

Abstract
The present study aimed at establishing feasibility of delivering short interfering RNA (siRNA) to target the coagulation cascade in rat and rabbit, two commonly used species for studying thrombosis and hemostasis. siRNAs that produced over 90% mRNA knockdown of rat plasma prekallikrein and rabbit Factor X (FX) were identified from in vitro screens. An ionizable amino lipid based lipid nanoparticle (LNP) formulation for siRNA in vivo delivery was characterized as tolerable and exerting no appreciable effect on coagulability at day 7 postdosing in both species. Both prekallikrein siRNA-LNP and FX siRNA-LNP resulted in dose-dependent and selective knockdown of target gene mRNA in the liver with maximum reduction of over 90% on day 7 following a single dose of siRNA-LNP. Knockdown of plasma prekallikrein was associated with modest clot weight reduction in the rat arteriovenous shunt thrombosis model and no increase in the cuticle bleeding time. Knockdown of FX in the rabbit was accompanied with prolongation in ex vivo clotting times. Results fit the expectations with both targets and demonstrate for the first time, the feasibility of targeting coagulation factors in rat, and, more broadly, targeting a gene of interest in rabbit, via systemic delivery of ionizable LNP formulated siRNA.
Keywords: coagulation, Factor X, lipid nanoparticle, plasma kallikrein, siRNA
Introduction
In recent years, the concept of using RNA interference (RNAi) for gene silencing has emerged as a promising tool for both disease treatment in humans1 and target validation in preclinical species.2,3,4,5 RNAi is an evolutionarily conserved biological process of gene silencing that employs double-stranded (ds) RNA molecules to direct target mRNA cleavage in a sequence-specific manner through the action of Argonaute2 (Ago2) endonuclease.6,7 Various oligonucleotide-based technologies, including short interfering RNA (siRNA), have been developed to successfully produce RNAi on target genes, particularly liver-based targets.2,3,7 While siRNA delivery platforms continue to evolve and be applied to various targets and disease areas, significant opportunities exist in utilizing this technology to aid early target discovery and validation in preclinical studies. siRNAs can be designed to have species-specific or cross-species silencing activity, can be utilized to target a single gene or through siRNA combinations to target multiple genes simultaneously, and can be fine-tuned via dose-titrations. Finally, siRNAs can be designed against targets that may be considered “undruggable” with traditional approaches. Application of siRNA in generating tools and models for thrombotic and hemostatic disorders has only been reported in mice and nonhuman primates so far, with the readouts being limited to coagulation biomarkers.8 Given that the coagulation cascade plays a central role in thrombosis and hemostasis,9,10 and that rat and rabbit are facile and informative preclinical species for thrombosis and hemostasis research,11,12 the present studies aimed at establishing feasibility for siRNA-mediated gene silencing of the liver-originated13 coagulation factors in rat and rabbit, using a previously described ionizable amino lipid-based lipid nanoparticle (LNP) delivery platform.2,3
For studies in rat, plasma kallikrein was selected as the target. In recent years, intrinsic cascade factors (factors IX and XI) and contact factors (factor XII and plasma kallikrein) are gaining attention as novel targets for antithrombotic therapies as they may offer antithrombotic efficacy with less bleeding risk compared to current standard-of-care anticoagulants.14,15,16 While numerous studies have established the pivotal roles of factors IX, XI, and XII in thrombotic processes in various preclinical models, contribution of plasma kallikrein to thrombosis and hemostasis is much less understood and so far could only be gleaned from the plasma kallikrein knockout (KO) mouse,17 the antisense oligo (ASO)-treated mouse,18 and a very small number of congenital deficient humans.19 In our studies with plasma kallikrein in rat, we aimed at examining tolerability, potency, selectivity, as well as exploring effects of plasma kallikrein knockdown on thrombosis in the arteriovenous (AV) shunt model and on hemostasis in the cuticle bleeding model.
For studies in rabbit, given that systemic delivery of siRNA for gene silencing in rabbit has not been reported, and that translational findings in the development of active factor X (FXa) inhibitors and their reversal agents have been generated in rabbit thrombosis and hemostasis models,11,20,21 FX was selected as the target. FX exerts its function in the common pathway of the coagulation cascade10 and inhibition of FXa constitutes a key mechanism for the novel oral anticoagulants that are superior to vitamin K antagonists, which have been the mainstay of oral anticoagulation for many decades.22 Our choice of targeting independent factors in rat and rabbit thus allows us a broader coverage of the coagulation cascade in seeking proof-of-concept for our delivery platform as well as an opportunity to compare to historical results for plasma kallikrein in rodents and for FX in rabbit. Rabbit FX studies focused on tolerability, potency, selectivity, and translational pharmacodynamic markers (ex vivo clotting assays).
