

Abstract
Using pre-miR-451 as a model molecule, we have characterized the general molecular properties of small hairpin RNAs that are processed into potent small interfering RNAs (siRNA) by Argonaute2 (Ago2). The Ago2-sliced siRNAs (sli-siRNAs) have the same silencing potency as the classical Dicer diced siRNAs (di-siRNAs) but have dramatically reduced unwanted sense strand activities. We have built vectors with the constitutive or inducible U6 promoter that can express sli-siRNAs in mammalian cells, in which the sli-siRNAs can be correctly processed to repress target genes. As a proof of principle for potential applications of sli-siRNAs in vivo, we show that the expression of one Ago2 shRNA-1148 in HCT-116 colon cancer cells knocked down RRM2 expression and reduced the proliferation and invasiveness of the cells. The defined sli-siRNA model molecules and the expression systems established in this study will facilitate the design and application of sli-siRNAs as novel potent RNAi triggers with reduced off-target effects.
Keywords: Ago2, agshRNA, agsiRNA, RNAi, siRNA, sliced siRNA
Introduction
Gene silencing by siRNAs is a powerful technology for manipulating gene expression and a potential therapeutic strategy for treating human diseases. Canonical siRNAs are ~21-nucleotide (nt) small RNAs that mimic products of Dicer processed double strand RNAs and can be incorporated into the RNA-induced silencing complex (RISC) to trigger the degradation of mRNA targets that contain highly complementary sequences.1 Canonical siRNAs are designed to resemble the biogenesis intermediates of microRNAs (miRNA), a family of endogenous small RNAs that can repress the translation of target mRNAs that contain fully or partially complementary sequences. Therefore, siRNA and miRNA shared the same functional machineries in the cell.2,3 The majority of miRNAs use Dicer to process the precursor-miRNAs (pre-miRNAs) to create 21- to 23-nt duplex RNAs that consist of one strand from the 5′ arm (5p) and one strand from the 3′ arm (3p). The 3′ end of each strand has an overhang of two nt. This duplex RNA is also referred to as miRNA/miRNA*(the dominant strand/the less abundant strand). Accordingly, siRNAs are designed as duplexes of antisense strand/sense strand (guide strand/passenger strand) RNAs that are 21-nt long and have a 19 base pair dsRNA stem and an overhang of two nt at the 3′ end of each strand (siRNA, Figure 1a, top). In contrast, similar duplexes that have overhangs of two nt at the 5′ end (hereafter referred to as reverse siRNA or rsiRNA, Figure 1a, middle) are thought to be mostly inactive.1 DNA vector systems can also be used to express siRNAs as short hairpin RNAs (shRNAs, exp-shRNA), which can be used to express corresponding siRNAs in stable cell lines.4,5 Several recent publications have revealed critical roles for loops, length of stems, and base pairing in the stem in exp-shRNA processing and silencing potency.6,7,8 In vitro T7 transcribed or chemically synthesized shRNAs (syn-shRNAs) were also shown to be potent RNAi triggers.9 The functional structure of syn-shRNAs was further characterized, and the short-stem version was named as short shRNAs (sshRNA), which are Dicer independent.10,11 Despite its extensive application as an effective gene manipulation reagent in research, the bright future of RNAi therapeutics is shadowed by growing evidence that many siRNAs have toxic side effects due to off-target activities of both the sense and antisense strands. These off-target effects will also produce biased research data.12 Therefore, siRNA molecules that have a potent on-target effect and lack off-target activities are highly desirable for both clinical and research applications. Despite extensive works done in the past decade for this purpose, it remains a challenge to find an optimized siRNA for a special target. Many design rules, including sequence selection, base modifications, target site accessibility, and the end thermodynamics stability of di-siRNAs, have to be applied during design in order to find an ideal siRNA.13,14 Interestingly, miR-451 uses an elegant slicing biogenesis mechanism that involves Ago2, but not necessarily Dicer.15,16,17 This mechanism can be used to design shRNAs that can be loaded onto Ago2 to be processed only by Ago2 into functional siRNAs without sense strand activity (Figure 1a, bottom). Ago2 acts by first nicking the shRNA substrates in the middle of the 3p to produce a long fragment (bases 1 to 30, 30-nt long; hereafter referred to as L30) and a short fragment (bases 31 to 42, 12-nt long; hereafter referred to as 3L12). The 3′ end of L30 is then trimmed by other enzymes to produce a functional siRNA that is capable of gene silencing (Figure 1b).15,16,17 Although pre-miR-451 and mimic sequences can be loaded into RISCs formed by all Agos, they are exclusively processed by Ago2.18,19 This mechanism could also explain the mystery behind the potency of syn-shRNAs related to the length of its stems and the choice of strand used to implement antisense strand.11 This mechanism was applied to design shRNAs with short stems which were characterized as Dicer-independent and Ago2-prone processing agoshRNA.20 Despite published results of detailed parameters regarding miR-451 biogenesis and the fact that pre-miR-451 mimic sequences have reduced sense strand activities and may avoid the competition with endogenous miRNAs for processing machinery,18,19,20,21 the application of sli-siRNAs is still very limited compared to traditional siRNAs. One reason is the lack of general rules to design sli-siRNAs. In addition, there are no versatile vectors that are specifically constructed to express sli-siRNAs. Furthermore, the effects of sli-siRNAs on their targets, endogenous miRNA pathways in cells, and other response of the host immune system to their presence must be addressed in order for them to have a broad usage and serve as clinical therapeutic molecules.
Figure 1.

sli-siRNA molecules can be processed by Ago2. (a) Schematic of the secondary structures of siRNA with a 3′ end overhang of two nt (upper), rsiRNA with a 5′ end overhang of two nt (middle), and agsiRNA (lower). (b) Upper panel: Secondary structure of human pre-miR-451. Mature miR-451 is shown in red, the short fragment generated by Ago2 from 3p is shown in blue (3L12); gray bases were trimmed during miR-451 maturation. Middle and lower panels: the predicted tertiary structures of hsa-pre-miR-451 without the last 3′ UC bases. (c) Alignment of pre-miR-451 from 18 species found in miRBase 19. (d) Structures predicted by mFold for agsiRNA-887, agsiRNA-887-mut, and agshRNA-887. (e) Northern blot analysis of agshRNA-887 and agsiRNA-887 in Dicer-/-, Ago2-/-, and wild-type MEFs. c, control sli-siRNA with a scrambled sequence; m, mutant; w, wild-type. Both U2 and U6 snoRNAs were used as RNA loading controls. (f) Reporter assays of sli-siRNA-887, rsiRNA-887, and siRNA-887 in Dicer-/-, Ago2-/-, and wild-type MEFs. mmp7, mismatch for base #7 with #34; wb, GU has mutated base #33 from C to U. (g) Antisense strand (5p) reporter assays in HCT-116 cells. rsi, rsiRNA-887; si, siRNA-887; L30-3L12 is agsiRNA-887 that was reconstituted by annealing 3L12 with L30. Rluc/Fluc ratios are plotted. Error bars represent the SD. h. Sense strand (3p) reporter assays of agsiRNA-887 (wt), rsiRNA-887 (rsi), and siRNA-887 (si) in HCT-116 cells. Rluc/Fluc ratios are plotted. Error bars represent the SD. MEF, mouse embryonic fibroblast.
