

Abstract
Peptide Nucleic Acids (PNAs) are artificial DNA mimics with superior nucleic acid binding capabilities. T7 RNA polymerase transcription upon encountering PNA bound to the non-template DNA strand was studied in vitro. A characteristic pattern of blockage signals was observed, extending downstream from the PNA binding site, similar to that produced by G-rich homopurine-homopyrimidine sequences and likely caused by R-loop formation. Since blocked transcription complexes in association with stable R-loops may interfere with replication and in some cases trigger apoptosis, targeted R-loop formation might be employed to inactivate selected cells, such as those in tumors, based upon their unique complement of expressed genes.
Keywords: Triplex, RNA-DNA hybrids, gene targeting, R-loop toxicity, transcription blockage
Peptide Nucleic Acids (PNAs) are constructed with electrically neutral peptide-like linkages that have been substituted for the natural negatively charged phosphodiester backbone, while the bases (and consequent rules of complementarity) are the same as those for DNA or RNA [1]. PNAs have very strong nucleic acid binding properties relative to those of natural nucleic acids, and under certain conditions they can invade double-stranded DNA by displacing the homologous DNA strand and binding the complementary one. The unique nucleic acid binding capability of PNA, as well as its stability against enzymatic degradation, makes it attractive for many biological applications, including gene-specific transcription blockage (reviewed in [2,3]). An important example of an especially robust PNA-DNA hybridization is when homopyrimidine PNA is targeted to a homopurine/homopyrimidine region in double-stranded DNA. In this case either two PNA molecules or two halves of one PNA molecule can form a triplex with the complementary homopurine DNA strand to leave the homopyrimidine strand unpaired (reviewed in [2,3]).
We have reported the effect upon transcription by T7 RNA polymerase (T7 RNAP) of the latter type of PNA, targeted to Friedreich’s ataxia triplet repeats (GAA/CTT)n [4]. When PNA was bound to the template DNA strand, there was a significant reduction in full length transcription products and well-pronounced blockage signals in a narrow region at the beginning of the repeat, consistent with the view that the RNAP is “bumping” into the PNA-DNA-PNA triplex on the template strand and attempting to displace the PNA to render that strand available for transcription. Surprisingly, for long repeats (n=39) there was also a strong reduction in the amount of full-length transcription product when PNA was bound to the non-template strand; however, in that case there was no well-defined narrow area of blockage signals; rather, there was a “smear” presumably comprising unresolved blockage signals beginning somewhere within the insert and continuing further downstream with no defined end-point. The nature of this effect of PNA binding to the non-template strand was initially unclear.
Unexpectedly, an insight into the phenomenon arose from our studies of transcription blockage due to G-rich homopurine-homopyrimidine sequences. These sequences only cause transcription blockage for the orientation in which the homopyrimidine (hPy) strand serves as a template (i.e. the RNA is homopurine (hPu)), [5,6]. Importantly, this orientation corresponds to the situation in which an especially stable RNA/DNA duplex would form (e.g. [7]). This observation, together with our analysis of the effects of sequence variations and buffer conditions upon the blockage led to our conclusion that the most likely cause of blockage is co-transcriptional re-hybridization of the nascent RNA with the DNA template, i.e. R-loop formation [6]. This conclusion was further supported by the observation that a nick in the non-template strand (which strongly facilitates R-loop formation [8]) exacerbates the blockage produced by G-rich sequences and even produces detectable blockage with random sequences [9]. Remarkably, the blockage signals produced are localized predominantly at the downstream flank of the causative sequence and extend further downstream; thus, most blockages occur when the RNAP has already passed the causative sequence (although there are a few blockages inside and upstream of the causative sequence). For a nick in the non-template strand, the blockages were also localized downstream from the site of the nick, providing further support for the conclusion that this pattern of blockage is associated with R-loop formation [9]. These observations lead us to propose that the main explanation for R-loop-induced transcription blockage is that an R-loop destabilizes the transcription complex (e.g. by disrupting some interactions between the nascent RNA and RNAP), thus “sensitizing” it to very weak (“hidden”) pausing/termination signals (i.e. short nucleotide stretches for which the probability of transcription blockage is somewhat greater than average; statistically these may occur within any DNA sequence [9], reviewed in [10]). That could account for the distinct bands in the blockage pattern. For the transcription complex destabilized by R-loop formation the probability of blockage would be much higher than that for a “normal” transcription complex; however, the exact sites at which this probability is maximal would be determined by some features of the DNA sequence.