Results
Identification of rat Klkb1siRNA leads
A total of 42 siRNAs were designed to target rat Klkb1 mRNA. Primary in vitro screening was performed using a luciferase reporter assay at a final concentration of 10 nmol/l for each siRNA. Twenty of the 42 siRNAs tested showed over 80% target knockdown at 48 hours post-transfection (Supplementary Figure S1). The 12 best performing siRNAs were selected for dose–response experiments to determine IC50 values. In agreement with the primary screen, all 12 oligos demonstrated >85% silencing activity) with subnanomolar calculated IC50 values (Supplementary Table S1). Based on the maximum knockdown and calculated IC50 values, we selected four of the siRNAs (sequences shown in Supplementary Table S2) for larger scale synthesis and formulation into the LNP for in vivo qualification.
The LNP formulation was well tolerated with minimal effect on coagulation in rat
The effects of the LNP formulation on circulating markers for tolerability and coagulability were evaluated in a pilot study with the nontargeting control (nt control) siRNA-LNP (Table 1). Sprague Dawley (SD) rats were dosed with either the vehicle or with siRNA-LNP formulations at 1 mg/kg siRNA dose. Circulating biomarkers were assessed at day 1, 3, and 7 postdosing. 1 mg/kg siRNA dose level was selected based on historical internal data demonstrating that this dose level is generally highly efficacious and well-tolerated (data not shown). In this pilot study, although a small number of serum chemistry parameters (blood urea nitrogen, creatinine, albumin) displayed a statistically significant change compared to vehicle control, the level of the changes was very modest (within 12%) and returned to baseline values on day 7. The liver hepatocellular leakage parameters (aspartate transaminase (AST) and alanine transaminase (ALT)) and a muscle injury parameter (creatine kinase) exhibited no difference from vehicle control at any of the tested time points. Fibrinogen showed a slight to moderate increase at day 1 and day 3 (32 and 52%, respectively) and returned to a level similar to vehicle by day 7. PT and aPTT values were essentially indistinguishable in the LNP and vehicle treated animals, other than a modest (10.7%) prolongation of aPTT at day 1 in the LNP-treated group. These results suggest that the LNP at 1 mg/kg was well tolerated in rats, and did not exert an appreciable effect on the blood coagulation profile with only a minor and reversible aPTT prolongation seen at 24 hours postdose.
Table 1. Circulating markers for liver and renal function and coagulability in rats following lipid nanopartilce dosing.

In vivo study on the potency of Klkb1 siRNAs in rat
Four lead Klkb1 siRNAs were formulated into LNP, evaluated in in vitro dose–response studies in rat hepatocytes to confirm activity (data not shown), and tested in SD rats. The siRNA-LNPs were delivered via a single tail vein (i.v.) injection at 0.5 mg/kg and hepatic Klkb1 mRNA expression levels were examined on days 3 and 7 postdosing (Supplementary Figure S2). All four siRNAs led to significant reductions in liver Klkb1 mRNA levels with maximum silencing (>90%) observed on day 3. Klkb1:(287) and Klkb1:(1461) demonstrated better duration of silencing at day 7 than the other two sequences tested. Levels of hepatic Klkb1 mRNA in the nontargeting siRNA group were not different from the vehicle control at both day 3 and 7 postdosing. Based on in vitro and in vivo activity and potency data, Klkb1:(287) was selected for further studies in rat.
Specificity of the rat Klkb1 lead siRNA
A comprehensive study was conducted to further evaluate the in vivo potency and selectivity of Klkb1:(287) and the effects of plasma kallikrein knockdown on thrombosis and hemostasis in rats. SD rats received a bolus dose of Klkb1:(287) siRNA-LNP at 0.018, 0.055, 0.166, 0.5 mg/kg, or nontargeting (nt) control siRNA-LNP at 0.5 mg/kg via tail vein injection at day 0. Liver samples were collected at the time of animal termination on day 7 for mRNA analyses for Klkb1 and coagulation Factors II, V, VII, VIII, IX, X, XI, XII (Figure 1). Klkb1:(287) treatment resulted in a dose-dependent decrease in liver Klkb1 mRNA level, and ~80% knockdown was observed at the highest siRNA dose level (0.5 mg/kg). None of the remaining factors displayed dose-dependent reduction in the mRNA levels. Factors VII, VIII, and XI showed slight increases (~0.5 ddCt relative to the control) for some of the treatment doses. Lack of dose-dependence and small magnitude of increase (less that 50%) suggest these increases are unlikely to be physiologically relevant.
