

Abstract
Background
RNA dot blot hybridization is a commonly used technique for gene expression assays. However, membrane based RNA dot/slot blot hybridization is time consuming, requires large amounts of RNA, and is less suited for parallel assays of more than one gene at a time. Here, we describe a glass-slide based miniaturized RNA dot blot (RNA array) procedure for rapid and parallel gene expression analysis using fluorescently labeled probes.
Results
RNA arrays were prepared by simple manual spotting of RNA onto amino-silane coated microarray glass slides, and used for two-color fluorescent hybridization with specific probes labeled with Cy3 and 18S ribosomal RNA house-keeping gene probe labeled with Cy5 fluorescent dyes. After hybridization, arrays were scanned on a fluorescent microarray scanner and images analyzed using microarray image analysis software. We demonstrate that this method gives comparable results to Northern blot analysis, and enables high throughput quantification of transcripts from nanogram quantities of total RNA in hundreds of samples.
Conclusion
RNA array on glass slide and detection by fluorescently labeled probes can be used for rapid and parallel gene expression analysis. The method is particularly well suited for gene expression assays that involve quantitation of many transcripts in large numbers of samples.
Background
Commonly used non-high throughput gene expression assay methods include, Real-time RT-PCR, RNA blot (Northern and dot/slot blot) hybridization and RNase protection assay. Quantitation of gene expression using real-time RT-PCR has the advantage of high sensitivity and requires only small amounts of RNA, but the method usually requires extensive optimization and validation [1]. Northern blotting, which is performed by agarose gel electrophoresis of RNA followed by transfer of the RNA onto porous solid supports [2], typically nylon or nitrocellulose membranes, is a robust technique for size determination and quantitation of transcripts. Quantitation of transcripts can also be done by dot/slot blot hybridization, performed after transfer of RNA to membranes directly without size separation [3]. Radioactive probes are still the most commonly used detection methods in RNA blot hybridizations, although non-radioactive labeling methods have also been developed [4]. Generally, RNA blot hybridization methods are robust and reliable procedures that require little optimization. However, membrane-based RNA dot/slot hybridizations are time-consuming, require large amounts of sample (usually at least 5 μg total RNA per sample), and are less suited for parallel analysis of many genes at a time.
New detection and imaging technologies have facilitated the development of microarray-based assays on glass slide such as DNA microarrays [5], protein microarrays [6], and tissue microarrays [7]. Here, we describe a glass slide-based RNA micro dot blot hybridization for rapid and parallel quantitation of specific transcripts in multiple samples using fluorescently labeled probes. The procedure involves preparation of RNA arrays by simple manual spotting of RNA onto amino-silane coated glass slides, and hybridization with two probes labeled with different color fluorescent dyes, followed by scanning and image analysis.
Results
Preparation of RNA arrays
RNA arrays were prepared by manually spotting total RNA on amino-silane coated glass slides. The RNA was dissolved in 50% Dimethyl sulfoxide (DMSO). Using a pipet tip with a narrow opening, it was possible to print 40–50 spots per μl of RNA, which corresponds to about 20–25 nl. At 0.5 μg/μl total RNA concentration, this corresponds to 10–12.5 ng RNA per spot. The spots were visible to the naked eye (typically about 400 μm average diameter) and were well isolated from each other (mean spot-to-spot distance about 800 μm). Slides containing 2 or 5 blocks of arrays were prepared. On one array pattern, 10 different liver total RNA samples from 5 controls and 5 drug treated rats were spotted, each in 10 replicates, making an array of 100 spots on an area of 18 × 18 mm2. Two such 100-spot replica arrays were made per slide for hybridization with two different cDNA probes at a time (Fig. 1A and 1B). For parallel hybridizations with 5 different cDNA probes at a time, a second pattern of arrays was prepared by dividing a slide into 5 slots (each with an area of 8 × 18 mm2). On such slides, 10 RNA samples were spotted in quadruplicates (10 × 4 spots) in each slot (Fig. 1C). Thus, it was demonstrated that, on a single slide, an array of at least 200 different samples can be prepared by simple manual spotting of as little as 10 ng total RNA per sample.
Figure 1.
