornblum C, Oteyza AC, Walter J, Konidari I, Hulme W, Speziani F, Schöls L, Züchner S, Schüle R. Mutations in Phospholipase DDHD2 cause Autosomal Recessive Hereditary Spastic Paraplegia (SPG54) Eur J Hum Genet. 2013 doi: 10.1038/ejhg.2013.29. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Schule R, Smets K, Rastetter A, Boukhris A, Loureiro JL, Gonzalez MA, Mundwiller E, Deconinck T, Wessner M, Jornea L, Caballero Oteyza AC, Durr A, Martin JJ, Schols L, Mhiri C, Lamari F, Züchner S, De Jonghe P, Kabashi E, Brice A, Stevanin G. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am J Hum Genet. doi: 10.1016/j.ajhg.2012.11.021. Accepted, 2012. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorquodale DS, 3rd, Montenegro G, Peguero A, Carlson N, Speziani F, Price J, Taylor SW, Melanson M, Vance JM, Zuchner S. Mutation screening of mitofusin 2 in charcot-marie-tooth disease type 2. J Neurol. 2011 doi: 10.1007/s00415-011-5910-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenegro G, Powell E, Huang J, Edwards YJK, Beecham G, Hulme W, Siskind C, Vance J, Shy M, Züchner S. Exome sequencing allows for rapid gene identification in a charcot-marie-tooth disease family. Annals of Neurology. 2011;3:464–70. doi: 10.1002/ana.22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenegro G, Rebelo AP, Connell J, Allison R, Babalini C, D’Aloia M, Montieri P, Schule R, Ishiura H, Price J, Strickland A, Gonzalez MA, Baumbach-Reardon L, Deconinck T, Huang J, Bernardi G, Vance JM, Rogers MT, Tsuji S, De Jonghe P, Pericak-Vance MA, Schols L, Orlacchio A, Reid E, Zuchner S. Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J Clin Invest. 2012 doi: 10.1172/JCI60560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton N, Robertson PD, Rieder MJ, Zuchner S, Rampersaud E, Martin E, Li D, Nickerson DA, Hershberger RE on behalf of the National Heart, Lung and Blood Institute GO Exome Sequencing Project. Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circ Cardiovasc Genet. 2012;5:167–174. doi: 10.1161/CIRCGENETICS.111.961805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OMIM Online Mendelian Inheritance in Man. An online catalog of human genes and genetic disorders. Baltimore, MD: Johns Hopkins University; Dec, 2012. [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Jr, Conforti L, Coleman M, Tessier-Lavigne M, Zuchner S, Freeman MR. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W, Haussler D. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, Bademci G, Agolini E, Guo S, Konuk B, Kavaz A, Blanton S, Digilio MC, Dallapiccola B, Young J, Zuchner S, Tekin M. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011;89:289–294. doi: 10.1016/j.ajhg.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shaw K, Cooper DN. The human gene mutation database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr Protoc Bioinformatics. 2012;Chapter 1(Unit1.13) doi: 10.1002/0471250953.bi0113s39. [DOI] [PubMed] [Google Scholar]
- Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: Visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012;28:599–600. doi: 10.1093/bioinformatics/btr711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM, Broad GO, Seattle GO NHLBI Exome Sequencing Project. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesson C, Nawara M, Salih MA, Rossignol R, Zaki MS, Al Balwi M, Schule R, Mignot C, Obre E, Bouhouche A, Santorelli FM, Durand CM, Oteyza AC, El-Hachimi KH, Al Drees A, Bouslam N, Lamari F, Elmalik SA, Kabiraj MM, Seidahmed MZ, Esteves T, Gaussen M, Monin ML, Gyapay G, Lechner D, Gonzalez M, Depienne C, Mochel F, Lavie J, Schols L, Lacombe D, Yahyaoui M, Al Abdulkareem I, Zuchner S, Yamashita A, Benomar A, Goizet C, Durr A, Gleeson JG, Darios F, Brice A, Stevanin G. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am J Hum Genet. 2012;91:1051–1064. doi: 10.1016/j.ajhg.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velinov M, Dolzhanskaya N, Gonzalez M, Powell E, Konidari I, Hulme W, Staropoli JF, Xin W, Wen GY, Barone R, Coppel SH, Sims K, Brown WT, Zuchner S. Mutations in the gene DNAJC5 cause autosomal dominant kufs disease in a proportion of cases: Study of the parry family and 8 other families. PLoS One. 2012;7:e29729. doi: 10.1371/journal.pone.0029729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell M, Huff C, Hu H, Singleton M, Moore B, Xing J, Jorde LB, Reese MG. A probabilistic disease-gene finder for personal genomes. Genome Res. 2011;21:1529–1542. doi: 10.1101/gr.123158.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) The column manager allows for adding up to an additional 44 columns, deleting columns or resorting of columns. (B) An example of a public resource link-out in GEM.app is the STRING database.
1) Raw sequence reads are aligned to reference and variants are called with available tools resulting in VCF files (ie BWA/GATK).; 2) From VCF files GEDI extracts variants to annotate and backfill all previous samples in GEM.app. GEDI also performs standard calculations, such as genotype counts across GEM.app samples and segregation analysis.; 3) After the GEDI pipeline is completed, analysis can be performed using the GEM.app GUI.