Figure 1.
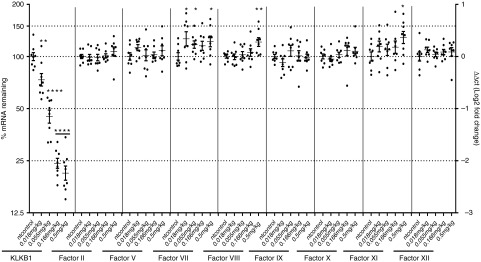
Klkb1 lead short interfering RNA (siRNA) demonstrates in vivo selectivity as well as potency. Sprague Dawley rats received a single administration of Klkb1:(287) siRNA-lipid nanoparticle at the specified doses or nontargeting (nt) control at 0.5 mg/kg (n = 8 per group). Hepatic mRNA levels of Klkb1 and other coagulation factors were determined at day 7. Individual animals and group means with standard deviations are shown. *P < 0.1, **P < 0.01, ****P < 0.0001 compared to nt control.
Effects of plasma kallikrein knockdown in rat thrombosis and bleeding studies
AV shunt thrombosis study and cuticle bleeding study were conducted in the rats at day 7 postdosing in the above-described study. Klkb1:(287) treatment resulted in a dose-dependent reduction in the circulating protein level of plasma prekallikrein, and 92% protein knockdown was achieved with the highest dose (0.5 mg/kg) (Figure 2a). In the ex vivo analysis of the coagulation markers, all four Klkb1:(287)-treated groups trended toward an increase in aPTT, with a statistically significant 16% increase observed for the 0.055 mg/kg group (Figure 2b). Klkb1:(287) at the highest dose (0.5 mg/kg) produced a subtle (6%) but statistically significant increase in PT, compared to nt control (Figure 2c). Klkb1:(287) at 0.018, 0.055, 0.166, and 0.5 mg/kg produced numerical clot weight reductions of 13, 25, 44, and 32%, respectively, compared to nt control, with the clot weight reduction at 0.166 mg/kg reaching statistical significance (Figure 2d). None of the Klkb1:(287)-treated groups produced any statistically significant changes in the cuticle bleeding time compared to nt control (Figure 2e). These results demonstrate that the siRNA treatment in the rats resulted in dose-dependent reduction in liver mRNA for plasma prekallikrein, decreased circulating zymogen for plasma kallikrein, and a modest antithrombotic effect in the AV shunt model with no appreciable increase in cuticle bleeding time. The treatment also produced a modest expected prolongation in aPTT.
Figure 2.
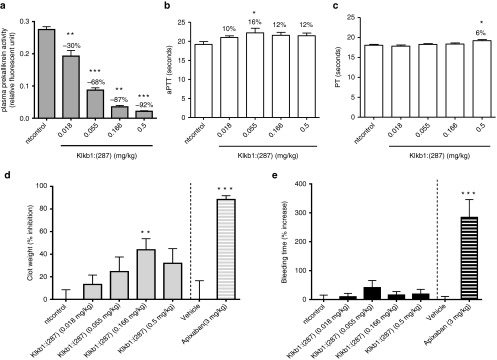
Effects of plasma kallikrein knockdown in rat models of arteriovenous (AV) shunt thrombosis and cuticle bleeding. AV shunt study and cuticle bleeding study were carried out on the rats at day 7 postdosing in the study described in Figure 1. Ex vivo measurements in plasma were also pursued. All results are expressed as mean and standard error of mean. (a) Circulating levels of plasma kallikrein zymogen (plasma prekallikrein) in the various treatment groups at day 7 were determined by the plasma kallikrein enzyme assay as described in Methods. Percent change compared to nt control is noted on the graph. (b) Ex vivo activated partial thromboplastin time (aPTT) at day 7. Percent increase compared to nt control is noted on the graph. (c) Ex vivo prothrombin time (PT) at day 7. Percent increase in the Klkb1:(287) 0.5 mg/kg treatment group compared to nt control is noted on the graph. (d) Percent inhibition of clot weight relative to nt control. Apixaban (3 mg/kg, n = 6 per group) was included as a benchmark. (e) Percent increase in bleeding time relative to nt control. Apixaban (3 mg/kg, n = 6 per group) was included as a benchmark. *P < 0.05, **P < 0.01, ***P < 0.001 compared to nt control for Klkb1:(287) or compared to vehicle control for apixaban.