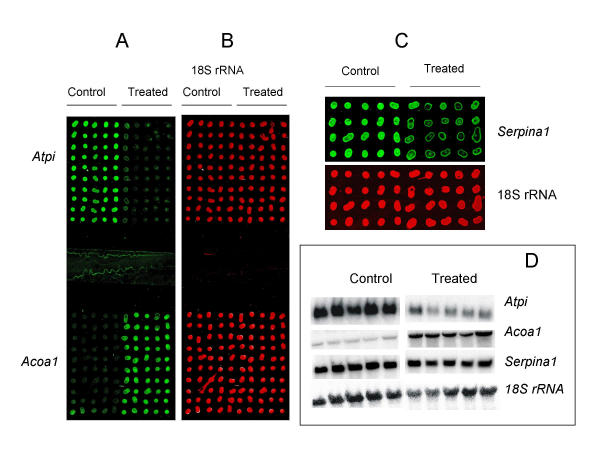
Quantitative gene expression analysis using RNA array. RNA samples from controls and drug treated rats were arrayed on a glass slide in two blocks of 100 spots consisting of 10 different samples (columns), each with 10 replicate spots (rows) and hybridized with fluorescently labeled probes (A and B). After hybridization and washing, the slide was scanned on a microarray scanner, resulting in two pseudo-color images for Cy3-labeled Atpi and Acoa1 (A) and Cy5-labeled 18S ribosomal RNA for normalization (B). A smaller array format with a block of 40 spots consisting of 10 different samples (columns), each with 4 replicate spots (rows) is shown in (C). Shown here (Serpina1) is one of 5 transcripts analyzed in parallel hybridizations on separate slots of a single slide. For comparison, Northern blot hybridization images for the Atpi, Acoa1, Serpina1 and 18SrRNA probes are shown in (D).
Array hybridization and image analysis
Each probe was labeled with Cy3-dCTP and spiked with Cy5-dCTP labeled 18S ribosomal RNA (rRNA) probe for two-color hybridization. After hybridization and washing, the slide was scanned using a microarray scanner, generating two color images showing spot intensities for the gene of interest and for 18S ribosomal RNA (Fig. 1A,1B,1C). The images were analyzed using microarray image analysis software. The Cy3 fluorescence intensity in each spot was quantified, normalized to corresponding Cy5 fluorescence intensity of 18S ribosomal RNA, and used to compare expression levels in the control and treated animals (Fig. 2). RNA array based analysis of Acoa1 and Atpi on a slide with two 100 spot arrays showed that they were significantly up-regulated and down-regulated, respectively (Figs. 1 and 2). To illustrate the possibility of increasing the throughput of the method, up to 5 different transcripts were quantified in parallel by dividing the slide into well isolated slots with separate blocks of arrays and hybridizing with different probes. An example of such a slot with a block of array is shown in Fig. 1C. Thus, 2–5 genes can be analyzed in parallel on a single slide.
Figure 2.
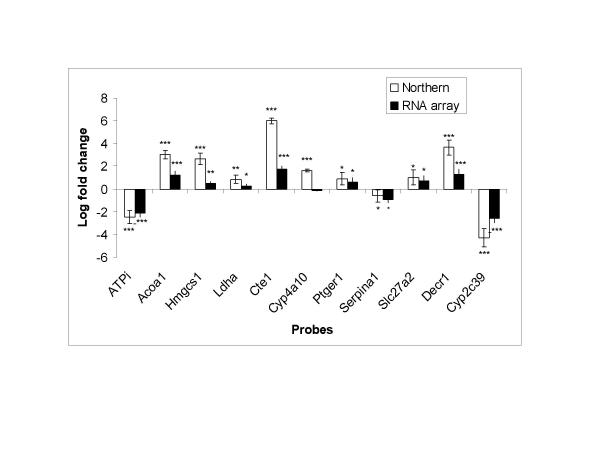
Comparison of differential expression levels using RNA array and Northern blotting. For RNA array and Northern blot, the normalized intensity of fluorescence or radioactivity signal was used to compare expression levels in controls and drug treated rats (n = 5 per group) for each gene. Expression levels were expressed in folds of control (fold changes) by dividing signal intensity of each drug treated rat by mean signal intensity of control rats. Log fold changes shown (y-axis) represent log2 transformed values for drug treated rats. The p-values are for comparison of expression levels in controls and drug treated rats. Data are presented as mean ± SD. Asterisk (*) indicate significance levels: * = p < 0.05; ** = p < 0.01; *** = p < 0.001.
