

Abstract
Helicobacter pylori infection causes gastric cancer, the third leading cause of cancer death worldwide. More than half of the world’s population is infected, making universal eradication impractical. Clinical trials suggest that antibiotic treatment only reduces gastric cancer risk in patients with non-atrophic gastritis (NAG), and is ineffective once preneoplastic lesions of multifocal atrophic gastritis (MAG) and intestinal metaplasia (IM) have occurred. Therefore, additional strategies for risk stratification and chemoprevention of gastric cancer are needed. We have implicated polyamines, generated by the rate limiting enzyme ornithine decarboxylase (ODC), in gastric carcinogenesis. During H. pylori infection, the enzyme spermine oxidase (SMOX) is induced, which generates hydrogen peroxide from the catabolism of the polyamine spermine. Herein, we assessed the role of SMOX in the increased gastric cancer risk in Colombia associated with the Andean mountain region when compared to the low risk region on the Pacific coast. When co-cultured with gastric epithelial cells, clinical strains of H. pylori from the high risk region induced more SMOX expression and oxidative DNA damage, and less apoptosis than low risk strains. These findings were not attributable to differences in the CagA oncoprotein. Gastric tissues from subjects from the high risk region exhibited greater levels of SMOX and oxidative DNA damage by immunohistochemistry and flow cytometry, and this occurred in NAG, MAG, and IM. In Mongolian gerbils, a prototype colonizing strain from the high risk region induced more SMOX, DNA damage, dysplasia and adenocarcinoma than a colonizing strain from the low risk region. Treatment of gerbils with either α-difluoromethylornithine (DFMO), an inhibitor of ODC, or MDL 72527, an inhibitor of SMOX, reduced gastric dysplasia and carcinoma, as well as apoptosis-resistant cells with DNA damage. These data indicate that aberrant activation of polyamine-driven oxidative stress is a marker of gastric cancer risk and a target for chemoprevention.
Keywords: Polyamines, DNA damage, oxidative stress, ornithine decarboxylase, DFMO, gerbils
Introduction
Gastric adenocarcinoma is currently the third leading cause of cancer-related death in the world, and approximately 723,000 persons die from this malignancy each year (globocan.iarc.fr). Chronic infection with Helicobacter pylori is the strongest known risk factor for gastric cancer,1,2 the leading global infection-associated cancer.3 H. pylori is a microaerophilic bacterium that selectively colonizes the stomach of the human host. Infection with H. pylori causes universal gastritis and the disease can progress through a histopathological cascade to atrophic gastritis, intestinal metaplasia, dysplasia, and gastric adenocarcinoma.4–7 Treatment with antibiotics is expensive and not completely effective, especially in high prevalence areas.8 Antibiotic-based eradication of H. pylori only reduces risk for cancer if given prior to the development of preneoplastic lesions.9–11 Moreover, epidemiologic data show a negative correlation between H. pylori infection and asthma, esophageal reflux disease, and eosinophilic esophagitis.12,13 These observations argue against universal antibiotic treatment.
Despite a very high prevalence of H. pylori infection, the incidence rates of gastric cancer differ greatly in high versus low altitude regions of Latin America.14,15 This has been well described in Colombia; in the state of Nariño, inhabitants of the Andes mountains have very high incidence rates of gastric cancer, as high as 150/100,000, compared to 6/100,000 in inhabitants of the Pacific coast, despite the fact that the two locations have similar high H. pylori prevalence of approximately 90% and are only 200 km apart.7,16,17 In the Andean region, there is a higher prevalence of precancerous lesions, namely multifocal atrophic gastritis (MAG), and intestinal metaplasia (IM), when compared to the low risk coastal region7,18. Thus, Colombia is a vital natural laboratory for understanding gastric carcinogenesis.
Numerous studies have focused on mechanisms of failed immune responses, the injection of cytotoxin associated gene A (CagA) protein by a type IV secretion system, and the injurious effects of the vacuolating toxin A (VacA), all of which have been linked to gastric cancer risk,19–23 including in Colombia.18 However, the limited efficacy of H. pylori eradication strategies emphasizes the need for other pharmacologic approaches to gastric cancer chemoprevention. We have shown that H. pylori infection results in increased levels of polyamines, natural polycations that are synthesized by the rate-limiting enzyme ornithine decarboxylase (ODC).24,25 Infection also increases the level of spermine oxidase (SMOX), which catabolizes spermine and produces hydrogen peroxide (H2O2) and leads to DNA damage in gastric epithelial cells, a key event in the process of gastric carcinogenesis.26–29 In the present study we demonstrate that H. pylori strains from the high risk region of Nariño, Colombia induce more SMOX and associated DNA damage in vitro and in vivo. We used inhibitors of ODC and SMOX and found that polyamine synthesis and oxidation drive H. pylori-associated gastric carcinogenesis in a gerbil model of gastric cancer.
Results
H. pylori clinical isolates from the high cancer region of Colombia induce more SMOX expression, H2O2 production, and DNA damage than isolates from the low risk region
We have reported that laboratory strains of H. pylori induce spermine oxidase (SMOX) that causes DNA damage in gastric epithelial cells.26,27,29 Herein we compared the ability of 10 clinical isolates from the low risk coastal area and 10 isolates from the high risk mountain area to induce pathogenic responses in AGS gastric epithelial cells. These strains were all positive for the virulence gene cagA and for the vacA s1m1 gene alleles that have been linked to gastric cancer risk.21–23 All 10 high risk clinical isolates caused significantly increased SMOX mRNA expression compared to uninfected cells, but only three low risk clinical isolates caused a significant increase (Figure 1a). Overall, the high risk strains induced a greater increase in SMOX mRNA expression (11-fold) than the low risk strains (3.7-fold) when compared to uninfected control cells (Figure 1b). Similarly, there was a greater increase in SMOX protein expression in the AGS cells, measured as the percentage of SMOX-positive cells by flow cytometry, with the high risk versus low risk strains (Figure 1c).
Figure 1.

Clinical isolates from the high risk region of Colombia induce higher levels of SMOX expression, H2O2, and DNA damage. AGS cells were co-cultured with clinical isolates from the low and high risk regions. (a and b) After 6 h, levels of SMOX mRNA were measured by real-time PCR. (c) Percentage of SMOXhigh (SMOX-positive) cells after 24 h co-culture, assessed by flow cytometry. (d) Levels of H2O2 in cell culture supernatants after 24 h by Amplex Red assay. (e) Representative histograms for SMOX and 8-oxoguanosine. (f–h) Cells were transfected with scrambled (Scr) siRNA or SMO siRNA and activated with strains from each risk region for 24 h, and summary data is shown. (f) SMOX protein levels in mean fluorescence units assessed by flow cytometry. (g) Levels of H2O2 in cell culture supernatants. (h) DNA damage measured by flow cytometry for 8-oxoguanosine levels. In a-h, data is from at least three experiments in duplicate. For a-d, *P < 0.05, **P < 0.01, ***P < 0.001 versus control (Ctrl); §§§P < .001 versus low risk. For f–h, *P < .05, ***P < 0.001 versus Scr control; §§§P < 0.001 versus Scr low risk or Scr high risk. ###P < 0.001 versus Scr low risk. For a, b, c, and e, 10 strains from each region were used; for d, 7 strains from each region were used, and for f–h, 4 strains from each region were used.