In view of the modest effects of plasma kallikrein knockdown on hemostasis and thrombosis in our models, we wanted to ensure that the models are sensitive to anticoagulant agents. Apixaban, a new oral anticoagulant therapy targeting FXa,22 was studied in the same models to help illustrate their dynamic range. Apixaban at a clinically relevant dose, 3 mg/kg, produced a more pronounced (88%) clot weight reduction (Figure 2d) and a more robust and significant increase (284%) in cuticle bleeding time (Figure 2e), compared to the vehicle control. Mean ± standard error of mean of aPTT and PT prolongation in the apixaban treated rats were 81 ± 3.1% and 245 ± 7.3%, respectively.
In vitro identification of rabbit FX siRNA leads
To achieve Factor X knockdown experiments in rabbit, siRNA oligo design, in vitro and in vivo studies were carried out in a similar workflow as the rat plasma kallikrein studies. In brief, 42 siRNAs were designed against rabbit FX mRNA, and screened using the luciferase reporter assay (Supplementary Figure S3). The two best performing oligos (FX:(335) and FX:(336), resulting in >80% knockdown, sequences shown in Supplementary Table S3) were selected for scale-up, LNP formulation, and further characterization in vivo.
In vivo characterization of LNP-formulated FX siRNAs in rabbit
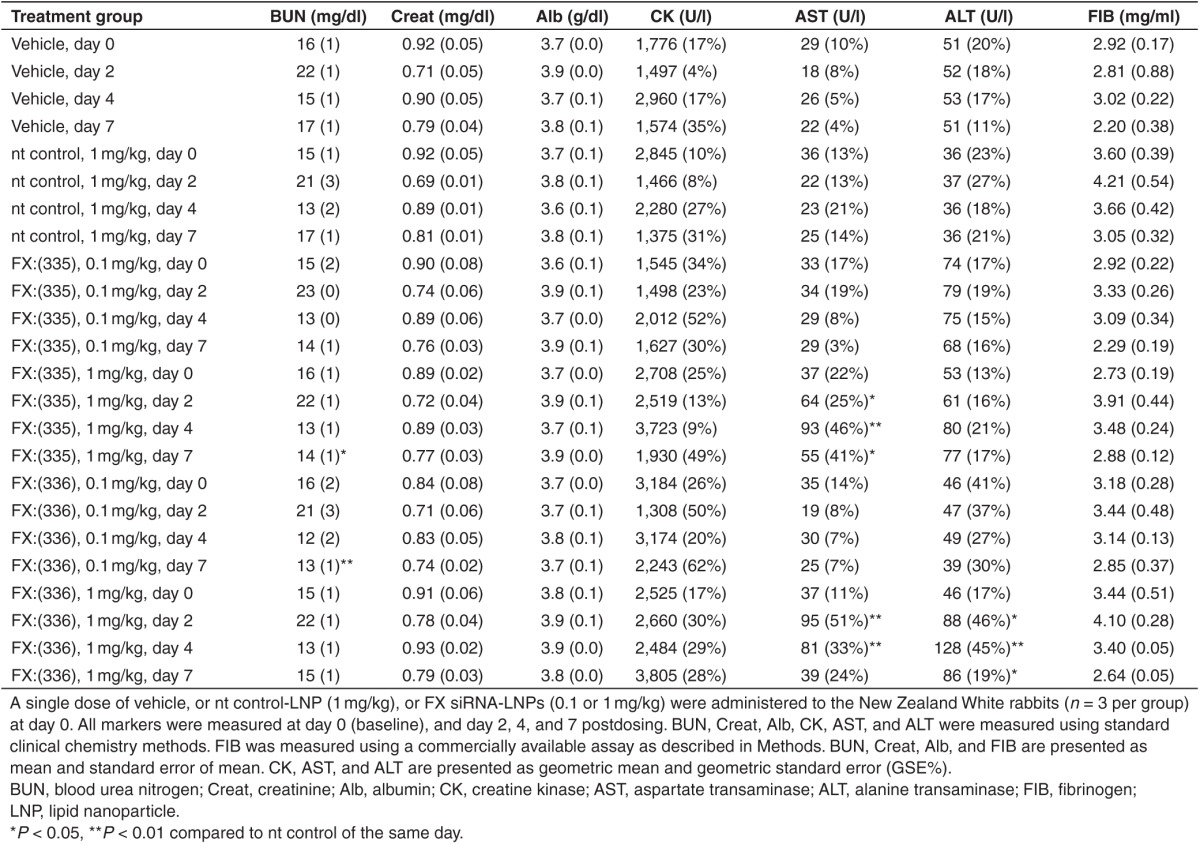
New Zealand White rabbits received a bolus dose of vehicle control, or nt control siRNA-LNP (1 mg/kg siRNA dose), or FX:(335) siRNA-LNP (0.1 or 1 mg/kg siRNA dose), or FX:(336) siRNA-LNP (0.1 or 1 mg/kg siRNA dose) at day 0 via intravenous (i.v.) infusion. Serial bleeds from day 0, 2, and 4 and terminal bleed at day 7 were subjected to various analyses. Table 2 shows the results for circulating markers for safety and tolerability. At all time points tested, for all parameters, nt control displayed no statistically significant difference compared to vehicle, suggesting the 1 mg/kg siRNA-LNP formulation is safe and well tolerated in rabbit. For comparisons between the FX siRNAs and the nt control, while FX:(335) and FX:(336) at low dose (0.1 mg/kg) did not produce any change in serum AST or ALT, FX:(335) at high dose (1 mg/kg) produced two- to fourfold increase in AST at all time points tested postdosing, and FX:(336) at 