To this end, we have experimentally defined general sequence parameters used to design sli-siRNAs that will be preferentially processed by Ago2 into potent siRNAs. We were able to modify the popular U6 shRNA expression promoter (U6m), but not the H1 or U1 promoter, to express sli-siRNAs in mammalian cells both constitutively and conditionally. Through lengthy characterization of the substrate properties of sli-siRNAs, we have defined the canonical structure that will produce potent sli-siRNAs. We have named the synthetic version of sli-siRNA “agsiRNA” and the expressed version of sli-siRNA “agshRNA.” Although agsiRNA and agshRNA model molecules are similar to the previously reported model molecules, sshRNA and agoshRNA, in that they all use Ago2 for processing and function, the major difference between these new types of small RNAs and the previously reported molecules lies in how the loop is designed: sli-siRNAs use 4 nt of the antisense as loop; sshRNA used UU to link 19 nt antisense strand with 19 nt sense strand; agoshRNA used 5–7 nt universal loops.10,20 Although sli-siRNAs generally have similar silencing potency to di-siRNAs, sli-siRNAs have dramatically reduced sense strand activities, thus much less off-target effect. We also showed our U6m promoter can express sli-siRNAs in mammalian cells, where they can be correctly processed to repress the expression of their targets. As a proof of principle for their potential in vivo applications, we showed the expression of agshRNA-1148 in HCT-116 colon cancer cells knocked down RRM2 expression and reduced the proliferation and invasiveness of the cells. Therefore, sli-siRNAs will be a viable option for developing novel potent RNAi triggers with much off-target effects.
Results
Define canonical sli-siRNA
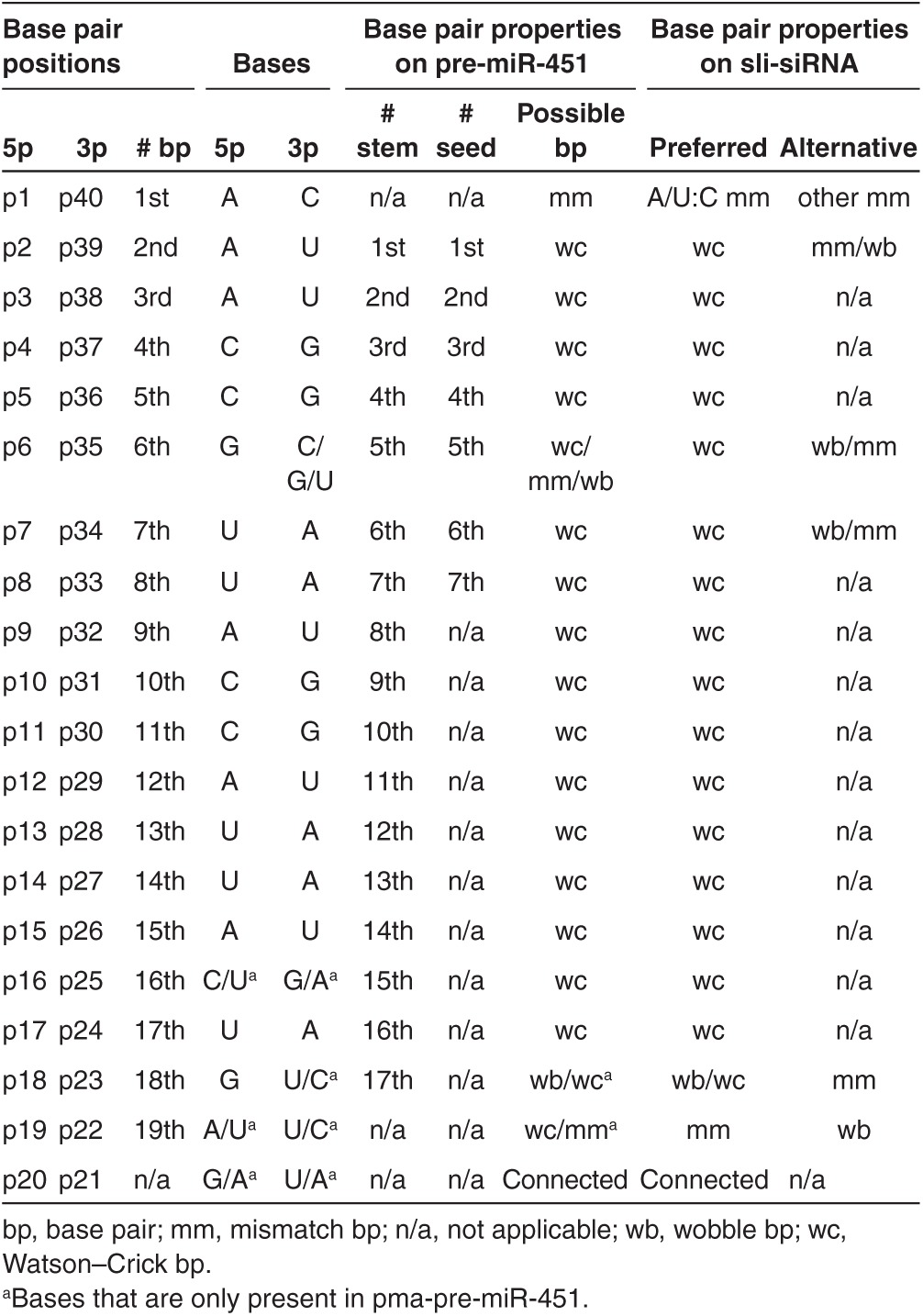
To characterize the structural properties of pre-miR-451 that are required for processing into the mature miR-451 by Ago2, we aligned all documented pre-miR-451 sequences from 18 species in miRBase 19 and found that pre-miR-451 sequences were highly conserved in 17 of the 18 species. Among all species, the 35th base (p35) was almost equally a C, U, or G, so it could form perfect GC pair, or GU wobble, or mismatch with the G at p6, respectively, indicating flexibility for this base pairing may have been maintained during evolution by an unknown selection mechanism (Figure 1c, Supplementary Figure S1, and Table 1). We observed that pre-miR-451 was 42 nt long and could be expressed by a RNA pol III promoter if we replace the last two pre-miRNA nt with an UU. Therefore, pre-miR-451 that lacks the last two nt is 40 nt long; the anchor base A (p1) is mismatched with the end base C (p40) and forms a small fork; nt p2 to p18 and p23 to p39 form a 17-nt stem (S17); and nt p19 to p22 form a small loop of four nt (L4). Furthermore, the Mid domain of Ago2 has a much higher binding affinity for substrates that have an A or U at the 5′ end, as opposed to a C or G.22,23 Therefore, the 5′ nt should be an A or U. We next reasoned that the CUC 3′ overhang of pre-miR-451 are products of Drosha/DGCR8 complex and may not be essential for Ago2 processing and subsequent silencing function because it will be degraded as part of 3L12 but that the C at p40 may be used for end base modifications to prevent the RNA from degrading from the 3′ end. Accordingly, we defined our canonical agsiRNA structure as the 40 nt structure A/U-S17-L4-C (L40, Figure 1a bottom, 3L12 becomes 3L10 from p31 to p40).
Table 1. Properties of pre-miR-451 versus sliced siRNA molecules.
To convert agsiRNA to agshRNA, the choice of promoters and their transcription start sites will be critical for their function since agshRNA will default 5p as antisense strand. We designed several agshRNAs that would target the M2 subunit of ribonucleotide reductase RRM2 gene (R2). The antisense strands were defined as 22 nt long (L22: the first 18 nt plus 4 nt in the loop), and hairpins were expressed by the U6m promoter (Supplementary Figure S2). At least half of the agshRNAs efficiently reduced R2 protein levels and knocked down expression of a Renilla luciferase reporter gene that had the human R2 cDNA sequences inserted into its 3′ UTR (Supplementary Figures S3 and S4). Thus, our U6m agshRNA expression vector can express this type of shRNA. To address the concern about how U6m will start its transcripts if the first nt of L22 is not a G, we sequenced products from eight R2 agshRNAs using small RNA deep sequencing.24 In most cases, U6m mainly used bases A and C to initiate agshRNA transcripts, but the transcription will be initiated at the upstream C of U6m or the second base in agshRNA if the first base of the agshRNA sequence was a T. In addition, Ago2 processed these agshRNAs into L30, but there are many uridylated and trimmed intermediate products were also derived from L30. The length distributions of these L30-derived intermediates resembled that for isomiR-451 forms that are documented in miRBase, suggesting that agshRNA transcripts expressed from the U6m promoter are processed by Ago2, similar to the way miR-451 is processed (Supplementary Table S2). Accordingly, we defined our canonical agshRNA structure as the 40 nt structure A-S17-L4-C.