In addition, blockage could occur when an RNAP encounters an R-loop created in the “wake” of another RNAP, similar to the case reported by Tous and Aguilera [11]. That could account for some blockage signals localized upstream from the causative sequence. The latter mechanism, however, has only a secondary role, since most blockages occur downstream of the causative sequence, and those would be difficult to ascribe to collisions with previously formed R-loops.
The similarity between the blockage patterns (with respect to the downstream extension) produced by G-rich homopurine/homopyrimidine sequences and nicks in the non-template strand on the one hand [6,9], and by PNA binding to the non-template strand on the other hand [4], suggest a common mechanism (i.e. R-loop formation). However, direct comparison of the blockage patterns reported in reference [6,9] and in reference [4] is difficult because of the different insert lengths and different flanking sequences. To address this issue, we compared the PNA-binding insert (GAA)10 GA and a G-rich homopurine-homopyrimidine insert (G5A)5 GG with identical lengths and flanking regions. As an additional control for the sequence-specificity of the PNA effect upon transcription, an A32 insert was also examined. The results of the transcription experiment for these sequences, with and without PNA, are shown in Fig.1.
Fig. 1. Sequence-specific transcription blockage by PNA binding to the non-template strand.

Linearized plasmids with the respective insert sequences are shown at the top of the gel. All inserts are 32 nt long, and start 252 nt downstream from the promoter. Position of the insert on gel (shown as thick black line) is estimated according to denatured DNA ladders with steps of 10 nucleotides and 100 nucleotides, designated by vertical 10 and 100, respectively. See Reference [9] for detailed description of the plasmids. The plasmids were pre-incubated with or without PNA Lys-TTJTTJTT-OOO-TTCTTCTT-Lys (J stands for pseudoisocytosine (a cytosine analog that forms non-protonated Hoogsteen base pairing with G, thus relaxing the pH-dependence for triplex formation), and O stands for 8-amino-3,6-dioxaoctonic acid residues, which form a flexible linker), and used in the T7 RNAP in vitro transcription experiment. Conditions for hybridization with PNA and for transcription are the same as in [4] and in [9], respectively, except that after pre-incubation with (or without) PNA, the samples were diluted up to 1 ng/μl in buffer containing 50 mM potassium acetate, 20 mM Tris-acetate (pH 7.9), 10 mM magnesium acetate, 1 mM dithiothreitol, and only 1 ng instead of 10 ng were used in the transcription reaction. The scheme of the transcription assay and the PNA-DNA hybrid substrate are shown to the right of the gel. The insert is expected to bind roughly three PNA molecules. Briefly, transcription was performed with NTPs and radioactive CTP, and the products were analyzed on a sequencing gel. Undisrupted transcription produced full-length product (“run-off”), while transcription blockage is indicated by the presence of truncated transcription products and a reduced amount of runoff. The strongest PNA-induced and intrinsic blockage bands are shown by white and gray block arrows, respectively.
It is seen that for the (GAA)10 GA insert (lanes 3 and 4), in the presence of PNA the total amount of run-off product is strongly reduced (about 16-fold relative to PNA-minus control with the same sequence), and well-pronounced blockage signals appear (shown by white block arrows), with the strongest one (shown by largest arrow) localized at the downstream flank of the insert. Additional blockage signals form an irregular ladder extending further downstream, and there are also weaker blockage signals upstream from the insert.