Evaluation of sensitivity and dynamic range of RNA array method
To evaluate the sensitivity and dynamic range of the method, a 10-fold dilution series (0.000625 pg/μl – 625 pg/μl) of three different RNAs (prepared from Arabidopsis thaliana genomic DNA fragments by in vitro transcription) were spiked into 0.5 μg/μl rat liver total RNA, and arrays were prepared for hybridization with the respective Cy3 labeled probes (Fig. 3A). Since the synthetic RNAs were diluted in total RNA, Cy5 labeled 18S rRNA was also used for normalization of differences in RNA quantities per spot (not shown). The three RNAs (2F9, 2H4 and 2G6) have 30 nt poly(A) tail to mimic cellular poly(A)+ RNAs, and were prepared for use as spiking controls in DNA microarray experiments (Yadetie et al., manuscript in preparation). As shown in Fig. 3A and 3B, the 0.625–62.5 pg/μl dilutions of the three RNAs show linear trends in log-log plots of fluorescent signals versus RNA concentrations. The highest concentration (625 pg/μl) resulted in signal saturations, as is most apparent for the 2H4 RNA (Fig. 3A and 3B). Thus, the linear dynamic range of the method was approximated to be two to three orders of magnitude. From figure 3A and 3B, the detection limit was approximated to be 0.0625–0.625 pg/μl spiked RNA in 0.5 μg total RNA. The 0.0625 pg/μl dilution corresponds to 1:8,000,000 spiked RNA to total RNA, or 1:400,000 spiked RNA to poly(A)+ RNA mass ratio [assuming 5% of total RNA mass as poly(A)+ RNA)].
Figure 3.
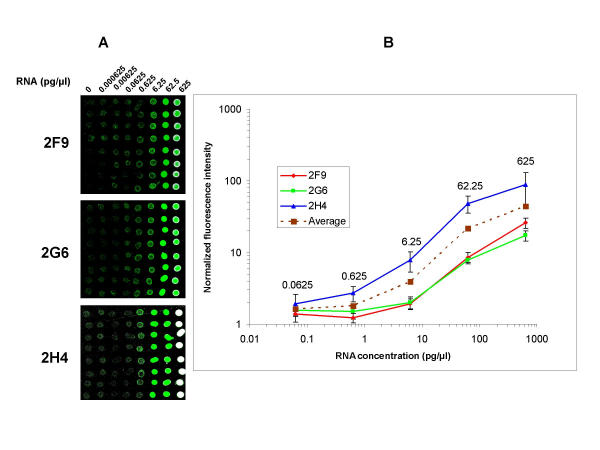
Evaluation of dynamic range and sensitivity of RNA Arrays. Images from RNA arrays prepared from 10-fold dilution series of three external control RNAs (2F9, 2G6 and 2H4) and hybridized with Cy3 labeled probes (A). Each of the three plant RNAs were spiked into rat liver total RNA (0.5 μg/μl) with 10 fold dilution series ranging from 0.000625 – 625 pg/μl. The first column is negative control (liver total RNA with no spiked in RNA). Log-log plots of normalized average fluorescence intensity of 8 replicate spots per dilution versus RNA concentration in the dilution series of the three transcripts (B). The two lowest concentrations 0.000625 and 0.00625 pg/μl did not result in signals over background levels and were omitted in the plots. Data are presented as mean ± SD.
In the experiments to assay gene expression in rat liver, the average amount of total RNA deposited per spot shown in Fig. 1A,1B,1C was 10–12.5 ng (using 0.5 μg/μl total RNA for spotting). Hybridization of RNA arrays prepared by spotting a dilution series of total RNA from control rat liver showed that a gene that appears to be moderately expressed (Atpi) could be detected in as little as 1 ng of total RNA per spot (data not shown).