The secondary structures of agshRNA and agsiRNA likely differ in how they form. Presumably, agshRNA is folded in vivo, and the single molecule folded form (agsiRNA in Figure 1a and Supplementary Figure S5a) is then exported to the cytoplasm. AgsiRNAs are artificially folded in vitro by denaturing and annealing. Thus, some agsiRNAs will be in the single molecule folded form, while some will be in dimer. Since the agsiRNAs in the dimer form is not necessary to be identical, we named them cross molecule hybridization form. Once the agsiRNAs are transfected into the cytoplasm, Dicer should be able to process the cross molecule hybridization form of agsiRNA into four products (A21, A19, 21C, and 19C in Supplementary Figure S5b,c), which will be similar to the products generated by RNases that nick the single molecule folded form of agsiRNA between p19 and p20 or p21 and p22. Based on this hypothesis, the L40 form of pre-miR-451 may be less likely to be processed by Dicer than the 42-nt pre-miR-451 form.
We further characterized agshRNA-887, -1148, and -1354, and their synthetic forms, agsiRNA-887, -1148, and -1354, all of which target R2 (Figure 1d and Supplementary Figures S3 and S4). We used sli-siRNA-887 for most of the studies because, among all of the R2 agshRNAs we constructed, agshRNA-887 exhibited moderate knockdown of R2 (Supplementary Figure S4a,b). We reasoned it would be easier to observe changes in the potency of this sli-siRNA in response to modifying its structure and base composition. The secondary structures of the agshRNA-887 forms used in our experiments are shown in Figure 2, and individual structures are listed in Supplementary Figure S6.
Figure 2.

Sli-siRNA-887 and variants. Variants of agsiRNA-887 used in this study were made by replacing nt in the backbone or adding extra nt to the 5′ end or 3′ end of the molecule. Based on the wt agsiRNA-887 backbone, modifications were made as following: replacing of the anchor “A” (p1) with “U” or a “C” to make U/C-S17-L4 forms; prefixing the anchor “A” (p1) with “a,” “aa,” “aaa,” or “aaaa” to make 5′ overhang variants a/aa/aaa/aaaa-A-S17-L4; mismatch base pairing at p6:35, p7:34, p8:33, p13:28, p14:27, and p15:26 to make the mmp6, mmp7, mmp8, mmp13, mmp14, and mmp15 forms, respectively; replacing the “C” at p33 with an “U” to make the GUp8 form; replacing the p31, p30, and p29 bases with the p10, p11, and p12 bases to make the mutant (mut) form; Replacing the “C” at p40 with a “ddC” to make the “ddC” form; adding one “U,” two “U”s, five “U”s, or two “dT”s to the “C” at p40, respectively, to make the U, 2U, 5U, and dTdT forms; replacing “G” at p39 with “C” to make mmp2 form; replacing “G” at p39 with “C” and “U” at p38 with “A” to make mmp2-3 form; replacing “U” at p23 with “C” to make p18GC; replacing the “U” at p23 with a “G” to make mmp18; replacing the “U” at p23 with a “G” and the “U” at p24 with an “A” to make mmp17-18; replacing the “U” at p23 with a “C”, an “U” at p24 with an “A”, and the “A” at p25 with an “U” to make mmp16-17-18. ddC, dideoxy C; dTdT, deoxy T deoxy T.
Sli-siRNAs are Ago2 specific and Dicer independent
We tested the processing of sli-siRNA-887 and its mutant that had mismatches at central bases (Figure 1d,e) in Dicer-knockout and Ago2-knockout mouse embryonic fibroblasts. Northern blot analysis and reporter assays showed that both agshRNA-887 and agsiRNA-887 were Ago2 dependent and Dicer independent and that Ago2 could not process the mutant form of sli-siRNA-887 (Figure 1d–f). Northern blot analysis also indicated that the cross molecule hybridization form of sli-siRNA probably exists only at very low levels, because the predicted Dicer processed forms (A19) were not detected (Figure 1e). Reporter assays showed that depleting Ago2 reduced the silencing efficacy of agsiRNA significantly more than that of siRNA or rsiRNA, suggesting that agsiRNA mainly functioned through Ago2 RISCs, whereas siRNA or rsiRNA could be loaded into other Ago RISCs to repress their targets (Figure 1f).
The potency of various concentrations of agsiRNA-887, rsiRNA-887, and siRNA-887 was also compared. An antisense reporter assay showed that agsiRNA-887 and siRNA-887 had similar potency, which was higher than that of rsiRNA-887 across all concentrations tested. Sense strand reporter assays showed that both siRNA-887 and rsiRNA-887 maintained strong sense strand activity (almost as potent as the antisense strand), but agsiRNA-887 had almost three orders of magnitude less sense strand activity (Figure 1g,h). These data suggest that the antisense and sense strands of certain di-siRNAs can be nearly equally loaded into mature RISCs, but RISCs mainly selected the antisense strand for sli-siRNAs.
5′ end modification
Phosphorylation of the 5′ end (5′p) increases the potency of di-siRNAs25 and is required for siRNA loading.26 It has also been proposed that 5′p will hold Ago2 in a special conformation.27 However, we did not observe obvious difference in the potency of agsiRNA-887 synthesized with or without the 5′p (Supplementary Figure S7a). This result agrees with the first bases replacement test in pre-miR-451.19 It is possible that similar to di-siRNA, agsiRNA is phosphorylated in vivo by hClp1.28 Accordingly, all agsiRNAs and siRNAs used in the following experiments were synthetic oligonucleotides without 5′p. Since 5′ end bases replacements showed the U-S17-L4-C form performed similarly to the canonical form, but not the C-S17-L4-C form, indicating that an U-S17-L4-C form can be easily expressed as an agshRNA that begin with an A (Supplementary Figure S7b). We also observed extra bases to the 5′end affected sli-siRNA potency. Adding one A, which made the 5′ overhang of the agsiRNA two nt long, slightly increased its potency, but adding two to four As reduced the potency (Supplementary Figure S7c). Northern blot analysis showed that the amount of mature agsiRNA-887 was reduced when extra bases were added to the 5′ end, implying that they are not Ago2 favorite substrates, and it may be difficult to anchor these molecules to the Mid domain of Ago2 or to fit them into the Ago2 substrate groove to trigger Ago2 slicer activity. The lengths of the long fragments and mature products were also increased, suggesting that the Ago2 slicing sites on 3p were not shifted by adding extra bases to the 5′ end and supporting a model that Ago2 slicing sites are defined by the stem region of agsiRNA (Figure 3a).
Figure 3.

Characterization of sli-siRNA-887. (a) Northern blot to detect the processed products from agsiRNA-887 that have extra bases on the 5′ or 3′ end in transfected HEK-293 cells. Ctrl, scrambled agsiRNA. U2 and U6 snoRNAs were used as RNA loading controls. (b) Northern blot to detect the processed products of base and loop modified agsiRNA-887 in transfected HEK-293 cells. U2 snoRNA was used as the RNA loading control. (c) Northern blot to detect the processed products of agshRNA-887 variants expressed by the U6m promoter in transfected HEK-293 cells. U6 snoRNA was used as the RNA loading control. (d) Northern blot to detect the processed products of stem variants of agshRNA-887 in transfected HEK-293 cells. Ctrl, scrambled agshRNA; S17, the wt agshRNA-887; mut, agshRNA-887 noncleavable mutant; U1, S17 driven by a modified U1 promoter; H1, S17 driven by a modified H1 promoter. All other agshRNAs were transcribed from U6m. U2 and U6 snoRNAs were used as RNA loading controls. (e) Northern blot to detect the processed products of stem variants of agsiRNA-887 in transfected HEK-293 cells. Ctrl, agsiRNA with a scrambled RNA sequence; S17, the wt agsiRNA-887; rsi, rsiRNA-887; si, siRNA-887. U2 and U6 snoRNAs were used as RNA loading controls. (f) Reporter assays of HCT-116 cells transfected with the agsiRNA-887 stem variants. Rluc/Fluc ratios are shown. Error bars represent the SD. (g) Northern blot to detect the processed products of agsiRNA-887 that have length variations in transfected HEK-293 cells. Ctrl, agsiRNA with a scrambled sequence; mut, agsiRNA-887-mut. The weak band in L38 was unintentionally caused by using only 1/10 of the molar concentration used for the others for transfection. U2 and U6 snoRNAs were used as RNA loading controls. (h) Reporter assays of HCT-116 cells transfected with agsiRNA-887 that have length variations. Rluc/Fluc ratios are shown. Error bars represent the SD.