There was also a greater increase in H2O2 production in culture supernatants of cells stimulated with high versus low risk strains (Figure 1d). Levels of SMOX protein abundance, measured as fluorescence intensity, were also further increased with the high risk versus low risk isolates (Figure 1e, upper panel). In parallel, cells infected with high risk isolates exhibited more intense staining with a FITC-labeled 8-oxoguanosine binding peptide, than cells infected with low risk isolates (Figure 1e, lower panel), indicating higher levels of oxidative DNA damage. In cells transfected with SMOX siRNA, levels of SMOX (Figure 1f), H2O2 (Figure 1g) and DNA damage (Figure 1h) were significantly reduced in parallel, indicating that the increased oxidative stress and DNA damage caused by the high risk strains is SMOX-mediated. Taken together, these data suggest that H. pylori strains from the high risk region exert a key effect by enhancing SMOX-dependent DNA damage in gastric epithelial cells.
High risk H. pylori clinical isolates induce less apoptosis despite increasing SMOX protein expression and DNA damage
To further assess the relationship between SMOX and DNA damage, we double-stained AGS cells for SMOX protein and 8-oxoguanosine, and observed a strong correlation (Figure 2a). High risk strains significantly increased the percentage of cells that exhibited both DNA damage (8-oxoguanosinehigh) and SMOX expression (SMOXhigh) compared to low risk clinical isolates or uninfected control cells (Figure 2b). Notably, the 8-oxoguanosinehigh cells derive from the SMOXhigh population. We have reported that SMOX induction causes apoptosis in gastric epithelial cells.26,27 In the current studies, strains from both regions induced apoptosis, but the high risk isolates induced less apoptosis than low risk strains (Figure 2c). We have previously implicated the anti-apoptotic protein Bcl-2 in the regulation of apoptosis in response to H. pylori in gastric epithelial cells27. When measured by flow cytometry, levels of Bcl-2 were significantly increased by high risk strains compared to uninfected control cells or to cells infected with low risk strains (Figure 2d). These data indicate that exposure of cells to clinical isolates from the high risk region results in high levels of SMOX expression and DNA damage, yet promotion of cell survival due to increased levels of Bcl-2.
Figure 2.

Correlation between SMOX and DNA damage, and levels of apoptosis and Bcl-2 in AGS cells co-cultured with clinical isolates for 24 h. (a) Correlation (Pearson coefficient) between the percentage of cells positive for SMOX and 8-oxoguanosine levels determined by flow cytometry. (b) Percentage of SMOX+, 8-oxoguanosine+ double-positive cells by flow cytometry. (c) Summary data for apoptosis measured by annexin V and propidium iodide (PI) using flow cytometry. (d) Summary data for levels of Bcl-2 measured by flow cytometry. For ad, at least three experiments were performed in duplicate. In b–d, *P < 0.05, ***P < 0.001 versus control (Ctrl); §§P < 0.01, §§§P < 0.001 versus low risk. Ten strains from each risk group were used.
Effects of high risk versus low risk strains are not attributable to differences in CagA or CagA phosphorylation
CagA, the product of the H. pylori cagA virulence gene, is considered an oncoprotein because persons infected with cagA-positive strains have higher relative risk for gastric cancer, and CagA induces aberrant epithelial cell signaling.19,20,30,31 The number of type C EPIYA (glutamic acid-proline-isoleucine-tyrosine-alanine) motifs within the gene sequence has been linked to risk for gastric cancer.32–34 However, when we analyzed the 3′-ends of the cagA sequences from multiple clinical isolates we found no increase in the number of type C EPIYA motifs in the high risk strains (Figure 3a).35 When strains from both risk regions were co-cultured with AGS cells, all were found to translocate CagA that was subsequently phosphorylated; two of the low risk strains and one of the high risk strains demonstrated a high degree of CagA phosphorylation (Figure 3a). These data suggest that differences in induction of SMOX and DNA damage between high and low risk strains are not due to differences in CagA translocation and phosphorylation.
Figure 3.

CagA in high and low risk strains. Upper panel: analysis of EPIYA motifs of clinical isolates; numbers of type C motifs are indicated. Lower panels: AGS cells were co-cultured with clinical strains for 4 h and Western blotting was performed for phosphorylated CagA and total CagA using antibodies to pY99, CagA, and β-actin.
SMOX expression and oxidative DNA damage are increased in H. pylori gastritis tissues from high risk versus low risk Colombian subjects
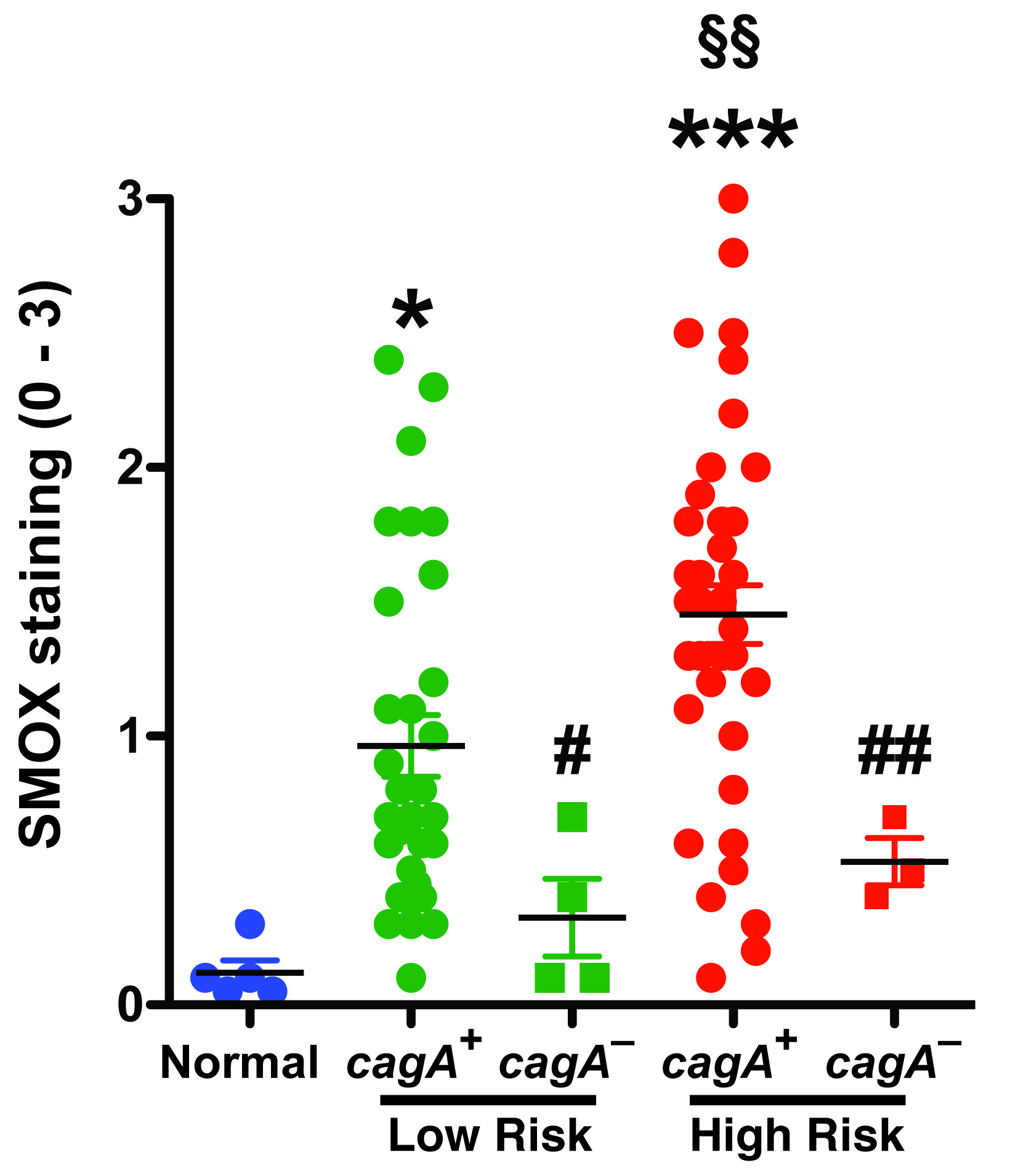
Because we identified differences in the ability of H. pylori strains from high and low risk subjects to induce SMOX in vitro, we examined gastric biopsies from the source patients. Biopsies from subjects in the high risk region demonstrated SMOX immunostaining in the gastric epithelium of non-atrophic gastritis (NAG) that was greater than that in NAG of low risk subjects, and was strongly increased in metaplastic glands in the IM cases (Figure 4a, upper panel). There was also increased staining for 8-OHdG, a marker for oxidative DNA damage, in epithelial nuclei in areas of NAG and IM in biopsies from the high risk region, that was less apparent in tissues from the low risk region (Figure 4a, lower panel). When quantified, SMOX staining was significantly increased in high risk versus low risk subjects (Figure 4b). Further, at each stage of NAG, MAG, and IM, the level of SMOX staining was higher in the high versus low risk tissues and peaked in the IM tissues (Figure 4c).