Validation of RNA array
Since our aim was to test the RNA array method, 11 rat genes shown to be regulated by ciprofibrate or other peroxisome proliferator-activated receptor-alpha agonists [8,9] were selected (Table 1). Attempts were made to include genes representing wide range of differential expression. To evaluate the performance of the method, the expression levels obtained using Northern blotting and RNA array were compared (Fig. 1A,1B,1C,1D and Fig. 2). As shown in Fig. 2, RNA array gave qualitatively similar fold changes in expression to Northern blot for 10 of the 11 genes. Only one gene (Cyp4a10) that appeared up-regulated by Northern blot analysis (p < 0.001) was recorded as unchanged (p = 0.14) by RNA array analysis. The observed differential expressions between the control and drug treated rats detected by Northern blotting were also detected as significant changes (p < 0.05) by the RNA array method for all the genes analyzed by both methods apart from one gene (Cyp4a10) (Fig. 2). Quantitative comparison of individual expression levels obtained by the two methods indicates that the RNA array method tends to underestimate fold changes. The compression of fold changes obtained by RNA array was more apparent for genes with high fold changes such as Cte1 and Decr1 (Fig. 2). However, the lower range fold changes were less compressed, and the RNA array method could detect differential expressions for the genes Ldha, Ptger1, Serpina1 and Slc27a2 that changed by 1.5–2 folds in Northern blot analysis.
Table 1.
cDNA clones used for preparation of probes
| Name | Symbol | I.M.A.G.E. Consortium Clone ID | Accession number |
| Acyl COA oxidase | Acoa1 | UI-R-A1-ea-c-06-0-UI | AA924697 |
| ATPase inhibitor | Atpi | UI-R-A0-ab-f-01-0-UI | |
| 3-Hydroxy-3-methylglutaryl-Coenzyme A synthase 1 | Hmgcs1 | UI-R-A1-eb-h-10-0-UI | AA924800 |
| Lactate dehydrogenase A | Ldha | UI-R-C2-ng-d-04-0-UI | |
| Cytosolic acyl-CoA thioesterase 1 | Cte1 | UI-R-A1-eh-d-03-0-UI | AA925003 |
| Prostaglandin E receptor 1 | Ptger1 | UI-R-E0-bo-d-07-0-UI | AA858510 |
| Cytochrome P450, 4a10 | Cyp4a10 | UI-R-A0-au-a-12-0-UI | AA818499 |
| Serine (or cysteine) proteinase inhibitor, clade A, member 1 | Serpina1 | UI-R-A0-bk-d-02-0-UI | AA819467 |
| 2,4-Dienoyl CoA reductase 1, mitochondrial | Decr1 | UI-R-E0-ce-b-01-0-UI | AA875267 |
| Solute carrier family 27 (fatty acid transporter) | Slc27a2 | UI-R-A1-es-e-04-0-UI | AA925897 |
| Cytochrome P450, 2c39 | Cyp2c39 | UI-R-A0-ai-c-04-0-UI | AA818043 |
Discussion
We have developed a miniaturized fluorescent RNA dot blot (RNA array) method for simple and rapid quantitation of many transcripts in parallel. The results show that very small quantities of total RNA from hundreds of samples can be efficiently immobilized onto glass slides by simple manual spotting, and hybridized to specific probes labeled with Cy3 and Cy5 fluorescent dyes. The hybridization, scanning and image analysis procedures are similar to two-color fluorescent DNA microarray protocols, obviating the need for additional equipment in laboratories with access to DNA microarray facilities.
The method was tested for analysis of differential expression of 11 genes in control and drug treated rats. The RNA array approach gave comparable results with membrane based Northern blot hybridizations. RNA array and Northern blot gave concordant results for 10 of the 11 genes compared. One gene (Cyp 4a10) was up-regulated as detected by Northern blotting, but not by RNA array. This discordance is probably because of cross-hybridization. Although the RNA array method resulted in compressed fold changes compared to Northern blot, particularly for genes with high range of fold changes, it could reliably detect as low as 1.5 fold differential expressions. In contrast to dot/slot blot hybridizations, size fractionation of the RNA in Northern blotting offers the advantage of minimizing background such as possible signals from cross-hybridization of the probe to other sequence related genes. Fold change compressions have also been observed in other fluorescent based detection methods compared to radioactive Northern blotting [10].