3′ end overhangs
The original pre-miR-451 has a 3′ overhang of CUC that arises from Drosha processing. When we designed the agsiRNAs, we assumed the last two bases would not be required for agsiRNA-mediated gene silencing. Firstly, because they would be degraded as part of the 3L12 after Ago2 nicks the substrate and secondly, because experiments with the R2 agshRNAs showed that the UC bases could be replaced by UU. However, these bases could maintain the structure of the substrate to allow efficient Ago2 binding and processing or protect pre-miR-451 from being degraded from the 3′ end. To test the function of different overhangs at the 3′ end of agsiRNA, we replaced these bases with modified bases that were resistant to RNases to prevent agsiRNA degradation from the 3′ end. We created 3′ end variants of agsiRNA-887 by attaching U, UU, or UUUUU, or deoxy T deoxy T (dTdT) to the last C, or by converting the last C to a dideoxy C. Northern blot analysis showed that the UUUUU form produced fewer mature products, indicating increased degradation of this agsiRNA. Both the U and UU mutants produced slightly less product than the canonical form, and the deoxy T deoxy T and dideoxy C forms produced mature products in similar amount to wild type (“wt” in Figure 3a,b). Reporter assays showed that the modified 3′ end bases, which were removed together with the short fragments, had little effect on the silencing potency of the mature agsiRNAs (Supplementary Figure S7c,d). Because the dideoxy C modification can prevent degradation from the 3′ end, it would be a good addition to the design of agsiRNAs to increase their stability in vivo.
Base pairs in the stem region
The stem region can be divided into the seed, central, and 3′ supplementary (3′supp) regions (Figure 1a). Because the central bases are critical for the slicing reaction, we only introduced mismatches, GU wobbles, and bulges into the seed and 3′supp regions to determine their effects on sli-siRNA processing and silencing potency. In contrary to the reported miR-451 processing data that G:G mismatch for p6:35 enhances miR-451 function,19 our northern blot analysis showed that agsiRNA-887 with mmp6 (base #6 mismatched with #35) and mmp7 modifications produced fewer mature products than wild-type agsiRNA-887, and the effects on agshRNA were even stronger (Figure 3b,c). AgshRNA-887-mmp8, -mmp13, -mmp14, -mmp15, -GU-p8, and -bulge-p7 were also processed poorly; the amounts of mature products were dramatically reduced (Figure 3c). While agshRNA-887-mmp13, 14, and 15 data agreed with the published miR-451 mutation data, agshRNA-887-mmp8, -GU-p8, and buldge-p7 showed much stronger effect on the silencing potency and processing efficiency of sli-siRNA-887 than miR-451 mutations at these positions.19 These data indicate that base pair modifications in the stem region affect agshRNA more severely than agsiRNA and also suggest that the seed region has more flexibility for mismatches and wobble base pairs, but bulges are not favored. It is possible that a mismatch (flexible for nt at any position) or a wobble base pair (context dependent, only for “G” or “U”) in the seed bases could help release the 3L10 from the L30 and facilitate the binding of products trimmed from L30 to their targets.19 Despite having the similar knockdown potency at higher concentrations, there was a several fold drop in the activity of the mmp6 and mmp7 mutants at lower concentrations compared to wt (Supplementary Figure S7g). We also revisited the published result that mismatch of p6:35 (G:G) enhances miR-451 potency.19 Our reporter assays using three natural existing miR-451 forms, the human (hmiR-451, G:U wobble pair for p6:p35), mouse (mmiR-451, G:C pair for p6:p35), and zebra fish miR-451 (dmiR-451, G:G mismatch pair for p6:p35), revealed that hmiR-451 was the most potent and dmiR-451 was the least potent in both target cleavage and repression (Supplementary Figures S1a and Figure 4a,b). We also observed that the three forms of pre-miR-451 exist almost equally in nature. There are six G:G, six G:C, and five G:U paired pre-miR-451s in 18 species that was documented in miRBase 19 (Supplementary Figure S1a). Therefore, both our sli-siRNA-887 and pre-miR-45 results argue against the conclusion that the flexible base pairing of p6 with p35 in pre-miR-451s enhanced their potency. But, they support the hypothesis that the flexible base pairing of p6 with p35 in pre-miR-451s may arise from natural selection in balancing short fragment release and mature 5p binding to targets.19 The p6 G of miR-451 may act as a “pivot” residue for target recognition like the p6 C of miR-124.29
Figure 4.
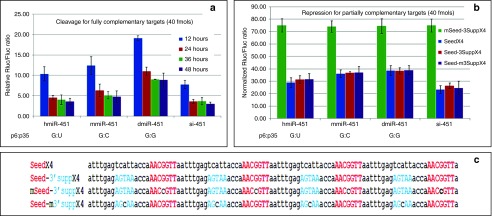
Cleavage of fully complementary targets and the repression of partially complementary targets by agsiRNA-451 or siRNA-451. (a) Time course analyses of target knockdown when the target sequence is a perfect complement. Three miRNA-451 variants (hmiR-451, mmiR-451, and dmiR-451) and siRNA-451 (si-451) were compared. Reporters and siRNAs (80 pmol/l) were cotransfected into HCT-116 cells. Rluc/Fluc ratios are shown. Error bars represent SD. (b) Reporter assays showing repression of partially complementary targets by hmiR-451 and siRNA-451 (80 pmol/l). Rluc/Fluc ratios are bar plotted and grouped by RNAi molecules. Error bars represent SD. (c) Sequence of four types of repression reporters. Each vector carried four copies of the same target sequence in tandem: (i) miR-451 seed sequence (SeedX4); (ii) Seed plus sequence that base paired with the 3′supp region (Seed-3SuppX4); (iii) Seed that had the middle base mutated plus 3Supp (mSeed-3SuppX4); and (iv) Seed plus 3Supp that had the middle base mutated (Seed-m3SuppX4).
Optimal stem length
If one more mismatch bp was added to the 5′/3′ ends of the sli-siRNA hairpin (mmp2, 16 nt stem), the structure behaved like the canonical form. But, if we opened two more bps (mmp2-3, 15 nt stem), the production and function of the mature form were negatively affected (Figure 3b and Supplementary Figure S7f). If we opened the stem from the loop region, which increased the loop size to 6 nt (mmp18, 16 nt stem), 8 nt (mmp17-18, 15 nt stem), or 10 nt (mmp16-17-18, 14 nt stem), only the mmp18 behaved like the canonical form (Figure 3b and Supplementary Figure S7g). Next, we removed or added extra nt at the end of the agshRNA-887 stem (p18), but only the S18 (18 nt stem) was processed like the S17. Both the S19 and S20 forms were processed into less mature products, and the processing generated multiple products or intermediates (Figure 3d). This agrees with the published results that Dicer and Ago2 will compete for processing shRNAs with stems of this range.6,20 For the short-stem variants, only S16 was processed like the canonical S17; the rest produced less mature product for both agshRNA and agsiRNA, and these data correlated well with the reporter assay results (Figure 3d–f). Northern blot data for both agshRNA-887 and agsiRNA-887 showed the most noticeable processing defect that mature products were mostly lost when the stem was shortened from 15 to 14 bases (Figure 3d,e). Therefore, it is likely that Ago2 cannot efficiently process substrates with stems shorter than 15 bases. Similar results were reported in sshRNA study and agsiRNA study.10,21 This result supports the conclusion that the optimal length of the stem or dsRNA needed to fit into the Ago2 groove and trigger the Ago2 slicer activity is ~16 bases and agrees well with the length of the dsRNA region in the molecules of several potent siRNA variants reported.30,31,32
The small loop makes a difference
To test whether the four nt loop is required for silencing, we paired the 5′ and 3′ ends, paired p19 with p22, and replaced p20-p21 with UU to convert the agsiRNA-887 into sshRNA-887.10 Next, we made a no-loop version (NL) of both agshRNA-887 and agsiRNA-887 by directly connecting the first 19 nt of sli-siRNA-887 with its complementary strand. The major mature products from sshRNA-887 was less and shorter than agsiRNA-887, and there are many products longer or shorter than the major band on the blot (Figure 3b). If we consider RRM2 agshRNA-1111 as a special case of sshRNA because its p20-21 bases are UU, our deep sequencing data showed this agshRNA has produced almost equal amount of 5p and 3p products with a clear cleavage by unknown RNase between the UU (Supplementary Table S2), and both 5p and 3p reads from this agshRNA actually are low compared to other seven agshRNAs being sequenced. The processing of NL was affected, and the silencing activity of the NL form was several folds lower than that of the wt (Figure 1g). There were much less mature products for both the agsiRNA and agshRNA versions of NL (Figure 3b,c,e).