In the case of the control sequences A32 and (G5A)5 GG, no additional blockage signals appear in the presence of PNA, and the intensity of the predominant intrinsic blockage signal in (G5A)5 GG insert (relative to run-off product) was the same (2.5%) with and without PNA; for comparison, in the case of the PNA-bound -sequence (GAA)10 GA the predominant blockage signal alone comprises about 40% of the run-off product, while intrinsic blockage signals for this sequence without PNA are barely detectable. However, there is roughly a 2-fold reduction in the amount of run-off products for the control sequences in the presence of PNA. The origin of this non-specific effect (which is an order-of-magnitude weaker than the respective effect for the PNA-bound specific sequence) is unknown; but, since it is not associated with any detectable blockages, it doesn’t affect our analysis.
Importantly, the locations of the most pronounced PNA-induced blockage signals (shown by white block arrow) at the downstream flank of the insert coincide with the location of the predominant intrinsic blockage signals for the G-rich hPu-hPy insert (G5A)5 GG, the “repeat-exiting signal” (shown by gray block arrow). However, for the latter insert, the further downstream blockage signals were poorly pronounced. To make them more robust, we took advantage of the strong exacerbation of transcription blockage induced by G-rich homopurine-homopyrimidine inserts, when the DNA is negatively supercoiled [6]. Fig.2 shows a strong correlation (shown by double-headed arrows) between the locations of the intrinsic blockage signals produced by the (G5A)5 GG insert in negatively supercoiled DNA, and by the (GAA)10 GA insert upon PNA binding to the non-template strand. (In supercoiled DNA the (GAA)10 GA insert also produces detectable intrinsic blockage signals, but they are much weaker than those for the (G5A)5 GG insert. Note that for the (GAA)-repeats the intrinsic transcription blockage could also be caused by intramolecular triplex formation, although that occurs in repeats much longer than the ones used in the present work [12]).
Fig. 2. Correlation between patterns of transcription blockage signals induced by PNA binding to the non-template strand, and by a G-rich hPu-hPy insert.
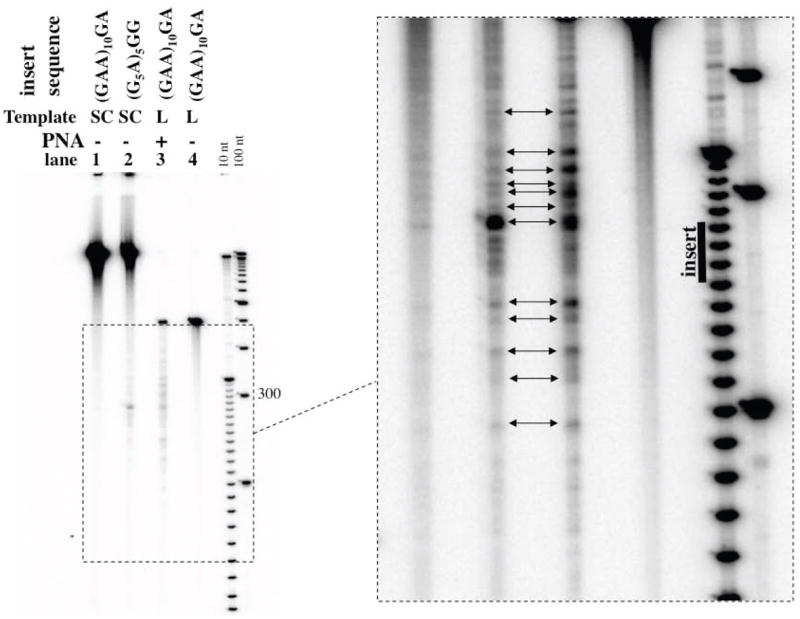
The transcription experiment was performed as for Fig.1. The higher exposure for the region of interest (in dashed-border box) is shown in the right panel. Double-headed arrows show correlation between blockage signals in lanes 2 and 3.The purpose of the experiment presented in this figure is to demonstrate the strong similarity between the locations of the blockage signals produced by two seemingly unrelated factors: “free” G-rich hPu-hPy insert in negatively supercoiled DNA and PNA-bound A-rich hPu-hPy insert in linear DNA. The correct comparison of percentage of blockage requires substrates of the same topology (e.g. linear), as done for the experiments presented in Fig.1.