Figure 4.

Levels of SMOX and DNA damage in Colombian human subjects. (a) Immunohistochemistry for SMOX and 8-OHdG at 200X and 600X magnification. (b) Scoring of SMOX staining intensity in epithelial cells from immunohistochemistry analysis. (c) SMOX staining determined in uninfected controls with normal histology, and subjects with NAG, MAG, and IM. (d) Correlation (Spearman coefficient) between SMOX and 8-OHdG levels in immunohistochemistry. (e–h) Gastric epithelial cells were isolated from gastric biopsies and flow cytometry was performed for SMOX and 8-oxoguanosine. (e) Representative histograms for levels of SMOX and 8-oxoguanosine. (f) Representative dot plots for SMOX and 8-oxoguanosine with the percent of cells in each quadrant marked. (h) Summary data for percentage of cells that were SMOX+, 8-oxoguanosine+. In b, **P < 0.01 versus low risk; in c, **P < 0.05, P < 0.01, P < 0.001 versus normal; §P < 0.05, §§P < 0.01 for high risk versus low risk; (h) §§P < 0.01 versus low risk. For a-d, 29 low risk and 35 high risk subjects were studied. For e–h, 10 low risk and 10 high risk subjects were used.
When we compared our published data for 8-OHdG staining in these subjects7 with SMOX staining, a strong correlation was observed (Figure 4d). We performed flow cytometry on isolated gastric epithelial cells from cases where frozen biopsies were available, and found a significant increase in SMOX (Figure 4e) and 8-oxoguanosine (Figure 4f) levels, and percentage of positive cells (Figure 4g) in high risk versus low risk subjects. Nearly all of the 8-oxoguanosinehigh cells were SMOXhigh (Figure 4g) and this double-positive population was increased in the high risk subjects (Figure 4h).
Increased carcinogenesis with high risk versus low risk H. pylori in gerbils
We next employed a model of H. pylori-induced gastric cancer in Mongolian gerbils that closely recapitulates the human disease.36–38 We infected groups of five gerbils with five different high risk (PZ5056, PZ5069, PZ5086, PZ5097, and PZ5114) or five low risk (PZ5009, PZ5010, PZ5024, PZ5033, and PZ5047) clinical isolates. Two low risk clinical isolates, PZ5009 (5/5 gerbils) and PZ5010 (1/5 gerbils), and one high risk strain, PZ5056 (4/5 gerbils), were recovered from the gerbil stomachs. The colonization with H. pylori was confirmed by Steiner staining of tissues (Figure 5a). While the low risk strains only induced gastritis, the high risk PZ5056 strain induced hyperplasia and dysplasia (Figure 5a). There was minimal SMOX staining in stomachs infected with the low risk PZ5009 isolate (Figure 5a, upper panel), but there was increased SMOX staining in the PZ5056-infected gerbils that was predominantly localized to dysplastic glands (Figure 5a, lower panel). Having identified these in vivo differences, we also tested strains PZ5009 and PZ5056 in an additional gastric epithelial cell line, namely murine conditionally-immortalized stomach cells (ImSt). The high risk PZ5056 strain induced higher levels of SMOX, H2O2, and DNA damage in these gastric epithelial cells when compared to the low risk PZ5009 strain (Supplementary Figure S1).
Figure 5.

Increased oncogenic potential of H. pylori from the high risk region in the gerbil model. (a) Upper and lower panels: photomicrographs from gastric tissues from animals infected with low risk strain PZ5009 and high risk strain PZ5056, respectively. Left panels: Steiner (modified silver) staining of H. pylori. Middle panels: H&E staining, with hyperplasia and dysplastic glands shown for strain PZ5056. Right panels: SMOX immunohistochemical staining, with increase in dysplastic glands of PZ5056-infected gerbil. (b) Frequency of high grade dysplasia or carcinoma in gerbils infected with gerbil-adapted low risk (PZ5009G) and high risk (PZ5056G) strains. n = 19 and 20 gerbils were infected with PZ5009G and PZ5056G, respectively. (c) Left panel and middle panels: H&E staining of gerbil stomach tissues, showing invasive malignant-appearing glands with PZ5056G, and diffuse gastritis with both strains. Right panels: Staining for SMOX showing more intense staining in dysplastic glands with PZ5056G infection. (d–f) Flow cytometry performed on gastric epithelial cells isolated from stomach tissues. (d) SMOX protein levels. (e) 8-oxoguanosine levels. (f) Correlation (Spearman coefficient) between SMOX-positive cells and 8-oxoguanosine levels. (g) Brightfield photomicrographs of cells from soft agar assays of cells isolated from gerbils infected with strains PZ5009G and PZ5056G. (h) Summary data for number of cells counted in 6 fields at 100X. n = 4 gerbils were infected with each strain for 4 weeks and gastric epithelial cells were isolated and plated on three different soft agar coated plates per stomach. In d and e, **P < 0.01, ***P < 0.001 versus control (Ctrl); §§P < 0.01, §§§P < 0.001 versus PZ5009G. For h, ***P < 0.001 versus PZ5009G.
Based on prior studies demonstrating increased oncogenic potential and colonization efficiency with a gerbil-adapted strain,37,38 we tested gerbil-passaged low risk PZ5009 and high risk PZ5056 output strains, which we termed PZ5009G and PZ5056G, respectively. These strains were utilized as prototype high and low risk strains. Both strains colonized the gerbils and induced a similar level of gastritis, indicating adaptation to the gerbil host. However, there was an increased frequency of both dysplasia and invasive adenocarcinoma in gerbils colonized with the high risk PZ5056G compared to the low risk PZ5009G (Figure 5b). There was diffuse gastritis induced by both strains, and invasive adenocarcinoma with strain PZ5056G (Figure 5c), including invasion of malignant glands into the submucosa (Figure 5c). Further, there was increased SMOX staining in dysplastic glands, particularly in invasive glands characteristic of carcinoma (Figure 5c). Flow cytometry performed on isolated gastric epithelial cells revealed a significant increase in both SMOX (Figure 5d) and 8-oxoguanosine levels (Figure 5e) in gerbils infected with PZ5056G versus PZ5009G. There was also a significant correlation of SMOX and DNA damage in these cells (Figure 5f).