We also tested if the sequence context of the loop affected its potency by changing the bases from p18 to p23 (tail bases) in agsiRNA and then tested the effects on gene silencing. We replaced the tail bases of agsiRNA-887, -1148, and -1354 with the tail bases from miR-451 (GAGUUU: LP451), which caused a fivefold reduction in potency for agsiRNA-887, whereas the potency of agsiRNA-1148 increased, and the potency of agsiRNA-1354 showed no difference (Supplementary Figure S8a,c,d). The tail bases of agsiRNA-1148, agsiRNA-1354, and hmiR-451 were then replaced with the tail bases of agsiRNA-887 (GGAUGU: LP887). This change led to a slight increase in potency of agsiRNA-1148, whereas the potency of agsiRNA-1354 was not changed, and the potency of hmiR-451 was reduced about fivefold (Supplementary Figure S8b–d). These data indicate the loop sequence (p18 to p23) may influence the silencing potency of sli-siRNA (Supplementary Figure S8). Therefore, the native sequence of the target should be used for the loop.
Activity of the L30
It has been showed that the L30 of miR-451 is inactive.19 To test the activity of L30 of agsiRNA-887, we removed nt from the 3′ end to generate L39 (39 nt, wt is L40), L38, L37, L35, L30, L29, L27, and L25 forms of agsiRNA-887. Both L39 and L38 behaved like L40. However, the amount of mature processed products from L37 and L35 was dramatically reduced, as was their gene silencing activity in reporter assays (Figure 3g,h). We did not observe any mature products processed from L30 or the other shorter forms, instead unprocessed cross molecule hybridization forms of these molecules were detected on northern blots (Figure 3g). More importantly, when L30 annealed with 3L12, it mimicked the intermediate products that were sliced from an agsiRNA by Ago2 and showed a approximately twofold increase in activity over L30, which was hundreds of folds lower than the activity of L40 (Figure 1g). These data indicate that, unlike di-siRNA, which can use a segmented passenger strand,33 sli-siRNA need the intact hairpin to be potent.19
Target cleavage and repression by sli-siRNAs and di-siRNAs
Next, we compared silencing potency, both target cleavage and repression activities, by the two types of RNAi triggers using reporter assays.
For the target cleavage activity, we cotransfected a reporter carried one copy of the perfectly matched target sequence of miR-451 with hmiR-451, mmiR-451, dmiR-451, or siRNA-451 (si-451: di-siRNA containing L21 of the miR-451 sequence). Time course experiments showed that si-451 silenced the reporter in significantly less time than all the miR-451 genes. Knockdown by si-451 peaked at ~24 hours posttransfection, whereas knockdown by miR-451 peaked at ~36 hours posttransfection. However, similar silencing levels were obtained using hmiR-451 and si-451 36 hours posttransfection (Figure 4a). It may indicate the Ago2 processing step or maturation step slowed down the onset action of sli-siRNA because there are many processing intermediates 1 day after transfection on northern blots (Supplementary Figure S9).
For target repression activity, we created four reporters with miR-451 seed or sequences that base paired with the 3′ supp region, or both as partially complementary targets. Each reporter had four copies of the target sequences in tandem in order to see the cooperative binding effect of multiple RISCs (Figure 4c). We found that although mismatches in the seed were somewhat tolerated in gene silencing mediated by target cleavage, they were not well tolerated by either sli-RISC or di-RISC for translational repression. However, mismatches in the sequences that base paired with the 3′supp region were well tolerated by both sli-RISC and di-RISC. There was no significant difference between the silencing effect on targets that had only the seed or the seed plus sequences that base paired with the 3′supp region. There was a significantly higher repression activity for all three reporters that carried the intact seed by si-451 compared to the three miR-451 species. This difference could be due to the ability of nonslicing Agos to participate in si-451–mediated repression, but not in miR-451–mediated repression, which functions solely through Ago2 (Figure 4a,b).
In vivo expression and potential applications of sli-siRNAs
We first examined the ability of sli-siRNAs to activate the innate immune response and found it was very low, which agrees with the reported results from sshRNA study (Supplementary Figure S10).34 We then put our design parameters into practice to generate sli-siRNAs that would target other endogenous genes. We were able to knockdown the R2 partners R1 and R2B by using sli-siRNAs (Supplementary Figure S11). However, the major concern regarding their usage, especially in vivo, is whether sli-siRNAs would saturate the endogenous miRNA pathways because they require Ago2 for processing and function. This concern is due to the toxicity of traditional shRNAs in that some of them could jeopardize the nuclear export of endogenous miRNAs by Exportin-5 and compete with endogenous miRNAs for Ago proteins.35 We built stable, constitutive, or inducible agshRNA expression systems using lentiviruses (Supplementary Figure S2). Sli-siRNA-1148 was chosen for these experiments because it has a 6 nt loop (mmp18) and two GC pair sites that can be converted into two GU wobble sites to better resemble the canonical pre-miRNA structure to compete with endogenous pre-miRNAs (Supplementary Figures S3 and S4).
First, we transiently transfected these lentiviral constructs into HEK-293 cells to evaluate their expression and processing. Wt, mmp7, and GU transcribed from U6m, and wt transcribed from U6TO (a Doxycycline [Dox]-inducible U6m) were strongly expressed and easily detected on northern blots. Wt and mmp7 had the most mature species, and the GU form had more unprocessed products, probably because of the double G:U bps introduced into the structure. There was no observable difference between mature miR-21 levels in the transfected cells (Supplementary Figure S12a). Next, we constructed lentiviral vectors that contain restriction sites engineered for cloning U6 driven agshRNA expression cassettes (vector). We constructed agshRNA with scrambled sequences as the negative control (ctrl), agshRNA-1148-mutant (mut; nt at p10-12 were swapped with their base pair partners on 3p; it can still be processed by Ago2), -1148 wild-type (wt), -1148-mmp7 (mmp7), and -1148-GUp27p36 (GU; Cs at p27 and p36 both replaced with Us to create wobble bps at these positions) into the lentiviral vectors (Supplementary Figures S3 and S4).
We made cell lines that stably expressed wt, mmp7, GU, or mut of agshRNA-1148. Both R2 protein and mRNA were reduced in the cell lines expressing wt, mmp7, or GU (Figure 5a,b). Northern blot analysis revealed that processed products were present in these cell lines, and there were no observable changes in the levels of either the pre-miRNA-21 or mature miRNAs of miR-21 and miR-31 (Figure 5c, Supplementary Figure S12a). We also measured the levels of miR-21 (high expression), miR-31 (medium expression), and miR-143 (low expression) in the stable cell lines by miRNA qPCR. There were no significant changes in miR-21 or miR-31 levels across all samples. There was some variation in miR-143 levels in the mmp7 and GU samples, but this may be due to technical variations that can occur when using qPCR to quantify miRNAs that have very low expression levels (Supplementary Figure S12b). We also evaluated the inducible expression of wt and mut agshRNA driven by the Dox inducible U6TO promoter. After adding Dox, products processed from the wt and mut agshRNA-1148 were detected on northern blots, and R2 levels were reduced in the cells expressing wt agshRNA-1148. Very low amount of processed product could be detected in wt samples that were not treated with Dox, indicating that the U6TO promoter was slightly leaky. The leakage should not be a concern because it will not produce enough amount of mature agshRNA for effective target knockdown (Figure 5d).