1 mg/kg produced about fourfold increase in AST at day 2 and 4 postdosing, and two- to fourfold increase in ALT at all time points postdosing. FX:(335) at 1mg/kg and FX:(336) at 0.1 mg/kg produced a statistically significant decrease in blood urea nitrogen, but the magnitude was very small and was not considered to be toxicologically significant. The remaining measured parameters (creatinine, albumin, creatine kinase, and fibrinogen) in the FX siRNA-treated groups did not exhibit any difference from nt control at any time points. FX:(335) and FX:(336) treatment in rabbits with the high dose (1 mg/kg siRNA dose) resulted in slight increases in hepatocellular leakage markers (AST and ALT) that peaked on day 4 and were returning to predose levels on day 7, suggesting a transient hepatocellular injury. Histopathologic evaluation of the liver would be needed to confirm this; however, it was not part of the study design.
Table 2. Circulating markers for liver and renal function in rabbits following dosing with lipid nanopartilce formulated nt control or FX siRNAs.
To examine potency and selectivity of FX:(335) and FX:(336) in vivo, liver specimens were collected at study termination (day 7) for mRNA expression analyses for all coagulation factors (Figure 3). FX:(335) at 0.1 and 1 mg/kg displayed ~40 and 5% mRNA remaining, compared to nt control, respectively, at day 7. FX:(336) at 1 mg/kg displayed ~30% mRNA remaining with no appreciable knockdown observed at 0.1 mg/kg. None of the remaining factors displayed a statistically significant change compared to nt control in their mRNA levels. Results in aggregate suggest that both FX:(335) and FX:(336) delivered selective knockdown on FX with the former being more potent.
Figure 3.
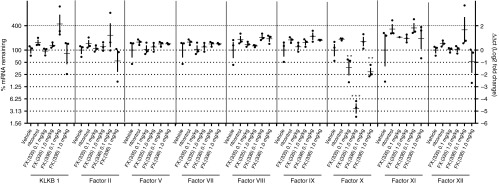
FX lead short interfering RNAs (siRNAs) demonstrate in vivo selectivity as well as potency. New Zealand White rabbits received a single administration of vehicle, or nt control siRNA-LNP (1 mg/kg), or FX:(335) siRNA-LNP or FX:(336) siRNA-LNP (0.1 or 1 mg/kg) at day 0 (n = 3 per group). Hepatic mRNA levels of FX and other coagulation factors were determined at day 7. Individual animals and group means with standard deviations are shown. **P < 0.01, ***P < 0.001 compared to nt control.