To further test the oncogenic potential of the two gerbil-adapted strains, epithelial cells were isolated from gerbils 4 weeks post-inoculation and soft agar assays performed. Cells from gerbils infected with PZ5056G grew in an anchorage-independent manner on soft agar, but this did not occur in cells from gerbils infected with PZ5009G (Figure 5g and 5h). These data indicate the presence of transformed cells in response to infection with the gerbil-adapted high risk strain early in the infection course.
Together these data indicate that there are differences in oncogenic potential of high versus low risk strains and implicate SMOX-dependent oxidative DNA damage in these responses.
Role of CagA in vivo in induction of gastric cancer, SMOX and DNA damage
CagA has been directly linked to gastric carcinogenesis, and we have reported that laboratory strains with cagA deletion induce less SMOX and DNA damage. We therefore constructed a cagA isogenic mutant of PZ5056G, which was verified by Western blotting (Supplementary Figure S2a). Compared to the wild-type strain, the cagA− strain caused less histologic gastritis (Figure 6a) and higher colonization levels (Figure 6b). There was complete loss of gastric dysplasia or cancer development with the cagA mutant (Figure 6c). Gerbils infected with PZ5056G cagA− exhibited markedly attenuated SMOX (Figure 6d) and 8-oxoguanosine (Figure 6e) levels in gastric epithelial cells compared to those infected with PZ5056G. Similarly, there was less SMOX staining in gastric biopsies from human subjects infected with cagA− strains, compared to subjects infected with cagA+ strains (Supplementary Figure S3). Together, these data indicate that in vivo, CagA is necessary for increased levels of SMOX, DNA damage, and gastric carcinogenesis.
Figure 6.

Diminished gastric pathology in gerbils infected with cagA isogenic mutant of oncogenic strain PZ5056G. Gerbils were infected with the PZ5056G parental strain or the cagA isogenic mutant for 16 weeks. (a) Gastric inflammation in tissues assessed by H&E staining and scoring of the levels of acute and chronic gastritis in antrum and corpus. (b) Colonization by serial dilution and culture. (c) Frequency of gastric dysplasia and invasive carcinoma. Note that with the cagA− strain there were no cases of dysplasia or carcinoma. (d and e) Gastric epithelial cells were isolated from stomach tissues and flow cytometry performed. (d) SMOX protein levels. (e) 8-oxoguanosine levels. In a, data are from 19 and 13 gerbils infected with PZ5056G and the PZ5056G cagA isogenic mutant, respectively. In d and e, stomach tissues from gerbils infected with PZ5056G (n = 9) and PZ5056G cagA− (n = 10) were used. For a and b, §§P < 0.01 and §§P < 0.001 versus PZ5009G. For d and e, *P < 0.05, **P < 0.01 versus control (Ctrl); §§P < 0.01, §§§P < 0.001 versus PZ5009G.
We also assessed the effect of cagA deletion in PZ5056G and in the PZ5056 pre-adapted clinical strain in vitro. There was a partial inhibition of the stimulated SMOX mRNA expression in AGS gastric epithelial cells with the cagA− strains (Supplementary Figure S2a). We also assessed the effect of cagA deletion in two additional high risk clinical strains, and there was a similar effect (Supplementary Figure S2b).
Inhibition of polyamine synthesis or metabolism reduces gastritis and gastric carcinogenesis in infected gerbils
To provide further translational relevance, gerbils infected with the cancer-inducing strain PZ5056G were treated with the ODC inhibitor, α-difluoromethylornithine (DFMO), the SMOX inhibitor, MDL 72527, or the combination. We verified in a subset of gerbils that DFMO reduced levels of the polyamines putrescine, spermidine, and spermine in infected animals (Figure 7a), thus resulting in decreased substrate availability for the SMOX enzyme. We also found that there was a positive correlation between polyamine levels and both gastric inflammation scores (Figure 7b) and DNA damage (Figure 7c).
Figure 7.

Levels of polyamines in gerbils. Gerbils were infected with PZ5056G or sham gavaged with broth. Animals were treated with DFMO (1% w/v given continuously in the drinking water) and MDL72527 (20 mg/kg given in 100 μl intraperitoneal injections, 3 times per week), with both beginning 7 days prior to inoculation with H. pylori and treatments continued for the duration of the experiment. After 16 weeks of infection, polyamines were measured from gastric tissues by HPLC. (a) Levels of putrescine, spermidine, spermine, and total polyamines. n = 10 gerbils per group, ***P < 0.001 versus control (uninfected); §P < 0.05, §§P < 0.01, §§§P < 0.001 versus non-treated. (b and c) Correlation (Spearman coefficient) between total polyamines and gastritis score (b) or 8-oxoguanosine-positive cells (c). n = 20 gerbils from the untreated, infected group were used.
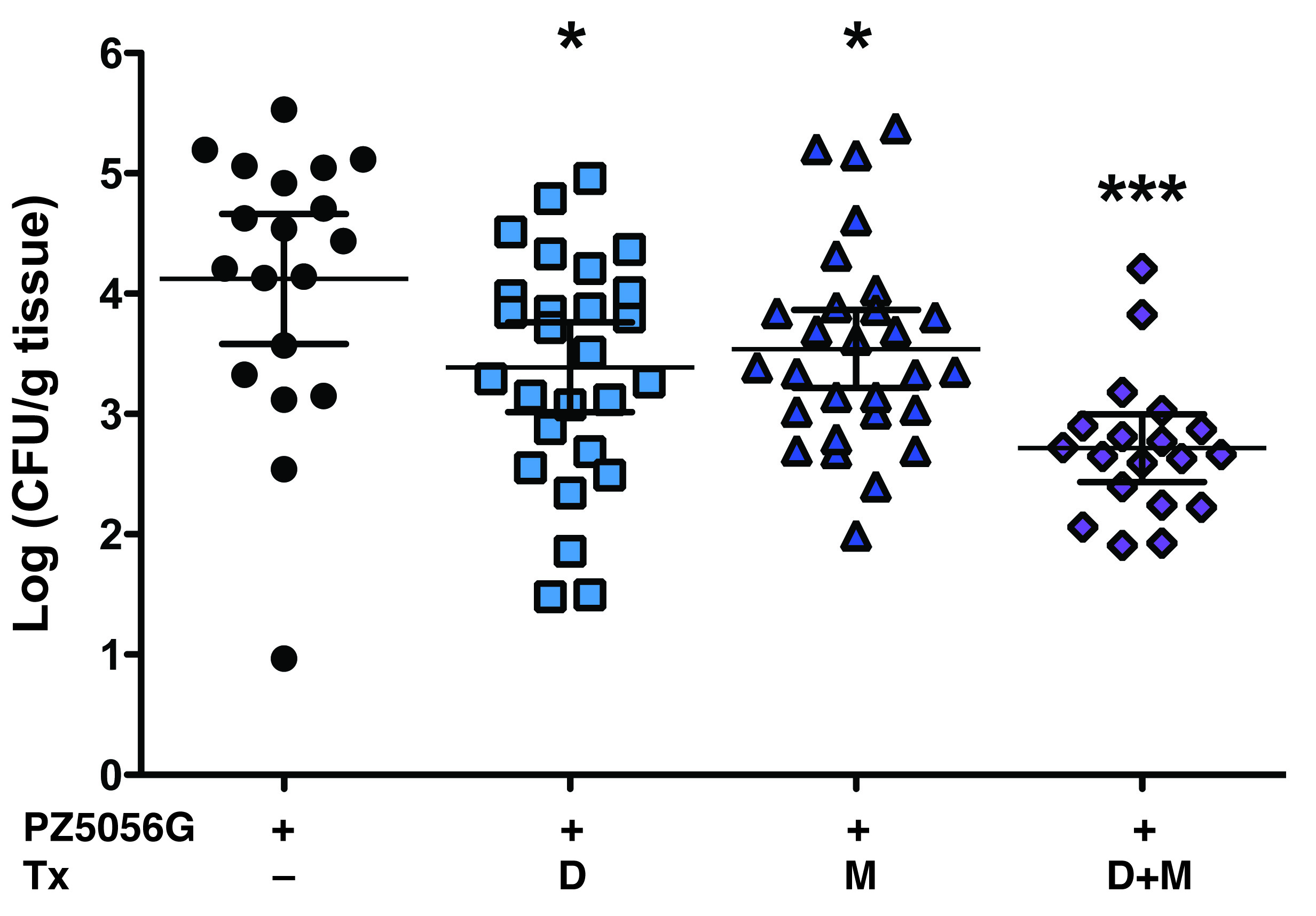
Administration of either of the inhibitors alone or in combination resulted in significant, but modest decreases in gastritis scores (Figure 8a). Further, in each of the groups, the highest levels of gastritis were associated with dysplasia or carcinoma (Figure 8a). We also observed a modest, but significant inhibitory effect on levels of H. pylori colonization with the inhibitors (Supplementary Figure S4).