It not known why in the case of PNA binding-induced blockages (Fig.2, lane 3) the decrease in blockage signal intensities downstream from the repeat-exiting signal is more gradual than that for the intrinsic blockages produced by the G-rich insert (Fig.2, lane 2). In this respect, the former pattern of blockage signals is reminiscent of that produced by a nick in the non-template strand [9].) However, the remarkable correlation between blockages induced in the surrounding sequences by two seemingly unrelated factors strongly suggests a common blockage mechanism. Since there is extensive evidence that the transcription blockage by G-rich hPu-hPy inserts is mediated by R-loop formation [6,9], the same mechanism is likely to be involved in blockage induced by PNA binding to the non-template DNA strand. As mentioned above, the discrete pattern of blockage signal is consistent with our hypothesis that very weak “hidden” sequence-specific blockage signals are exacerbated by R-loop formation.
The facilitation of R-loop formation by PNA binding to the non-template DNA strand can be easily explained by the fact that re-hybridization of the non-template DNA strand with the template DNA strand behind the transcription complex must compete with R-loop formation [8]; and, consequently, sequestration of the non-template DNA strand by PNA would prevent its hybridization with the template DNA strand, thus facilitating R-loop formation, as shown schematically in Fig.3.
Fig.3. Model for PNA-mediated R-loop formation followed by transcription blockage.

DNA, RNA and PNA are shown in light gray, dark gray and black, respectively. For simplicity, only one PNA molecule is shown. RNAP is shown as a light-gray oval with dotted borders, and the area within the RNAP that is involved in interactions with exiting nascent RNA, (which presumably stabilizes the transcription complex), is shown as a white patch with dashed borders. From top to bottom: the transcribing RNAP moves toward the PNA/DNA complex. Because the homologous region within the template strand is rendered single-stranded by PNA binding, it tends to hybridize with the nascent RNA after the RNAP passes this region. This hybridization could begin as “threading back” and then continue further in both directions, extending the R-loop upstream, and partially disrupting interactions between the nascent RNA and the RNAP, thus destabilizing the transcription complex and likely blocking transcription. Note that we don’t imply that the blockage necessarily occurs immediately after the R-loop forms. Rather, the transcription could continue in the R-loop mode, but it becomes more prone to spontaneous blockage with a probability depending upon the transcribing sequence (for more detailed discussion of the model for R-loop-mediated transcription blockage see [9]). In addition, R-loops, which extend upstream from the causative sequence could block a following RNAP (not shown). As in the case of downstream blockages, the blockage in this case could be a statistical event, with the probability depending upon the local properties of the sequence (e.g. increased stability of RNA-DNA hybrids within R-loop, which makes them more difficult to displace during transcription).
R-loop formation could be monitored directly by transcribing unlabeled plasmid DNA in the presence of radioactive NTP, followed by treatment with single-strand specific RNase and resolving the products on neutral agarose gels. Sequestration of radioactive RNA within a plasmid (e.g. in the form of R-loops) leads to the appearance of a slower migrating plasmid-size radioactive band, while free RNA fragmented by single-strand specific RNases migrates much faster. The presence of RNA-DNA hybrids within these slower migrating species can be confirmed by RNaseH treatment, which degrades RNA within the RNA-DNA hybrid, leading to loss of these species. The results from this approach (Fig.4) confirm that PNA binding to the non-template DNA strand facilitates R-loop formation.
Fig. 4. PNA enhances R-loop formation in a sequence-specific manner.