Circulating FX zymogen levels in the FX siRNA-treated animals at day 7 were measured using the enzyme assay described in Materials and Methods (Figure 4a). FX:(335) treatment resulted in marked knockdown in the circulating FX protein in a dose- and time-dependent manner, reaching 88, 96, and 98% knockdown at day 2, 4, and 7, respectively, for the 1 mg/kg treatment group. FX:(336) produced discernible level of FX protein knockdown (75, 84, and 77% at day 2, 4, and 7, respectively) only at high dose (1 mg/kg). Ex vivo PT (Figure 4b) and aPTT (Figure 4c) clotting assays were further performed as these are established pharmacodynamic markers for FX loss-of-function.20 FX:(335) produced a dose- and time-dependent increase in PT compared to nt control group, with the 1 mg/kg dose eliciting ~200% PT prolongation at day 7. FX:(336) at 1 mg/kg produced appreciable level of PT prolongation (41 and 50% increase over nt control at day 2 and 4, respectively) (Figure 4b). Effect of the FX siRNAs on aPTT was less pronounced with only FX:(335) at 1 mg/kg producing a significant prolongation (96 and 103% increase over nt control at day 4 and 7, respectively) (Figure 4c). Results demonstrate that the FX siRNA treatment in the rabbits resulted in FX protein knockdown that tracked with mRNA reduction, and the expected hypocoagulability as reflected in ex vivo PT and aPTT prolongations. No difference was observed between the nt control and vehicle-treated groups for FX protein level, aPTT, and PT at any of the time points examined.
Figure 4.

Effects of FX knockdown on circulating levels of FX zymogen and the ex vivo clotting assays. Serial blood sampling at day 0 (baseline), and day 2, 4, and 7 postdosing were performed in the in vivo study described in Figure 3. Circulating levels of FX zymogen (as determined by the FXa enzyme assay) (a), ex vivo prothrombin time (PT) (b), and ex vivo activated partial thromboplastin time (aPTT) (c) in the various treatment groups at all time points were examined. All results are expressed as mean and standard error of mean. *P < 0.05, **P < 0.01, ***P < 0.001 compared to nt control of the respective time point. Percent change from the nt control where there is a statistical significance is noted on the graph.
Discussion
Of the various siRNA in vivo delivery platforms which include LNPs, polymer conjugates, and simple conjugates, the LNP platform is the most advanced and widely used for systemic delivery.7 We and others have demonstrated that siRNA-LNP can be successfully used for target gene silencing across a wide range of targets and in a number of preclinical species.1,2,3,5,23 In this report, we sought to expand our earlier siRNA efforts and develop a siRNA-LNP delivery platform for the coagulation factors in rat and rabbit with proof-of-concept studies on plasma kallikrein and FX respectively.
We have previously shown that ionizable LNPs, similar to the one described here, primarily target liver in the biodistribution studies,24 which renders the LNP platform particularly suitable for the liver-originated coagulation factors. We set out by first assessing effects of LNP formulation on safety parameters and coagulation readouts, given that liver production of hemostatic components can be altered during acute liver response to foreign agents.25 In the pilot study with the nt control siRNA-LNP in rat (Table 1), key liver and renal function markers displayed no difference from vehicle control at all three tested time points (day 1, 3, and 7) postbolus dose of LNP. Additional serum chemistry markers did exhibit a statistically significant difference from vehicle control particularly at the earlier time points (day 1 and/or day 3) but the magnitude of the changes was relatively modest and unlikely to be physiologically relevant. Of note is the significant increase at day 1 and 3 in fibrinogen, which is known to often increase in acute-phase conditions,26 and a subtle prolongation in aPTT at day 1 which subsequently returned to normal levels. In the rabbit study (Table 2 and Figure 4), no difference was observed between LNP-nt control and vehicle-treated groups on any of the liver or renal parameters, fibrinogen, or aPTT and PT at day 2, 4, and 7 postdosing. The data in aggregate suggest that the siRNA- LNP formulation at the selected dose of 1 mg/kg elicited a minor and transient response in the liver but was overall tolerable. We therefore deemed it appropriate to further examine target-specific effects of the siRNA-LNPs on thrombosis and hemostasis at day 7 postdosing.
Consistent with historical siRNA effort for other targets and species, our rat plasma kallikrein siRNA lead and rabbit FX siRNA leads were able to achieve dose-dependent and robust knockdown with long duration. At its highest dose (0.5 mg/kg), Klkb1:(287) resulted in more than 80% (Figure 1) and more than 90% (Figure 2) knockdown of plasma kallikrein mRNA and protein, respectively, at day 7 post-single dose. Likewise, FX:(335) produced >95% mRNA and protein knockdown (Figures 3 and 4) at day 7 post a single dose of 1 mg/kg. These results thus open up the avenue for using siRNAs to study coagulation factors in chronic models and in a titratable fashion in rat and rabbit.