The dynamic range and sensitivity of the approach was also evaluated using in vitro transcribed plant RNAs. The dynamic range of the method (at least two orders of magnitude) is within the range reported for other fluorescent labeling methods [11,12]. Assuming 360,000 poly(A)+ RNA copies per cell [13], the detection limit 0.0625–0.625 pg in 0.5 μg total RNA background is equivalent to 1–9 transcripts per cell, which suggests that the method enables detection of most cellular transcripts in total RNA preparations. However, it may be necessary to use poly(A)+RNA for quantitative detection of rare transcripts.
The RNA array method described here has important improvements over membrane-based RNA dot/slot blot hybridizations. One of its main advantages is that it can be performed using only nanogram quantities of total RNA, which is about three orders of magnitude less than the amounts often required for membrane based dot blot or Northern blot assays. Thus, the method can be useful for analysis of gene expression in very small samples, for example from Laser Capture Microdissection [14] and small clinical biopsies. Fluorescently labeled probes can also be stored for a long time and are more convenient to use than radioactive probes. A further advantage is that the throughput of the method can be increased by using multiple hybridization slots per slide, and probes labeled with different color fluorescence to detect multiple transcripts in one hybridization mixture. As demonstrated here, using simple manual spotting, 5 genes could be analyzed at a time on a single slide (divided into 5 slots) with two-color labeling. Therefore, using four-color fluorescent labeling and simultaneous hybridization, it should be possible to analyze up to 15 genes in parallel on a single slide.
In addition to minimizing the amount of RNA needed for analysis, the miniaturization allows quantitation of expression of many genes in hundreds of samples on a single slide. Although arrays can be easily prepared by manual spotting for a small number of samples, a microarray printing robot or commercially available hand-held spotting devices may be used when large numbers of samples are to be analyzed.
Therefore, the method should find application as an alternative approach of gene expression assay, in particular when large numbers of samples and small quantities of RNAs are to be analyzed. For example, since the RNA array procedure described here can be performed in DNA microarray facilities without extra equipment, one possible application of the method is in validation of DNA microarray results. Results obtained using large scale gene expression assay by DNA microarrays are currently validated using more time consuming techniques such as real-time RT-PCR, Northern blotting, in-situ hybridization and ribonuclease protection assay [15].
Conclusions
RNA array on glass slide and quantitation of gene expression by hybridization with fluorescently labeled probes is a reliable, simple and rapid method of gene expression assay. The method can be used for quantitation of transcript levels in very small quantities of total RNA, and allows analysis of a large number of samples at a time.
Methods
Reagents and chemicals
Fluorescently labeled deoxyribonucleotides Cy5-dCTP and Cy3-dCTP, radioisotope [α-32P] dCTP (3000 mCi/mmol), and random hexanucleotide primers were purchased from Amersham Biosciences (Little Chalfont, UK). DNA polymerase (Klenow fragment) was from Promega (Madison, WI). Amino-silane coated UltraGAPS glass slides were from Corning (Corning, NY). Gel loading pipet tips (Prot/Elec Tips, catalog # 223-9915EDU) were purchased from Bio-Rad Laboratories (Bio-Rad Laboratories, Hercules, CA)
RNA preparation
RNA samples were from liver of control and combined ciprofibrate and octreotide dosed rats described previously [16]. Briefly, total liver RNA was isolated from animals dosed with vehicle (control, n = 5) or drug (n = 5) by ultracentrifugation on a cesium chloride cushion and further purified using TRIZOL®Reagent according to manufacturer's protocols (GIBCO BRL Life Technologies Inc., New York, NY).
Preparation of RNA arrays
The RNA samples were dissolved in spotting solution (50% DMSO in RNAse free water) at a concentration of 0.5 μg/μl. For spotting RNAs, the array pattern was drawn on a paper in a rectangular sketch with dimension of a glass slide (25 × 75 mm) indicating the positions of all spots and blocks. A freshly unpacked 25 × 75 mm amino-silane coated UltraGAPS glass slide was positioned on the rectangular sketch and fixed to the paper at each corner using a piece of adhesive tape or glue. Then, using narrow-bore gel loading pipet tip (Prot/Elec Tips; Bio-Rad Laboratories), 0.5–2 μl of the RNA solution was taken up by capillary action, and manually spotted by gently touching the slide surface at the spot positions indicated by the sketch on the paper underneath. The slide was then left to dry at room temperature for a few minutes, before the RNA was immobilized by baking for 20–30 min at 60°C in an oven followed by UV irradiation at 200 mJoules using a UV Crossliker (Hoefer Scientific Instruments, San Francisco CA).