A: The specific and control targets were supercoiled plasmids (sc), containing the sequences (GAA)10 GATCT (the PNA-binding sequence is in bold) and C8G4, respectively; the rest of the sequence (about 3.6 kb total) is the same for the two targets. To obtain PNA/DNA hybrids, a mixture containing 0.1μg/μl of the plasmid, 10 μM of PNA, 40 mM NaCl, 9 mM TrisHCl (pH 7.9) and 0.9 mM EDTA were incubated for 2 hours at 37°C (for PNA-minus control, all procedures were the same, except without PNA). Hybrids purified from excess of PNA by using QIAquick PCR purification kit (Qiagen), recovered in EB buffer (10 mM TrisHCl, pH 8.5), supplemented with 0.1 mM EDTA, and used for in vitro transcription reaction. The transcription reaction mixture (60 μl) containing ~10 ng/μl of the PNA/DNA hybrids, 33 mM TrisHCl (pH 7.9), 5 mM MgCl2, 8.3 mM NaCl, 1.7 mM spermidine, 4.2 mM DTT, 0.17 mM of each ATP, GTC, UTP, CTP, 10 μCi (α-32P)CTP (which corresponds to final concentration of about 0.0003 mM), and 1.7 units/μl of T7 RNA polymerase (Promega corp, Madison, WI) was incubated for 1 hour at 37°C. Transcription was stopped by addition of EDTA (up to 10 mM), diluted up to 100 μl by TE buffer (pH 7.9), and 5 μl of single-stranded RNA-specific RNase mixture containing 0.5 units/μl of RNase A and 2 units/μl of RNase T1 (Ambion) was added; incubation continued for 1 hour at 37°C. After that, samples were purified by PCR purification kit, recovered in 50 μl EB buffer, diluted twice with TE buffer, supplemented with NaCl (final concentration 50 mM); the RNase A and T1 digestion step was repeated with the same concentration of RNases for 1.5 hours at 37°C. After that, samples were finally purified by PCR purification kit and recovered in 30 μl EB buffer supplemented by 0.1 mM EDTA. For RNase H digest, 7 μl of these samples (~ 0.1 μg) were incubated with 7.5 units of RNase H (NEB) in 15 μl of RNase H buffer (NEB) for 2 hours at 37°C (The RNaseH-minus control was incubated under the same conditions). Samples were analyzed on agarose gels, first by ethidium bromide staining (bottom panel), and then (after drying on DE 81 paper) by phosphorimaging (top panel). Retention of radioactive material within slow migrating fractions with position on the gel roughly consistent with position of the plasmid DNA indicates R-loop formation. The R-loop formation was much greater in the presence of PNA and with the PNA-binding insert in the plasmid (lane 1). A low background of R-loops forms in the absence of PNA, as expected for negatively supercoiled templates. Note that in the absence of PNA, the background amount of R-loops in the control plasmid (lane 4) is larger than that in the GAA plasmid (lane 2), probably because of the higher G/C-rich insert or greater superhelicity. However, this background is not affected by PNA (lane 3 versus lane 4), and is much less than the PNA-induced R-loops in the GAA plasmid (lane 1). These data additionally emphasize the sequence-specificity of PNA-induced R-loop formation. B: RNaseH treatment of the sample similar to the one in lane 1 in A leads to disappearance of the slower migrating radioactive species, confirming that they contain RNA-DNA hybrids (i.e. R-loops).
Partial transcription blockage by PNA binding to the non-template strand has been previously observed for RNA polymerase II in HeLa cell nuclear extracts [13] and in human prostatic cancer cells [14]; and it is possible that a mechanism similar to that suggested here might be responsible for the blockage.
We believe that our suggested mechanism, of robust transcription blockage mediated by R-loop formation induced by PNA binding to the non-template strand (Fig.3) may have practical applications. The R-loop formation, in addition to blocking transcription, could interfere with many cellular processes (reviewed in [15]) and in some cases trigger apoptosis [16]. Thus, the deleterious effects of R-loops could extend far beyond the depletion of a specific gene product: actually, their very presence in the cell might be deleterious. Since the R-loops would be induced only in a transcribed genes by the mechanism suggested from our work, we speculate that targeted R-loop formation could render the expression of a given gene “toxic” for the cell; and consequently one could selectively inactivate a designated sub-population of cells (e.g. cancer cells) by targeting PNA to particular genes transcribed only in this sub-population, regardless of how important the transcription products of these genes might be for those cells. In addition, the non-template strand is likely to be much more accessible for hybridization with PNA in an actively transcribed gene than in a silent gene, because of the transient opening of DNA during transcription. That should further enhance the selectivity of this approach for cells, in which a particular gene is expressed. Thus, targeted R-loop formation is worth considering for possible anticancer strategies based upon the elucidated differences in transcription profiles between the normal and tumor cells (e.g. [17]).