Figure 5.

In vivo knocks down R2 in HCT-116 cells by agshRNA-1148. (a) Western blot analysis of R2 in HCT-116 cell lines constitutively expressing agshRNA-1148 and its variants. Actin was used as loading control. (b) qPCR of R2 mRNAs in HCT-116 cell lines constitutively expressing agshRNA-1148 and its variants. Data was normalized to GAPDH, and then to the vector. Error bars represent SD. (c) Northern blots of the processed products in HCT-116 cell lines constitutively expressing agshRNA-1148 or variants. U2 and U6 snoRNAs were used as RNA loading controls. (d) Northern blots of processed products in HCT-116 cell lines that were induced by Dox to express agshRNA-1148 and corresponding western blots of the R2 protein levels. U6 snoRNA was used as the RNA loading control, and GAPDH was used as the cell extract loading control.
We compared the proliferation rates, invasiveness, and wound-healing abilities of the above stable cell lines. Real-time cell proliferation experiments showed that wt and mmp7 grew much more slowly than the other variants (Supplementary Figure S12c). Matrigel invasion assay showed that cells expressing wt, mmp7, or GU were less invasive than other variants (Supplementary Figure S12d). In addition, wound-healing assays showed that cells expressing wt, mmp7, or GU did not close the wound gaps as quickly as the other variants (Supplementary Figure S12e). Nontransduced cells, cells transduced with vector only, or ctrl RNA had similar proliferation rates, indicating that scrambled agshRNA did not titrate Ago2 protein away to affect cell growth. Therefore, agshRNA have potential for in vivo applications to target genes involved in the pathogenesis of human diseases, such as cancer.
Discussion
We have defined the structure parameters for designing and expressing sli-siRNAs that are as potent as classical di-siRNAs, but have much less sense strand activity, and demonstrated their potential for physiological use in mammalian cells. Sli-siRNAs can be effectively expressed by a modified U6 promoter to mount a potent target knockdown, but not H1 or U1 promoter with similar modification, presumably due to much weaker transcription by H1 or U1 promoter upon modification (Figure 3d, Supplementary Figures S12a and S13). Sli-siRNAs not only have fewer off-target effects by the sense strand but also are easier to design than di-siRNA because they have 5p as antisense strand as default and can avoid the concern of end thermodynamics stability in di-siRNA design.36 Because of similar function mode and molecular structure between sshRNAs and agsiRNAs, the effect of chemical modification on sshRNA should also be applicable to agsiRNA.34 The biogenesis mechanism of sli-siRNAs also assumes that incorporation of the sli-siRNAs into nonslicing Ago RISCs will be limited and will avoid the competition for Dicer with endogenous miRNAs.18,19,21 Therefore, it is possible that sli-siRNAs may also reduce the RNAi off-target effects that are caused by strands being loaded onto nonslicing Agos.37 Nevertheless, agsiRNAs clearly have advantages over di-siRNAs, including being single stranded and self-destroyed passenger strand during maturation, as well as needing only one synthetic setup procedure, one purification procedure, and fewer nt modifications. Therefore, sli-siRNAs are a viable option for developing novel, potent RNAi triggers.
Although sli-siRNAs and di-siRNAs have similar potency in both target cleavage and repression, there are some differences in their functional mechanisms and may deserve further studies. First, di-siRNAs can use any of the Agos, whereas sli-siRNAs only use Ago2. Second, there is an uridylation and 3′ trimming step during sli-siRNA maturation, and it is expected that the rate for this step will be sequence dependent, e.g., uridylating at U is not necessary, and the trimming rate for different nt is not known. It has been shown that GC rich sequences in the trimming region will result in poor potency.19 The maturation step may cause sli-siRNAs to have a slower silencing rate at the onset. Third, di-siRNAs need go through strand selection, passenger strand displacement, and conformational change for guide strand loaded di-RISC to activate RISCs, whereas sli-siRNAs activates RISCs during its maturation step. The sli-siRNA maturation step may also be able to couple with its silencing function.
In summary, because the sli-siRNA molecule itself enables superb antisense strand selection, we strongly believe that sli-siRNA will be a viable option as potent RNAi triggers.
Materials and methods
Antisense sequence selection. The sequences for the L22 forms of sli-siRNAs that targeted the M2 subunit of ribonucleotide reductase (RRM2 or R2) were selected using SiRNA Site Selector (siDuplex), which calculates the theoretical difference in thermodynamic stability of the ends of an siRNA duplex, and the relative accessibility of the target sites for optimal siRNA design (http://infosci.coh.org/HPCDispatcher/Default.aspx).38 The length of the duplex region was changed to 20 nt, and 2 nt from the native sequence were used as the 3′ overhang. The sequences for the L22 forms of the sli-siRNAs that targeted R1 and R2B were selected using the Si-ShRNA Selector set at the default settings, except the length of the duplex was changed to 20 nt. Si-ShRNA Selector uses a different algorithm from siDuplex for selection of antisense strands. It was designed to use the same antisense sequence for both the siRNA and shRNAs and takes GU pairs and accessibility into consideration.39
Cell lines and cell culture. HEK-293 cells, HCT-116 cells, Ago2-knockout mouse embryonic fibroblasts, and Dicer-knockout mouse embryonic fibroblasts were maintained in high glucose (4.5 g/l) Dulbecco's modified Eagle's medium supplemented with 2 mmol/l glutamine, 10% fetal bovine serum, and 2 mmol/l penicillin/streptomycin. Cells were incubated at 37 °C, 5% CO2.
Transfection. For reporter assays, shRNA expression plasmids and reporter constructs were cotransfected into cells by using Lipofectamine 2000 (Life Technologies, Grand Island, NY). For each experiment, at least three independent transfections were performed in duplicate in 24-well plates. Cell were grown to 75 to 85% confluency in 500 µl medium and were transfected with reporter (50 ng), agshRNA, or differing amount of siRNA or agsiRNA (100 ng of U6-agshRNA vector as stuffer DNA, plus 1 µl of 5 µmol/l, 1 µmol/l, 200 nmol/l, 40 nmol/l, 8 nmol/l, 1.6 nmol/l, or 0.32 nmol/l siRNA or agsiRNA, and 1 µl of Lipofectamine 2000).
For RNA isolation and immunoblots, plasmids (4 µg) or 5 µl of 5 µmol/l siRNA or agsiRNA were transfected into cells in six-well plates, using 10 µl of Lipofectamine 2000 or 5 µl RNAiMAX per well. Prior to transfection, cells were grown to 75 to 85% confluency in 2 ml of culture medium.
Dual-luciferase reporter assays. All reporter assays were performed using psiCheck 2.0-based, dual-luciferase reporters from Promega that express both firefly luciferase (Fluc) and Renilla luciferase (Rluc). Reporters carried complementary target sequences that were constructed by inserting annealed oligonucleotides or digested PCR products into the XhoI/SpeI sites of the 3′ UTR of the Rluc gene in psiCheck2.2 vector.40 These reporters were used to quantify gene silencing. Forty-eight hours after transfection, cells were lysed with 100 μl passive lysis buffer and luciferase levels for 20 μl of lysate were determined (Dual-Luciferase reporter assay kit; Promega, Madison, WI). Changes in expression of Rluc (target) were calculated relative to Fluc (internal control) and normalized to the agshRNA expression vector (U6-agshRNA) or scramble agsiRNA control. The normalized relative ratios of Rluc/Fluc were used to measure the efficiency of silencing. Data were averaged from least three independent transfections, and each transfection had at least two replicates. Error bars indicate the SD.