Preparation of fluorescently labeled probes
Plasmids containing cDNA inserts of the analyzed genes (Table 1) were prepared from bacterial cultures of I.M.A.G.E. Consortium [LLNL] rat cDNA [17]http://image.llnl.gov obtained from a commercial supplier (Research Genetics, Huntsville, AL). Probe cDNA fragments were prepared by PCR amplifying cDNA inserts using M13 primers 5'-CTG CAA GGC GAT TAA GTT GGG TAA C and 5'-GTG AGC GGA TAA CAATTT CAC ACA GGA AAG. The PCR products were separated on agarose gel and purified using QIAQuick Gel Extraction kit (QIAGen, GmbH Max-Volmer-Strasse, Germany). The 18S ribosomal RNA (rRNA) probe has been described previously [16]. The purified cDNA fragments were labeled using random priming method described below.
The following reaction mix was made in a microcentrifuge tube on ice: 2 μl 10 × dNTP mix (2 mM each of dATP, dGTP, dTTP, and 0.5 mM dCTP), 2 μl 10 × Klenow reaction buffer, 0.5 μl 1 mM Cy3-dCTP or Cy5-dCTP, 10 U DNA polymerase (Klenow Fragment) and ddH2O, to a final volume of 10 μl.
In another microcentrifuge tube, 30–100 ng of the DNA fragment to be labeled was combined with 2.5 μg random hexamers in a volume of 10 μl and denatured by heating at 98°C for 3 min, and cooled on ice. This template and primer solution was mixed with the above reaction mix (reaction buffer, nucleotides and DNA polymerase) and incubated protected from light at room temperature for 1–2 hours. The reaction was stopped by adding 1 μl 0.5 mM EDTA, pH 8.0 or by heating at 70°C for 10 min.
The labeled probe was then separated from unincorporated nucleotides using AutoSeq sephadex G-50 spin column according to manufacturer's protocol (Amersham Bioscences). After elution the probe was concentrated to 5–10 μl using SpeedVac. In a typical hybridization, 10–25% of the Cy3-labeled cDNA probe was used. For 18S rRNA, 0.1–1% of the Cy5-labeled probe resulted in fluorescent signals within the dynamic range as determined in initial pilot hybridizations (not shown).
RNA array hybridization
The RNA array slides were prehybridized in 1% BSA, 3.5 × SSC, 0.1% SDS at 65°C for 20 min, then washed by dipping in ddH2O (5 times) and finally dipped in isopropranol (2 times) and air dried. The concentrated Cy3-labeled probe spiked with 0.1–1% of the labeled 18S rRNA probe (combined volume 1–2 μl) was added to 17 μl hybridization solution [(0.25 M NaH2PO4, 1 mM EDTA, 4.5% SDS, 1 × SSC, 2 × Denhardt's Solution, 10–20 μg tRNA, 1 μg poly(dA)40–60)]. The probe was then denatured by incubating for 2 min at 98°C, cooled to room temperature, transferred onto the array and covered by a 22 × 22 mm2 cover slip (for 10 × 10 spots arrays). For the 4 × 10 spot arrays, the amount of probe and hybridization buffer was reduced by half and a 22 × 11 mm2 cover slip was used. The slide was then put in a hybridization chamber (Corning) with 10 μl ddH2O on each side to avoid drying. The hybridization chamber was assembled and submerged in a water bath at 65°C for hybridization for 2–5 hours.
Post-hybridization washes were done once in 2 × SSC, 0.2% SDS at 65°C for 10 min, then in 2 × SSC at 55°C for 10 min, and finally with 0.2 × SSC at 55°C for 10 min. After the final wash, the slide was dried by centrifuging at 1000 rpm for 3 min.
Preparation of Arabidopsis RNAs for spiking
The preparation of RNAs for spiking controls is described in detail elsewhere (Yadetie et al., in preparation). Briefly, Arabidopsis thaliana genomic DNA fragments were cloned into a plasmid vector pSP64 poly(A) (Promega) and sequenced. No sequence similarity was detected with mammalian transcripts as checked by BLAST search, and no cross-hybridization was observed in microarray hybridization experiments with RNAs from mammalian tissues (Yadetie et al., in preparation).