Figure 8.

Effect of DFMO and MDL 72527 on PZ5056G-induced gastric pathology. Gerbils were infected with PZ5056G and treated with DFMO (D) or MDL 72527 (M) or the combination (D+M) as described in Figure 7. After 16 weeks gerbils were euthanized. (a) Gastritis score was assessed as in Figure 6. Each symbol represents a gerbil, with orange symbols indicating animals with dysplasia, and red symbols animals with carcinoma. All gerbils with no histologic gastritis were colonized when assessed by either culture or qPCR for the ureA gene. *P < 0.05 versus PZ5056G without treatment. (b) Frequency of advanced lesions. Left panel: dysplasia; right panel: carcinoma. (c) Representative H&E staining showing malignant appearing glands invading into the submucosa, top panels; and only gastritis in a gerbil treated with DFMO.
There was a marked inhibitory effect on gastric carcinogenesis with the disruption of polyamine synthesis by DFMO or spermine metabolism by MDL 72527. The incidence of dysplastic lesions was significantly reduced by 51% in gerbils treated with DFMO compared to non-treated gerbils (Figure 8b, left panel and Figure 8c). MDL 72527 also caused a 38% reduction in dysplasia (Figure 8b, left panel) and both inhibitors together produced a 60% reduction (Figure 8b, left panel). Similarly, gastric adenocarcinoma was significantly reduced by 58% with DFMO, 56% with MDL72527, and 71% with the combination treatment (Figure 8b, right panel).
Inhibition of polyamine synthesis or metabolism reduces gastric epithelial cells with DNA damage and cells with DNA damage that are resistant to apoptosis in infected gerbils
We have reported that H. pylori infection in gerbils increases gastric epithelial cells with DNA damage (8-oxoGhigh) and apoptosis (active caspase-3high), but also leads to a subpopulation of cells with DNA damage that are resistant to apoptosis (8-oxoGhigh, active caspase-3low) as assessed by flow cytometry.27 In the current studies, treatment with DFMO or MDL 72527 alone or in combination reduced the number of 8-oxoGhigh (Figure 9A) or active caspase-3high (Figure 9B) cells. The inhibitors also significantly reduced the 8-oxoGhigh, active caspase-3low cells (Figure 9C). Thus, inhibition of polyamine synthesis and oxidation can reduce cellular events related to gastric carcinogenesis.
Figure 9.

Percentage of epithelial cells with DNA damage, apoptosis, and DNA damage with apoptosis resistance in gerbil gastric tissues. Gerbils were infected with PZ5056G for 16 weeks and were treated with DFMO, MDL 72527 or the combination. Gastric epithelial cells were isolated from gerbil stomachs and stained for 8-oxoguanosine and active caspase-3, and flow cytometry was performed. (a) Percent DNA damage-positive cells, by gating on cells with high levels of 8-oxoguanosine. (b) Percent apoptosis-positive cells, by gating on cells with high levels of active caspase-3. (c) Percent DNA damagehigh, apoptosislow cells, measured by gating on 8-oxoguanosinehigh, active caspase-3low cells. In a-c, 7–10 gerbils were used in uninfected groups, and 20 gerbils were used in infected groups. For a–c, **P < 0.01, ***P < 0.001 versus control uninfected; §P < 0.05, §§P < 0.01, §§§P < 0.001 versus PZ5056G non-treated. Solid symbols represent gerbils with dysplasia or carcinoma.
Discussion
H. pylori-associated gastric cancer is a multifactorial process. In addition to host and environmental factors, H. pylori genotypes play a significant role in carcinogenesis. Epidemiological data suggest a link between H. pylori virulence factors, especially CagA, and gastric cancer.19–22 CagA has been implicated in H. pylori-induced apoptosis, proliferation, oxidative stress, and DNA damage in gastric epithelial cells.19,28,39 In the present study, all H. pylori clinical strains used in our cell culture models were positive for cagA, and for the vacA s1m1 allele, so we excluded potential differences in these virulence genes. Despite substantial increases in the ability of the high risk versus low risk strains to induce SMOX, oxidative stress, and DNA damage, these affects were not attributable to differences in their EPIYA motifs or the translocation and phosphorylation of CagA, which are linked to carcinogenesis.19,21,28,35 We have reported that in response to North American H. pylori clinical isolates, the induction of SMOX expression in gastric epithelial cells was greater in strains that expressed CagA.27 In the current study, the effect of cagA deletion in different strains on SMOX expression in gastric epithelial cells was only partial. Thus, a component of the ability of Colombian high risk strains to induce SMOX may be independent of CagA, at least in vitro.
Importantly, in the gerbil model, a cagA mutant of the cancer-inducing PZ5056G strain induced little inflammation and no dysplasia or carcinoma, despite colonizing well. These data suggest that CagA and inflammation are both required for gastric carcinogenesis, consistent with other studies in the gerbil model.27,37,38 Thus, CagA may be required for carcinogenesis, but alone it is not sufficient. Furthermore, our results in the gerbil model were obtained by initiating the studies with randomly selected high risk and low risk strains that were all cagA+, suggesting that our findings are representative. Moreover, of the high risk strains tested in gerbils, two strains (PZ5056 and PZ5097) induced levels of SMOX mRNA above the mean level in vitro, and the other 3 strains tested induced levels of SMOX mRNA below the mean level for the high risk strains in the co-culture assays, indicated the in vivo studies were conducted with representative strains. Colonization occurred with 20–40% of the tested clinical isolates. We subsequently tested an additional 8 strains from each region in gerbils, and we were able to recover H. pylori with 25% of the high risk strains and 50% of the low risk strains, indicating that our initial results were typical for these clinical isolates.