Acknowledgments
Grant Support
The work was supported by a grant, CA077712, to PCH from the National Cancer Institute, NIH.
Abbreviations
- PNA
Peptide Nucleic Acid
- RNAP
RNA polymerase
References
- 1.Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991;254(5037):1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 2.Nielsen PE. PNA Technology. Mol Biotechnol. 2004;26(3):233–248. doi: 10.1385/MB:26:3:233. [DOI] [PubMed] [Google Scholar]
- 3.Frank-Kamenetskii MD, Mirkin SM. Triplex DNA structures. Annu Rev Biochem. 1995;64:65–95. doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- 4.Belotserkovskii BP, Liu R, Hanawalt PC. Peptide nucleic acid (PNA) binding and its effect on in vitro transcription in Friedreich’s ataxia triplet repeats. Mol Carcinog. 2009;48(4):299–308. doi: 10.1002/mc.20486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krasilnikova MM, Samadashwily GM, Krasilnikov AS, Mirkin SM. Transcription through a simple DNA repeat blocks replication elongation. Embo J. 1998;17(17):5095–5102. doi: 10.1093/emboj/17.17.5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belotserkovskii BP, Liu R, Tornaletti S, Krasilnikova MM, Mirkin SM, Hanawalt PC. Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc Natl Acad Sci U S A. 2010;107(29):12816–12821. doi: 10.1073/pnas.1007580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts RW, Crothers DM. Stability and properties of double and triple helices: dramatic effects of RNA or DNA backbone composition. Science. 1992;258(5087):1463–1466. doi: 10.1126/science.1279808. [DOI] [PubMed] [Google Scholar]
- 8.Roy D, Zhang Z, Lu Z, Hsieh CL, Lieber MR. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site. Mol Cell Biol. 2010;30(1):146–159. doi: 10.1128/MCB.00897-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belotserkovskii BP, Neil AJ, Saleh SS, Shin JH, Mirkin SM, Hanawalt PC. Transcription blockage by homopurine DNA sequences: role of sequence composition and single-strand breaks. Nucleic Acids Res. 2013;41(3):1817–1828. doi: 10.1093/nar/gks1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belotserkovskii BP, Mirkin SM, Hanawalt PC. DNA sequences that interfere with transcription: implications for genome function and stability. Chem Rev. 2013;113(11):8620–8637. doi: 10.1021/cr400078y. [DOI] [PubMed] [Google Scholar]
- 11.Tous C, Aguilera A. Impairment of transcription elongation by R-loops in vitro. Biochem Biophys Res Commun. 2007;360(2):428–432. doi: 10.1016/j.bbrc.2007.06.098. [DOI] [PubMed] [Google Scholar]
- 12.Grabczyk E, Usdin K. The GAA*TTC triplet repeat expanded in Friedreich’s ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 2000;28(14):2815–2822. doi: 10.1093/nar/28.14.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanvey JC, Peffer NJ, Bisi JE, et al. Antisense and antigene properties of peptide nucleic acids. Science. 1992;258(5087):1481–1485. doi: 10.1126/science.1279811. [DOI] [PubMed] [Google Scholar]
- 14.Boffa LC, Morris PL, Carpaneto EM, Louissaint M, Allfrey VG. Invasion of the CAG triplet repeats by a complementary peptide nucleic acid inhibits transcription of the androgen receptor and TATA-binding protein genes and correlates with refolding of an active nucleosome containing a unique AR gene sequence. J Biol Chem. 1996;271(22):13228–13233. doi: 10.1074/jbc.271.22.13228. [DOI] [PubMed] [Google Scholar]
- 15.Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46(2):115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Lin Y, Wilson JH. Transcription-induced DNA toxicity at trinucleotide repeats: double bubble is trouble. Cell Cycle. 2011;10(4):611–618. doi: 10.4161/cc.10.4.14729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross DT, Scherf U, Eisen MB, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24(3):227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]