The genomic fragments were amplified by PCR from three plasmid clones (2F9, 2H4 and 2G6) using plasmid specific primers (forward: 5'-CAA-CAT-AAC-CTT-ATG-TAT-CAT-ACA-C and reverse: 5'-GAC-ATG-ATT-ACG-AAT-TCG-GTT). The PCR products were purified and labeled as described above. Purified PCR products were also used to synthesize poly(A)+RNA by in vitro transcription using Ambion's MEGAscript™ kit according to accompanying protocols (Ambion, Austin, TX). The size of the RNA product (in base), is 472, 811 and 713 for 2F9, 2H4 and 2G6, respectively. For each of the RNAs, a 10-fold dilution series was prepared in 0.5 μg/μl rat liver total RNA (in 50% DMSO) with the following concentrations (pg/μl): 0, 0.000625, 0.00625, 0.0625, 0.625, 6.25, 62.5 and 625. RNA arrays were prepared and hybridized with the corresponding Cy3 labeled probes as described above. Cy5 labeled 18S rRNA probe was also included in the hybridization for normalization of differences in RNA quantities per spot.
Scanning and image analysis of RNA arrays
Arrays were scanned with a ScanArray ExpressHT microarray scanner (Packard, Billerica, USA) at 10 μm resolution. Image analysis was done using GenePix Pro 4.1 software (Axon Instruments, Union City, USA). Spot identification and quantitation was performed as for a standard microarray image analysis, and the results were exported to an Excel spreadsheet. In order to compensate for variations in amounts of RNA deposited in spots, normalization was performed by dividing the background corrected mean fluorescence intensity of target cDNA probe (Cy3 channel) by background corrected mean fluorescence intensity of the house-keeping gene (18S rRNA) probe (Cy5 channel). Expression levels were expressed in folds of control (fold changes) by dividing signal intensity of each drug treated rat by mean signal intensity in control rats. The resulting ratios were log2 transformed for comparison of expression levels between controls and drug treated rats.
Northern blot hybridization and analysis
Preparation and purification of DNA probes is as described above for RNA arrays. The purified cDNA fragment was labeled using [α-32P] dCTP (3000 Ci/mmol) (Amersham Biosciences) by random priming method and purified on Sephadex G-50 columns (Amersham Biosciences). Total RNA samples (15 μg/lane) were separated on a 1.1% formaldehyde-agarose gel and blotted onto Hybond N nylon membrane (Roche Applied Science, Mannheim, Germany). After UV cross-linking, the membrane was prehybridized at 42°C for at least 3 h in 50% formamide, 5 × SSPE (1 × SSPE is 0.18 M NaCl, 10 mM NaH2PO4, 1 mM EDTA, pH 7.4), 5 × Denhardt's solution, 0.5% SDS, and 200 μg/ml salmon sperm DNA. After prehybridization, the purified labeled probe was added to the prehybridization buffer and the membrane was hybridized at 42°C for 12 – 18 h. Post-hybridization washes were done twice in 2 × SSPE, 0.1% SDS at room temperature for 20 min, and then once in 0.1 × SSPE, 0.1% SDS at 65°C for 20 min. The membrane was stripped by boiling in 0.1% SDS before rehybridization. The membrane was also hybridized with 18S rRNA house-keeping gene probe for normalization of differences in RNA quantities loaded in each lane. The membrane was exposed to a phosphor storage screen for 2–24 h, scanned and analyzed using the PhosphorImager system with the ImageQuant software (Molecular Dynamics, Kent, UK).
Statistical analysis
For RNA array and Northern blotting, mean values of normalized expression levels between control (n = 5) and drug treated rats (n = 5) were compared using Student's t-test. P < 0.05 was considered significant. Data are presented as mean ± SD.
Authors' contributions
FY designed and performed the experiments, and drafted the manuscript. HB and KN participated in the discussions of the experimental design, data analysis and review of the manuscript. AKS supervised the work and reviewed the manuscript. AL, coordinated and supervised the study, and reviewed the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We thank Sylvia Nome Kvam and Anne Kristensen for technical assistance. This study was supported by The Norwegian Research Council, The Cancer Fund at the University Hospital of Trondheim and The Norwegian Cancer Association.