Conflicting findings between cell culture and animal models pertaining to the role of CagA have been well-recognized.19 An important additional consideration is that inflammatory factors such as macrophage-derived TNF-α promote gastric tumorigenesis.40 Further, TNF-α induces SMOX in lung epithelial cells.41 Because the level of gastric inflammation was similar in gerbils infected with the high risk or low risk gerbil-adapted strains, it is apparent that inflammation alone is not sufficient alone for carcinogenesis. Thus, other factors are important and may involve differential expression of a variety of genes associated with virulence phenotypes that appear to be enhanced in high risk strains.42
Excessive gastric epithelial apoptosis has been considered to be highly pathogenic, since it can lead to barrier dysfunction, and compensatory cell proliferation.28,43,44 However, insufficient apoptosis in cells with DNA damage is problematic, as it can lead to failed clearance of cells with damaged DNA. We have previously demonstrated that in both the gerbil model of carcinogenesis and in hypergastrinemic mice with dysplasia, H. pylori infection is associated with a subpopulation of gastric epithelial cells with DNA damage that are resistant to apoptosis.27 In the present study, we showed that high risk strains induced more anti-apoptotic Bcl-2 and less apoptosis than low risk strains in vitro, and in parallel, the gerbil-adapted high risk strain caused generation of cells that were 8-oxoguanosinehigh, active caspase-3low in infected gerbils, indicative of cells that simultaneously exhibit DNA damage and resistance to apoptosis. We recently reported that phosphorylation of epidermal growth factor receptor (EGFR) and ERBB2 is associated with the resistance to apoptosis in infected gastric epithelial cells, and that levels of pEGFR and pERBB2 were increased at baseline in gastric tissues from subjects in the high risk region of Colombia that later had progression of gastric lesions in a longitudinal cohort of subjects distinct from the current cross-sectional study.29 Thus, studies of the effect of high risk versus low risk strains on activation of EGFR and ERBB2 are a target of our future investigations.
Recently our group has demonstrated that the greatest risk for advanced gastric histology in Colombia was associated with loss of co-evolution of H. pylori and their respective human hosts.45 In future studies, including in additional Latin American high and low risk sites, we plan to incorporate whole genome sequencing of H. pylori and high density human genetic analyses; through such methods we can prospectively determine the bacterial and human factors affecting pathways of carcinogenesis, such as polyamine-mediated oxidative DNA damage. Moreover, by using whole genome sequencing we expect to identify genetic differences between high and low risk strains that can be studied further; as candidate genes are elucidated, this can be addressed by using deletion mutants in H. pylori strains for mechanistic studies using our in vitro and in vivo models.
Another key point is that clinical trial data indicate that antibiotic eradication of H. pylori is effective in reducing risk for gastric cancer in subjects with NAG, but not in those with more advanced lesions.9–11 Because MAG and IM are very prevalent in high risk populations, such as in Colombia,7,46 additional treatment strategies are needed. Our data in the gerbil model provide a rationale for targeted chemoprevention in high risk subjects based on interference with polyamine-mediated oxidative stress. While our studies indicate that SMOX is both a disease marker and a key cause of oxidative DNA damage, there are no SMOX inhibitors available for human use. In contrast, DFMO has been successfully utilized in human Phase 2 clinical trials for prevention of colonic adenomatous polyps, where efficacy and safety has been demonstrated.47,48 Because our data show a benefit of DFMO in prevention of dysplasia and carcinoma in the gerbil model, we consider ODC to be a promising target in populations at high risk for gastric cancer, such as in mountainous areas of Latin America. Our finding of a modest additive benefit with DFMO plus MDL 72527 most likely reflects the incomplete enzyme inhibition with the respective monotherapies, but also suggests that other drugs in combination with DFMO such as future inhibitors of SMOX or inhibitors of polyamine uptake may be efficacious against gastric cancer.
Materials and Methods
Reagents
All reagents used for cell culture and RNA extraction were obtained from Invitrogen (Carlsbad, CA). Reagents for cDNA synthesis and real-time PCR were purchased from Bio-Rad (Hercules, CA). N′,N2-bis(2,3-butadienyl)-l,4-butanediamine (MDL 72527) was synthesized by PMW as described,49 and DFMO was provided by PMW. All other chemicals were purchased from Sigma.
Human subjects
Data in this study was derived from male subjects (ages 39–60) in Colombia from the high cancer risk region (Tuquerres) in the Andes Mountains and the low cancer risk region (Tumaco) on the Pacific Coast and were enrolled according to Institutional Review Board protocols approved by Vanderbilt University and Universidad del Valle Ethics Committees, all as described.7 Histopathology of gastric biopsies was assessed in the diagnostic categories of normal, non-atrophic gastritis (NAG), multifocal atrophic gastritis (MAG), intestinal metaplasia (IM), and dysplasia by two pathologists (MBP and PC), as described.7
H. pylori clinical strains and isogenic mutants
H. pylori was isolated from gastric biopsies.7 In the initial studies five H. pylori cagA+, vacA+ s1m1 clinical isolates from each region were randomly selected for both the in vitro and in vivo experiments. Subsequently, an additional five low risk and five high risk H. pylori cagA+, vacA+ s1m1 clinical isolates were used for the in vitro studies, for a total of 10 from each region. Isogenic cagA mutants were made by insertion mutagenesis using the pMC3::Km vector.50 For in vitro experiments, clinical isolates and isogenic mutants were grown on blood agar plates under microaerobic conditions.27
Cell and culture conditions
AGS cells were grown in F12 medium.26 Clinical isolates were co-cultured with AGS cells for 4, 6 or 24 h. In addition, conditionally-immortalized stomach cells derived from the immortomouse were infected under non-permissive conditions as described.27 A multiplicity of infection (MOI) of 200 was used for in vitro studies.
Real-time polymerase chain reaction
Isolation of RNA from AGS cells, cDNA synthesis, primer sequences for human SMOX and β-actin, real-time PCR using SYBR green, and relative expression of SMOX were all as described.26,27
Detection of SMOX protein, DNA damage (8-oxoguanosine) and Bcl-2 protein by flow cytometry
Assessment of each of these parameters was conducted as described.26,27,29
Measurement of H2O2
2.5 × 106 cells were plated in 6-well plates and H2O2 was measured in supernatants by Amplex Red assay.24,26
Transient transfection of SMOX siRNA
Small interfering RNA (siRNA) duplexes that targeted human SMOX and a scrambled small interfering RNA with no sequence homology to any known genes were transfected, as described.24,26
Apoptosis
Apoptosis was assayed using annexin V; 2.5 × 106 cells were stained and acquired by flow cytometry using a BD LSR II, and cells analyzed as described.24,26,27
Immunohistochemistry
Paraffin-embedded tissues were deparaffinized, antigen retrieval was performed, and tissues were stained with polyclonal rabbit anti-SMOX antibody (provided by RAC, dilution 1:10,000) and monoclonal mouse anti-8-hydroxy-2′-deoxyguanosine (8-OHdG; dilution 1:4000; Abcam, Cambridge, MA), and staining intensity was scored, all as described.7,27,29
Isolation of epithelial cells from gastric tissue
Gastric epithelial cells were isolated from frozen tissues by dissociation and dispersion.27 After washing, cells were fixed with 0.1% paraformaldehyde and used for flow cytometry.
Western blotting for total and phosphorylated CagA
Immunoblotting for CagA and pCagA was performed as described in AGS cells co-cultured with H. pylori strains grown overnight in brucella broth.27,51
Gerbil infection with H. pylori clinical isolates
Ten groups of 5 male Mongolian gerbils (Harlan Labs, Indianapolis, IN) aged 6–8 weeks, were each inoculated37,38 with one of the 5 low risk or 5 high risk strains. Gerbils were euthanized 12 weeks after challenge and stomachs were divided longitudinally into linear strips from the squamocolumnar junction to the duodenum.37,38 Strips were fixed, embedded and sections were stained with hematoxylin and eosin for histology,37,38 and with modified Steiner silver stain for detection of H. pylori.52 Inflammation (acute and chronic) were each scored on a 0–3 scale based on the Sydney System,25,53 and dysplasia and adenocarcinoma diagnosed as described.37,38 Strips were also used to culture H. pylori, which was quantified as described.25 In addition qPCR was performed for ureA.25 Recovered single colonies were isolated from gerbils and propagated. Additional gerbils were orogastrically challenged with gerbil-adapted strains and euthanized 16 weeks post-inoculation.