Identification of selective small molecule inhibitors for coagulation factors which belong to the broad class of serine proteases is a well-documented challenge.27,28 Through our established in vitro and in vivo screening methodologies, we were able to identify a lead Klkb1 siRNA and lead FX siRNAs that produced selective knockdown of the target in rat (Figure 1) and rabbit (Figure 3), respectively. We note that even though plasma kallikrein bears high sequence similarity to factor XI (FXI) and the two proteases are believed to result from a gene duplication event from a common ancestor,29 Klkb1:(287) treatment did not result in any reduction in the mRNA levels of FXI. In addition, even though the rabbit siRNAs were selected using a limited set of predicted rabbit transcript sequences (full sequence of the rabbit genome is currently not available), our siRNA design algorithms and screening methodologies enabled us to identify selective FX leads in rabbit. Results thus illustrate the high specificity of the siRNAs as an advantage of the platform and the potential for applying siRNA technology to additional species that are less well characterized.
To study the effect of plasma kallikrein knockdown in thrombosis in rat we selected the AV shunt model, since thrombus formation in this model is a predominantly contact-driven event and plasma kallikrein has not been studied in this model before. Klkb1:(287) produced a dose-dependent clot weight reduction across the range of 0.018–0.166 mg/kg in the rat AV shunt thrombosis model (Figure 2d). At 0.5 mg/kg, protein knockdown was 92%, and clot weight reduction was numerically smaller than though not statistically different from 0.166 mg/kg, suggesting that the effect of plasma kallikrein knockdown in this model may have reached a plateau. Apixaban, a marketed drug targeting FXa in the common pathway, produced marked (>80%) clot weight reduction (Figure 2d). More robust antithrombotic effect has also been observed in our hands in the same model for the FXIIa mechanism.30 We therefore regard the antithrombotic efficacy observed in this model with plasma kallikrein knockdown to be minor. In previously published studies, plasma kallikrein KO mice displayed ~50% clot weight reduction in 3.5% FeCl3-induced venous thrombosis model, and were only partially protected from occlusion in 5% FeCl3-induced arterial thrombosis.17 The modest antithrombotic effects observed with the KO mice are therefore in line with our results.
Klkb1:(287) did not produce any statistically significant changes in the cuticle bleeding times (Figure 2e), consistent with the low-to-no bleeding signal in the plasma kallikrein KO mouse or ASO rat models,17,18,31 and with the absence of bleeding diathesis in plasma kallikrein deficient humans,19 and in contrast with the FXa mechanism (Figure 2e). While these results reinforce the concept that targeting plasma kallikrein will not compromise hemostasis, the modest antithrombotic effect suggests it may not be the most attractive mechanism for developing novel antithrombotic therapies.
To study effect of the FX siRNA leads on coagulation in rabbit, ex vivo aPTT and PT assays were conducted, as it has been established in literature that PT prolongation to a large extent and aPTT prolongation to some extent track with FXa inhibition across multiple species including human and rabbit and can serve as translational markers for the FX mechanism.20,32 In our rabbit FX siRNA studies, consistent with the stack-ranking of mRNA and protein knockdown, FX:(336) at 1 mg/kg overall produced a similar magnitude (and numerically slightly higher level) of PT prolongation compared to FX:(335) at 0.1 mg/kg. FX:(335) at 1 mg/kg produced the most robust PT prolongation with its kinetics tracking well with protein knockdown (Figure 4a,b). Ex vivo aPTT displayed less pronounced prolongation than ex vivo PT, and only the FX:(335) 1 mg/kg group produced statistically significant and time-dependent aPTT prolongation (Figure 4c). Results thus demonstrated that the FX siRNAs produced expected pharmacodynamic effects in rabbit and suggest that our methodologies of mRNA and protein quantification and clotting assays are robust and informative.
It is to be noted that throughout our rabbit study, no spontaneous bleeding or abnormal bleeding was observed in any of the treated animals. It is known that FX KO mice have fatal bleeding33 and congenital deficiency of FX in human is very rare due to the severe defect on hemostasis.34 We thus hypothesize that the residual level of FX in our rabbit study (lowest level remaining was ~2% (Figure 4a)) was likely sufficient in preventing spontaneous or evident bleeding. The titratable knockdown from the siRNA platform thus allows one an opportunity to further study targets for which complete deficiency is incompatible with life. The discernible increase in serum AST and/or ALT associated with high-dose FX siRNA treatment (Table 2) is somewhat surprising and further studies are needed to better assess the reason.