Contributor Information
Fekadu Yadetie, Email: fekadu.yadetie@medisin.ntnu.no.
Arne K Sandvik, Email: arne.Sandvik@medisin.ntnu.no.
Hallgeir Bergum, Email: hallgeir.bergum@medisin.ntnu.no.
Kristin Norsett, Email: kristin.norsett@medisin.ntnu.no.
Astrid Laegreid, Email: astrid.lagreid@medisin.ntnu.no.
References
- Bustin SA. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol. 2002;29:23–39. doi: 10.1677/jme.0.0290023. [DOI] [PubMed] [Google Scholar]
- Alwine JC, Kemp DJ, Stark GR. Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc Natl Acad Sci U S A. 1977;74:5350–4. doi: 10.1073/pnas.74.12.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafatos FC, Jones CW, Efstratiadis A. Determination of nucleic acid sequence homologies and relative concentrations by a dot hybridization procedure. Nucleic Acids Res. 1979;7:1541–52. doi: 10.1093/nar/7.6.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler C. Non-radioactive analysis of biomolecules. J Biotechnol. 1994;35:165–89. doi: 10.1016/0168-1656(94)90034-5. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–70. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Haab BB, Dunham MJ, Brown PO. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001;2:RESEARCH0004. doi: 10.1186/gb-2001-2-2-research0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4:844–7. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–88. doi: 10.1210/er.20.5.649. [DOI] [PubMed] [Google Scholar]
- Yadetie F, Laegreid A, Bakke I, Kusnierczyk W, Komorowski J, Waldum HL, Sandvik AK. Liver gene expression in rats in response to the peroxisome proliferator-activated receptor-{alpha} agonist ciprofibrate. Physiol Genomics. 2003;15:9–19. doi: 10.1152/physiolgenomics.00064.2003. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Miura K, Iwao H, Yamanaka S. Quantitative assessment of DNA microarrays – comparison with Northern blot analyses. Genomics. 2001;71:34–9. doi: 10.1006/geno.2000.6427. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R, Dorris D, Lublinsky A, Nguyen A, Domanus M, Prokhorova A, Gieser L, Touma E, Lockner R, Tata M, et al. An assessment of Motorola CodeLinkTM microarray performance for gene expression profiling applications. Nucl Acids Res. 2002;30:e30. doi: 10.1093/nar/30.7.e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum M, Bielau S, Rittner N, Schmid K, Eggelbusch K, Dahms M, Schlauersbach A, Tahedl H, Beier M, Guimil R, et al. Validation of a novel, fully integrated and flexible microarray benchtop facility for gene expression profiling. Nucl Acids Res. 2003;31:e151. doi: 10.1093/nar/gng151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MD, Jatkoe TA, Stumpf CR, Lu J, Thomas JD, Madore SJ. Assessment of the sensitivity and specificity of oligonucleotide (50 mer) microarrays. Nucl Acids Res. 2000;28:4552–4557. doi: 10.1093/nar/28.22.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, Weiss RA, Liotta LA. Laser Capture Microdissection. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- Chuaqui RF, Bonner RF, Best CJ, Gillespie JW, Flaig MJ, Hewitt SM, Phillips JL, Krizman DB, Tangrea MA, Ahram M, et al. Post-analysis follow-up and validation of microarray experiments. Nat Genet. 2002;32:509–14. doi: 10.1038/ng1034. [DOI] [PubMed] [Google Scholar]
- Bakke I, Sandvik AK, Waldum HL. Octreotide inhibits the enterochromaffin-like cell but not peroxisome proliferator-induced hypergastrinemia. J Mol Endocrinol. 2000;25:109–19. doi: 10.1677/jme.0.0250109. [DOI] [PubMed] [Google Scholar]
- Lennon G, Auffray C, Polymeropoulos M, Soares MB. The I.M.A.G.E. Consortium: an integrated molecular analysis of genomes and their expression. Genomics. 1996;33:151–2. doi: 10.1006/geno.1996.0177. [DOI] [PubMed] [Google Scholar]