For intervention studies, gerbils were challenged with the gerbil-adapted high risk strain. Gerbils were treated with DFMO (1% w/v in drinking water)25 or with 100 μl (20 mg/kg) of MDL 72527 in PBS administered via intraperitoneal injection three times a week,54 both beginning 7 days prior to infection and continued for the duration of the experiment. Gerbils were euthanized 16 weeks after inoculation and studies were performed on gastric tissues as above.
Colony formation assay
Gerbil gastric epithelial cells were isolated and positively selected using a magnetic column and E-cadherin antibody (BD Biosciences, San Jose, CA). Purified cells were suspended in 0.3% agar with F12 medium supplemented with 10% fetal bovine serum, plated at a density of 5×104 cells in 60 mm dishes coated with 0.5% base agar, and maintained at 37°C. Fresh complete medium was added every five days. On day 21, the number of cells was counted at four locations in each dish.
Measurement of polyamine concentration
Polyamine levels were determined by high performance liquid chromatography as reported.25,55
Statistics
Statistical analyses were performed using Prism 5.0 (GraphPad Software, San Diego, CA). When comparisons between multiple groups were made, analysis of variance with the Student-Newman-Keuls posthoc multiple comparisons test was performed. Student’s t test was performed when comparisons between only 2 groups were made. Contingency analyses were performed using chi-square or Fisher’s exact test as appropriate. A P value of <0.05 was considered significant. In all figures, where histogram plots were used, means ± 95% confidence intervals are shown; for bar graphs, means ± standard errors are shown.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This study was supported by National Institutes of Health grants R01DK053620 and R01AT004821 (to K.T.W.), P01CA028842 (to K.T.W, J.G.F., and P.C.), P01CA116087 (to R.M.P. and K.T.W.), R01CA077955 and R01DK058587 (to R.M.P.), R01CA051085 and R01CA098454 (to R.A.C.), K01AT007324 (to R.C.), the Flow Cytometry Core of the Vanderbilt Digestive Disease Research Center grant (P30DK058404), the Flow Cytometry Core of the Vanderbilt Ingram Cancer Center (P30CA68485), UL1RR024975 (Vanderbilt CTSA, Pilot Project to K.T.W.), and Merit Review Grant 1I01BX001453 from the Office of Medical Research, Department of Veterans Affairs (to K.T.W.). D.M.H. was supported by T32GM008554.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991;325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 2.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 3.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 4.Correa P. A human model of gastric carcinogenesis. Cancer Res. 1988;48:3554–3560. [PubMed] [Google Scholar]
- 5.Correa P, Haenszel W, Cuello C, Zavala D, Fontham E, Zarama G, et al. Gastric precancerous process in a high risk population: cross-sectional studies. Cancer Res. 1990;50:4731–4736. [PubMed] [Google Scholar]
- 6.Correa P, Piazuelo MB, Wilson KT. Pathology of gastric intestinal metaplasia: clinical implications. Am J Gastroenterol. 2010;105:493–498. doi: 10.1038/ajg.2009.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Sablet T, Piazuelo MB, Shaffer CL, Schneider BG, Asim M, Chaturvedi R, et al. Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut. 2011;60:1189–1195. doi: 10.1136/gut.2010.234468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan DR, Torres J, Sexton R, Herrero R, Salazar-Martinez E, Greenberg ER, et al. Risk of recurrent Helicobacter pylori infection 1 year after initial eradication therapy in 7 Latin American communities. JAMA. 2013;309:578–586. doi: 10.1001/jama.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.You WC, Brown LM, Zhang L, Li JY, Jin ML, Chang YS, et al. Randomized double-blind factorial trial of three treatments to reduce the prevalence of precancerous gastric lesions. J Natl Cancer Inst. 2006;98:974–983. doi: 10.1093/jnci/djj264. [DOI] [PubMed] [Google Scholar]
- 10.Ma JL, Zhang L, Brown LM, Li JY, Shen L, Pan KF, et al. Fifteen-year effects of Helicobacter pylori, garlic, and vitamin treatments on gastric cancer incidence and mortality. J Natl Cancer Inst. 2012;104:488–492. doi: 10.1093/jnci/djs003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong BC, Lam SK, Wong WM, Chen JS, Zheng TT, Feng RE, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291:187–194. doi: 10.1001/jama.291.2.187. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Blaser MJ. Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis. 2008;198:553–560. doi: 10.1086/590158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koike T, Ohara S, Sekine H, Iijima K, Kato K, Shimosegawa T, et al. Helicobacter pylori infection inhibits reflux esophagitis by inducing atrophic gastritis. Am J Gastroenterol. 1999;94:3468–3472. doi: 10.1111/j.1572-0241.1999.01593.x. [DOI] [PubMed] [Google Scholar]
- 14.Torres J, Correa P, Ferreccio C, Hernandez-Suarez G, Herrero R, Cavazza-Porro M, et al. Gastric cancer incidence and mortality is associated with altitude in the mountainous regions of Pacific Latin America. Cancer Causes Control. 2013;24:249–256. doi: 10.1007/s10552-012-0114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dominguez RL, Crockett SD, Lund JL, Suazo LP, Heidt P, Martin C, et al. Gastric cancer incidence estimation in a resource-limited nation: use of endoscopy registry methodology. Cancer Causes Control. 2013;24:233–239. doi: 10.1007/s10552-012-0109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuello C, Correa P, Haenszel W, Gordillo G, Brown C, Archer M, et al. Gastric cancer in Colombia. I. Cancer risk and suspect environmental agents. J Natl Cancer Inst. 1976;57:1015–1020. doi: 10.1093/jnci/57.5.1015. [DOI] [PubMed] [Google Scholar]
- 17.Correa P, Cuello C, Duque E, Burbano LC, Garcia FT, Bolanos O, et al. Gastric cancer in Colombia. III. Natural history of precursor lesions. J Natl Cancer Inst. 1976;57:1027–1035. doi: 10.1093/jnci/57.5.1027. [DOI] [PubMed] [Google Scholar]
- 18.Bravo LE, van Doom LJ, Realpe JL, Correa P. Virulence-associated genotypes of Helicobacter pylori: do they explain the African enigma? Am J Gastroenterol. 2002;97:2839–2842. doi: 10.1111/j.1572-0241.2002.07031.x. [DOI] [PubMed] [Google Scholar]
- 19.Wroblewski LE, Peek RM, Jr, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23:713–739. doi: 10.1128/CMR.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peek RM, Jr, Fiske C, Wilson KT. Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol Rev. 2010;90:831–858. doi: 10.1152/physrev.00039.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basso D, Zambon CF, Letley DP, Stranges A, Marchet A, Rhead JL, et al. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology. 2008;135:91–99. doi: 10.1053/j.gastro.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 22.Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–2115. [PubMed] [Google Scholar]
- 23.Figueiredo C, Machado JC, Pharoah P, Seruca R, Sousa S, Carvalho R, et al. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94:1680–1687. doi: 10.1093/jnci/94.22.1680. [DOI] [PubMed] [Google Scholar]
- 24.Chaturvedi R, Cheng Y, Asim M, Bussiere FI, Xu H, Gobert AP, et al. Induction of polyamine oxidase 1 by Helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization. J Biol Chem. 2004;279:40161–40173. doi: 10.1074/jbc.M401370200. [DOI] [PubMed] [Google Scholar]