AgshRNA expression vectors. Design of both the constitutive (U6-agshRNA) and inducible (U6TO-agshRNA) expression vectors for agshRNAs was based on a previously reported shRNA expression vector that contains the U6 promoter.41 Constitutive expression was achieved by transducing cells with lentiviral vectors that expressed U6-agshRNA cassettes (Supplementary Figure S2). To create inducible vectors, part of the 3′ end of the U6 promoter sequence was mutated into a TetR binding sequence (U6TO) as previously described41 (Supplementary Figure S2). All shRNAs were cloned by ligating annealed oligonucleotides into BglII and Xho sites.
Lentiviral vector construction. The lentiviral vector pHIV7-EGFP42 was modified by replacing the EGFP expression cassette driven by the CMV promoter with a puromycin (Puro) expression cassette driven by the SSFV promoter to generate SSFVLV-Puro. The lentiviral vector pHIV7-TIG (Tet repressor-IRES-eGFP)41 was modified by replacing the EGFP gene cassette with the Puro gene cassette to generate the CMVLV-TIP (Tet repressor-IRES-Puromycin) vector (Supplementary Figure S2). U6-agshRNA was cloned into SSFVLV-Puro for stable, constitutive agshRNA expression, and U6TO-agshRNA was cloned into CMVLV-TIP for stable integration and inducible agshRNA expression.
Lentiviruses production. Lentiviruses were produced as described.43 Lentiviruses were used to infect HCT-116 cells, and positive clones were screened in media containing 1 ng/ml Puro. Expression of mature processed products was analyzed by northern blot.
RNA isolation and northern blot analysis. RNA isolation, northern blot analysis, and small RNA cloning were carried out as described.44 Briefly, RNA was extracted using Trizol, total RNA (20 µg) was separated on 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis/8% urea gels, and gels were blotted onto positive charged nylon membranes. A DNA oligonucleotide probe complementary to the target RNA sequence was labeled with γ-32P-ATP. The probe was hybridized to the membranes overnight in PerfectHyb Plus hybridization buffer (Sigma-Aldrich, St Louis, MO), after which membranes were washed once in 6× SSPE (NaCl, NaH2PO4-H2O, and EDTA)/0.1% sodium dodecyl sulfate for 10–30 minutes and twice in 6× SSC (NaCl and Na-citrate-2H2O)/0.1% sodium dodecyl sulfate for 10–30 minutes each. U2 or U6 snoRNAs were used as RNA loading controls.
Small RNA deep sequencing. Deep sequencing of small RNAs derived from agshRNA was performed using the HiSeq-2000 platform (Illumina, San Diego, CA). Small RNA library construction and sequence read analyses were conducted as described.24 Briefly, 1.0 µg of total RNA was used to construct small RNA libraries for single reads, flow cell cluster generation and 42 cycle (42-nt) sequencing.
Real-time cell proliferation assay. ACEA Biosciences RT-CES was used to monitor cell growth in real time. This system uses microelectronic cell sensor arrays that are integrated at the bottom of microtiter plates to monitor cell growth by measuring changes in electrode resistance. Measurements were taken every 30 minutes during the 3-day incubation.
Wound-healing assay. Cells were grown in 24-well plates to at least 90% confluency, scratched using pipette tips, washed with PBS, and then cultured in complete medium for about 2 days to allow cells to migrate into the wound areas or until the scratched areas in control cells were filled. Wound areas were photographed before and after the 2-day incubation.
Cell invasion assay. Cell invasion assays were performed with cell invasion chambers (BD Bioscience, Franklin Lakes, NJ), according to the manufacturer's instructions. Infiltrated cells were stained with Diff-Quik Stain Kit (Thermo Fisher Scientific, Waltham, MA) 24 or 48 hours after plating. Three random areas were chosen for analysis; cells that had infiltrated these areas were counted and averaged.
Bioinformatics analysis. RNA and DNA secondary structures were predicted by mFold (http://mfold.rna.albany.edu/?q=mfold),45 the Vienna RNA software package (http://rna.tbi.univie.ac.at),46 and RNAstructure (http://rna.urmc.rochester.edu/RNAstructureWeb/).47 CLUSTALW (http://www.clustal.org/clustal2/) and Jalview (http://www.jalview.org/)48 were used to perform multiple sequence alignments. Three-dimensional RNA structures were predicted using the MC-Fold|MC-Sym pipeline (http://www.major.iric.ca/MC-Pipeline/)49 and RNAcomposer (http://rnacomposer.ibch.poznan.pl/Home).50 3D structures were viewed using PyMOL (The PyMOL Molecular Graphics System, version 1.5.0.4; Schrödinger, Cambridge, MA).
Oligonucleotides. All oligonucleotides were synthesized by Integrated DNA Technologies; sequences are listed in Supplementary Table S1.
Immunoblotting. R2, R1, R2B, GAPDH, and β-actin antibodies were purchased from Santa Cruz Biotechnology. Western blot analyses were performed as previously described.51 Briefly, cells in six-well plates were washed with cold phosphate-buffered saline (2 ml) and lysed in 0.3 ml M-PER mammalian protein extraction reagent (Thermo Fisher Scientific, Waltham, MA). Samples were centrifuged at top speed for 10 minutes, and then supernatants were collected. A protease inhibitor cocktail (Roche, South San Francisco, CA) was added to the supernatants, and the protein concentration of each sample was quantified by Bradford assay (Bio-Rad, protein assay dye). Twenty micrograms of total protein from each sample was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis at 100 V for 2–3 hours, and then electroblotted at 15 V onto Hybond-P PVDF membranes (GE Healthcare, Buckinghamshire, UK) for 30 minutes. The membranes were blocked in TBS-T (0.05% Tween 20) plus 5% milk for at least 1 hour at 4 °C, and then probed with primary antibodies overnight at 4 °C. After washing, the membranes were probed with secondary antibodies for 1 hour at 4 °C and visualized using standard AP detection chemistry (ECL western blotting substrate; Thermo Fisher Scientific).
SUPPLEMENTARY MATERIAL Figure S1. Characterization of pre-miR-451. Figure S2. Schematic representations of agshRNA expression constructs. Figure S3. Secondary structures predicted by mFold for the agshRNAs designed to target R2. Figure S4. AgshRNAs that target R2. Figure S5. Single molecular folding form and cross molecular hybridization form of agsiRNA. Figure S6. Predicted secondary structures of sli-siRNA-887 variants used in this study. Figure S7. Reporter assays of agsiRNA-887 variants. Figure S8. Reporter assay of tail-base replacements in HCT-116 cells. Figure S9. Northern blot analysis to detect products processed from sli-siRNA-887 in HEK-293 cells. Figure S10. Immune response to sli-siRNA-887 in HEK-293 cells. Figure S11. Sli-siRNAs targeting R2 partners. Figure S12. Application of sli-siRNA-1148 in mammalian cells. Figure S13. Comparison of agshRNA-887, -1148 and -1354 expressed by the U6, U1 and H1 promoters. Table S1. Sequences of shRNAs, siRNAs, PCR primers, and probes used for northern blot analyses. Table S2. Small RNA deep sequencing summary for U6-agshRNAs-RRM2.
Acknowledgments
We thank Gregory Hannon for generously providing the mouse Ago2 knockout mouse embryonic fibroblasts, and the Dicer knockout mouse embryonic fibroblasts are generous gift from Alexander Tarakhovsky. We thank Keely Walker and Margaret Morgan for expert editing of the manuscript, and our City if Hope colleagues John Rossi, Mark Boldin, David Ann, and Art Riggs for suggestions, helpful discussion, and sharing of reagents and equipments.
The authors declare no competing financial interests.
G.S. and Y.Y. conceived and designed the experiments. G.S., S.Y.Y., C.W.-Y.Y., M.J.-Y.C., and B.S.H.Y. performed the experiments. G.S. and Y.Y. drafted the manuscript. All authors contributed to the final version of the manuscript.