- 25.Chaturvedi R, Asim M, Hoge S, Lewis ND, Singh K, Barry DP, et al. Polyamines impair immunity to Helicobacter pylori by inhibiting L-arginine uptake required for nitric oxide production. Gastroenterology. 2010;139:1686–1698. doi: 10.1053/j.gastro.2010.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu H, Chaturvedi R, Cheng Y, Bussiere FI, Asim M, Yao MD, et al. Spermine oxidation induced by Helicobacter pylori results in apoptosis and DNA damage: implications for gastric carcinogenesis. Cancer Res. 2004;64:8521–8525. doi: 10.1158/0008-5472.CAN-04-3511. [DOI] [PubMed] [Google Scholar]
- 27.Chaturvedi R, Asim M, Romero-Gallo J, Barry DP, Hoge S, de Sablet T, et al. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology. 2011;141:1696–1708. e1691–1692. doi: 10.1053/j.gastro.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardbower DM, de Sablet T, Chaturvedi R, Wilson KT. Chronic inflammation and oxidative stress: the smoking gun for Helicobacter pylori-induced gastric cancer? Gut microbes. 2013;4:475–481. doi: 10.4161/gmic.25583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaturvedi R, Asim M, Piazuelo MB, Yan F, Barry DP, Sierra JC, et al. Activation of EGFR and ERBB2 by Helicobacter pylori results in survival of gastric epithelial cells with DNA damage. Gastroenterology. 2014;146:1739–1751. e1714. doi: 10.1053/j.gastro.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tammer I, Brandt S, Hartig R, Konig W, Backert S. Activation of Abl by Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology. 2007;132:1309–1319. doi: 10.1053/j.gastro.2007.01.050. [DOI] [PubMed] [Google Scholar]
- 31.Backert S, Moese S, Selbach M, Brinkmann V, Meyer TF. Phosphorylation of tyrosine 972 of the Helicobacter pylori CagA protein is essential for induction of a scattering phenotype in gastric epithelial cells. Mol Microbiol. 2001;42:631–644. doi: 10.1046/j.1365-2958.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 32.Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, et al. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci U S A. 1993;90:5791–5795. doi: 10.1073/pnas.90.12.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, et al. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci U S A. 2002;99:14428–14433. doi: 10.1073/pnas.222375399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naito M, Yamazaki T, Tsutsumi R, Higashi H, Onoe K, Yamazaki S, et al. Influence of EPIYA-repeat polymorphism on the phosphorylation-dependent biological activity of Helicobacter pylori CagA. Gastroenterology. 2006;130:1181–1190. doi: 10.1053/j.gastro.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 35.Sicinschi LA, Correa P, Peek RM, Camargo MC, Piazuelo MB, Romero-Gallo J, et al. CagA C-terminal variations in Helicobacter pylori strains from Colombian patients with gastric precancerous lesions. Clin Microbiol Infect. 2010;16:369–378. doi: 10.1111/j.1469-0691.2009.02811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology. 1998;115:642–648. doi: 10.1016/s0016-5085(98)70143-x. [DOI] [PubMed] [Google Scholar]
- 37.Franco AT, Johnston E, Krishna U, Yamaoka Y, Israel DA, Nagy TA, et al. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008;68:379–387. doi: 10.1158/0008-5472.CAN-07-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, et al. Activation of {beta}-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci U S A. 2005;102:10646–10651. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M. Attenuation of Helicobacter pylori CagA x SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J Biol Chem. 2003;278:3664–3670. doi: 10.1074/jbc.M208155200. [DOI] [PubMed] [Google Scholar]
- 40.Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, et al. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J. 2008;27:1671–1681. doi: 10.1038/emboj.2008.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Babbar N, Casero RA., Jr Tumor necrosis factor-alpha increases reactive oxygen species by inducing spermine oxidase in human lung epithelial cells: a potential mechanism for inflammation-induced carcinogenesis. Cancer Res. 2006;66:11125–11130. doi: 10.1158/0008-5472.CAN-06-3174. [DOI] [PubMed] [Google Scholar]
- 42.Sheh A, Chaturvedi R, Merrell DS, Correa P, Wilson KT, Fox JG. Phylogeographic origin of Helicobacter pylori determines host-adaptive responses upon coculture with gastric epithelial cells. Infect Immun. 2013;81:2468–2477. doi: 10.1128/IAI.01182-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moss SF, Calam J, Agarwal B, Wang S, Holt PR. Induction of gastric epithelial apoptosis by Helicobacter pylori. Gut. 1996;38:498–501. doi: 10.1136/gut.38.4.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valentin-Vega YA, Okano H, Lozano G. The intestinal epithelium compensates for p53-mediated cell death and guarantees organismal survival. Cell Death Differ. 2008;15:1772–1781. doi: 10.1038/cdd.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kodaman N, Pazos A, Schneider BG, Piazuelo MB, Mera R, Sobota RS, et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci U S A. 2014;111:1455–1460. doi: 10.1073/pnas.1318093111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mera R, Fontham ET, Bravo LE, Bravo JC, Piazuelo MB, Camargo MC, et al. Long term follow up of patients treated for Helicobacter pylori infection. Gut. 2005;54:1536–1540. doi: 10.1136/gut.2005.072009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyskens FL, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, Hawk E, et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res Phila Pa. 2008;1:9–11. doi: 10.1158/1940-6207.CAPR-08-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zell JA, McLaren CE, Chen WP, Thompson PA, Gerner EW, Meyskens FL. Ornithine decarboxylase-1 polymorphism, chemoprevention with eflornithine and sulindac, and outcomes among colorectal adenoma patients. J Natl Cancer Inst. 2010;102:1513–1516. doi: 10.1093/jnci/djq325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Basu HS, Thompson TA, Church DR, Clower CC, Mehraein-Ghomi F, Amlong CA, et al. A small molecule polyamine oxidase inhibitor blocks androgen-induced oxidative stress and delays prostate cancer progression in the transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2009;69:7689–7695. doi: 10.1158/0008-5472.CAN-08-2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tummuru MK, Cover TL, Blaser MJ. Mutation of the cytotoxin-associated cagA gene does not affect the vacuolating cytotoxin activity of Helicobacter pylori. Infect Immun. 1994;62:2609–2613. doi: 10.1128/iai.62.6.2609-2613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagy TA, Frey MR, Yan F, Israel DA, Polk DB, Peek RM., Jr Helicobacter pylori regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J Infect Dis. 2009;199:641–651. doi: 10.1086/596660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zabaleta J, Camargo MC, Piazuelo MB, Fontham E, Schneider BG, Sicinschi LA, et al. Association of interleukin-1beta gene polymorphisms with precancerous gastric lesions in African Americans and Caucasians. Am J Gastroenterol. 2006;101:163–171. doi: 10.1111/j.1572-0241.2006.00387.x. [DOI] [PubMed] [Google Scholar]
- 53.Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. Am J Surg Pathol; International Workshop on the Histopathology of Gastritis; Houston. 1994 ; 1996. pp. 1161–1181. [DOI] [PubMed] [Google Scholar]
- 54.Goodwin AC, Destefano Shields CE, Wu S, Huso DL, Wu X, Murray-Stewart TR, et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci U S A. 2011;108:15354–15359. doi: 10.1073/pnas.1010203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Casero RA, Jr, Celano P, Ervin SJ, Porter CW, Bergeron RJ, Libby PR. Differential induction of spermidine/spermine N1-acetyltransferase in human lung cancer cells by the bis(ethyl)polyamine analogues. Cancer Res. 1989;49:3829–3833. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.