Supplementary Material
References
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Yi R, Cullen BR. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci USA. 2003;100:9779–9784. doi: 10.1073/pnas.1630797100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus MT, Petersen CP, Haines BB, Chen J, Sharp PA. Gene silencing using micro-RNA designed hairpins. RNA. 2002;8:842–850. doi: 10.1017/s1355838202024032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Gu S, Jin L, Zhang Y, Huang Y, Zhang F, Valdmanis PN.et al. (2012The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo Cell 151900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera-Carrillo E, Harwig A, Liu YP, Berkhout B. Probing the shRNA characteristics that hinder Dicer recognition and consequently allow Ago-mediated processing and AgoshRNA activity. RNA. 2014;20:1410–1418. doi: 10.1261/rna.043950.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre GJ, Yu YH, Lomas M, Fanning GC. The effects of stem length and core placement on shRNA activity. BMC Mol Biol. 2011;12:34. doi: 10.1186/1471-2199-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siolas D, Lerner C, Burchard J, Ge W, Linsley PS, Paddison PJ.et al. (2005Synthetic shRNAs as potent RNAi triggers Nat Biotechnol 23227–231. [DOI] [PubMed] [Google Scholar]
- Ge Q, Ilves H, Dallas A, Kumar P, Shorenstein J, Kazakov SA.et al. (2010Minimal-length short hairpin RNAs: the relationship of structure and RNAi activity RNA 16106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas A, Ilves H, Ge Q, Kumar P, Shorenstein J, Kazakov SA.et al. (2012Right- and left-loop short shRNAs have distinct and unusual mechanisms of gene silencing Nucleic Acids Res 409255–9271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M.et al. (2003Expression profiling reveals off-target gene regulation by RNAi Nat Biotechnol 21635–637. [DOI] [PubMed] [Google Scholar]
- Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- Petri S, Meister G. siRNA design principles and off-target effects. Methods Mol Biol. 2013;986:59–71. doi: 10.1007/978-1-62703-311-4_4. [DOI] [PubMed] [Google Scholar]
- Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465:584–589. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S.et al. (2010A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity Science 3281694–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R.et al. (2010Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis Proc Natl Acad Sci USA 10715163–15168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueck A, Ziegler C, Eichner A, Berezikov E, Meister G. microRNAs associated with the different human Argonaute proteins. Nucleic Acids Res. 2012;40:9850–9862. doi: 10.1093/nar/gks705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Maurin T, Lai EC. Functional parameters of Dicer-independent microRNA biogenesis. RNA. 2012;18:945–957. doi: 10.1261/rna.032938.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YP, Schopman NC, Berkhout B. Dicer-independent processing of short hairpin RNAs. Nucleic Acids Res. 2013;41:3723–3733. doi: 10.1093/nar/gkt036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Zhang J, Wu H. Designing Ago2-specific siRNA/shRNA to avoid competition with endogenous miRNAs. Mol Ther Nucleic Acids. 2014;3:e176. doi: 10.1038/mtna.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank F, Sonenberg N, Nagar B. Structural basis for 5'-nucleotide base-specific recognition of guide RNA by human AGO2. Nature. 2010;465:818–822. doi: 10.1038/nature09039. [DOI] [PubMed] [Google Scholar]
- Elkayam E, Kuhn CD, Tocilj A, Haase AD, Greene EM, Hannon GJ.et al. (2012The structure of human argonaute-2 in complex with miR-20a Cell 150100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, Wu X, Wang J, Li H, Li X, Gao H.et al. (2011A bias-reducing strategy in profiling small RNAs using Solexa RNA 172256–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Patkaniowska A, Urlaub H, Lührmann R, Tuschl T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell. 2002;110:563–574. doi: 10.1016/s0092-8674(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- Ma JB, Yuan YR, Meister G, Pei Y, Tuschl T, Patel DJ. Structural basis for 5'-end-specific recognition of guide RNA by the A. fulgidus Piwi protein. Nature. 2005;434:666–670. doi: 10.1038/nature03514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Shuman S, Schwer B. Human RNA 5'-kinase (hClp1) can function as a tRNA splicing enzyme in vivo. RNA. 2008;14:1737–1745. doi: 10.1261/rna.1142908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Hannon GJ, Darnell RB. An alternative mode of microRNA target recognition. Nat Struct Mol Biol. 2012;19:321–327. doi: 10.1038/nsmb.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Rogoff HA, Li CJ. Asymmetric RNA duplexes mediate RNA interference in mammalian cells. Nat Biotechnol. 2008;26:1379–1382. doi: 10.1038/nbt.1512. [DOI] [PubMed] [Google Scholar]
- Chang CI, Yoo JW, Hong SW, Lee SE, Kang HS, Sun X.et al. (2009Asymmetric shorter-duplex siRNA structures trigger efficient gene silencing with reduced nonspecific effects Mol Ther 17725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CY, Rana TM. Potent RNAi by short RNA triggers. RNA. 2008;14:1714–1719. doi: 10.1261/rna.1161908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramsen JB, Laursen MB, Damgaard CK, Lena SW, Babu BR, Wengel J.et al. (2007Improved silencing properties using small internally segmented interfering RNAs Nucleic Acids Res 355886–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Q, Dallas A, Ilves H, Shorenstein J, Behlke MA, Johnston BH. Effects of chemical modification on the potency, serum stability, and immunostimulatory properties of short shRNAs. RNA. 2010;16:118–130. doi: 10.1261/rna.1901810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Wang L, Lee JS, Schürmann N, Gu S, Börner K.et al. (2010Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver J Clin Invest 1203106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- Petri S, Dueck A, Lehmann G, Putz N, Rüdel S, Kremmer E.et al. (2011Increased siRNA duplex stability correlates with reduced off-target and elevated on-target effects RNA 17737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heale BS, Soifer HS, Bowers C, Rossi JJ. siRNA target site secondary structure predictions using local stable substructures. Nucleic Acids Res. 2005;33:e30. doi: 10.1093/nar/gni026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveeva OV, Kang Y, Spiridonov AN, Saetrom P, Nemtsov VA, Ogurtsov AY.et al. (2010Optimization of duplex stability and terminal asymmetry for shRNA design PLoS One 5e10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, Rossi JJ. Problems associated with reporter assays in RNAi studies. RNA Biol. 2009;6:406–411. doi: 10.4161/rna.6.4.9218. [DOI] [PubMed] [Google Scholar]
- Aagaard L, Amarzguioui M, Sun G, Santos LC, Ehsani A, Prydz H.et al. (2007A facile lentiviral vector system for expression of doxycycline-inducible shRNAs: knockdown of the pre-miRNA processing enzyme Drosha Mol Ther 15938–945. [DOI] [PubMed] [Google Scholar]
- Li MJ, Bauer G, Michienzi A, Yee JK, Lee NS, Kim J.et al. (2003Inhibition of HIV-1 infection by lentiviral vectors expressing Pol III-promoted anti-HIV RNAs Mol Ther 8196–206. [DOI] [PubMed] [Google Scholar]
- Li M, Rossi JJ. Lentiviral vector delivery of siRNA and shRNA encoding genes into cultured and primary hematopoietic cells. Methods Mol Biol. 2008;433:287–299. doi: 10.1007/978-1-59745-237-3_18. [DOI] [PubMed] [Google Scholar]
- Sun G, Yan J, Noltner K, Feng J, Li H, Sarkis DA.et al. (2009SNPs in human miRNA genes affect biogenesis and function RNA 151640–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- Popenda M, Szachniuk M, Antczak M, Purzycka KJ, Lukasiak P, Bartol N.et al. (2012Automated 3D structure composition for large RNAs Nucleic Acids Res 40e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, Li H, Rossi JJ. Sequence context outside the target region influences the effectiveness of miR-223 target sites in the RhoB 3'UTR. Nucleic Acids Res. 2010;38:239–252. doi: 10.1093/nar/gkp870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.