In summary, we set out to utilize LNP-based siRNA platform to target coagulation factors in rat and rabbit. Our results demonstrated that the current LNP formulation is well tolerated in rat and rabbit and does not alter blood coagulation beyond day 3 following single dose administrations. Plasma kallikrein lowering following siRNA-mediated silencing of hepatic mRNA generated modest antithrombotic efficacy in the rat AV shunt model with no bleeding in the rat cuticle bleeding model. FX lowering following siRNA-mediated silencing of hepatic mRNA generated robust PT prolongation and modest aPTT prolongation. Our results are largely consistent with previous findings on both targets, and offer proof-of-concept for using siRNA-LNP platform to target the coagulation cascade in rat and rabbit. Employing this platform to target additional liver-based hemostatic components may generate novel agents and models for study and treatment of thrombotic and hemostatic disorders. As systemic delivery of siRNA in rabbit has not been reported before, our findings mark the first effort of such and pave the way of applying the siRNA technology to rabbit on a broad range of targets and disease biology.
Materials and methods
siRNA oligo design, synthesis and formulation. A total of 42 siRNA oligonucleotides were designed against rat Klkb1 (NM_012725) and rabbit FX (NM_001082016) and synthesized using previously described methods.2,35 To ensure siRNA selectivity and selection of oligos with greatest predicted activity, the oligo design criteria invoked utilization of filters including off-target homology, predicted knockdown activity, exclusion of the repeat sequences, and exclusion of known microRNA sites. The best scored oligos were tested in vitro for target silencing and potency evaluations. The sequences of lead Klkb1siRNAs and nontargeting (nt) siRNA control employed in the rat studies is shown in Supplementary Table S2. The sequences of lead FX siRNAs and nontargeting (nt) siRNA control employed in the rabbit study is shown in Supplementary Table S3. For in vivo studies, siRNAs were formulated into ionizable LNPs, which were prepared via rapid precipitation process as previously described,36,37 In brief, LNPs were assembled by micro-mixing appropriate volumes of an organic solution of lipids with an aqueous solution containing siRNA duplexes. The lipid solution was prepared by dissolving ionizable amino lipid, cholesterol, distearoylphosphatidylcholine, and PEG 2000-DMG, with a molar ratio of 58:30:10:2, in ethanol. Additional detail regarding the chemical structure of ionizable amino lipid, LNP composition, and LNP tolerability are as reported in ref. 37. siRNA duplexes were prepared in an aqueous citrate buffer (pH 5) using a siRNA to cationic lipid molar ratio of 0.17. Reagent solutions were delivered at nearly equal volumetric flow rates to the inlet of a confined volume T-mixer device using syringe pumps (Harvard Apparatus PHD 2000, Holliston, MA). The mixed material was sequentially diluted into equal volumes of citrate buffer (pH 6) and vehicle (phosphate-buffered saline, pH 7.5) in a multistage in-line mixing process. Following buffer dilutions, LNPs were purified to remove free siRNA via anion exchange chromatography. The residual ethanol was removed and the buffer was exchanged into a buffer suitable for cryo-preservation via tangential flow diafiltration. Finally, LNPs were concentrated to the target siRNA concentration, sterilized by filtration via 0.2-μm sterile filter, vialed under aseptic conditions, and frozen for storage at −20 °C. LNP size and size distribution were determined following freeze/thaw by dynamic light scattering using a DynaPro particle sizer (Wyatt Technology, Santa Barbara, CA). For these LNP formulations, the average particle diameter was 80 ± 1 nm with a particle distribution index of 0.15 ± 0.03. The efficiency of siRNA encapsulation in LNPs was determined by the SYBR gold fluorimetric method and was greater than 90% for all preparations.
In vitro siRNA screening. In vitro screening of siRNA oligos was performed using a luciferase-reporter assay in Hepa1-6 cells. All luciferase reporter plasmids used were derived from psiCHECK2 vector (Promega, Madison, WI) through de novo synthesis of the full-length transcript of rat Klkb1 or rabbit FX cloned downstream of Renilla reporter gene in a genetic fusion. Hepa1-6 cells were seeded in 96-well plates at a density of 10,000 cells per well and incubated at 37 °C overnight. For primary screens, siRNAs were transfected at 10 nmol/l final concentration as described before.2 The cells were cotransfected with siRNAs and luciferase reporter plasmids using Lipofectamine 2000 reagent (Life Technologies, Grand Island, NY) on the following day. At 48 hours post-transfection, reporter Renilla luciferase and control firefly luciferase activities were measured using Dual-Glo Luciferase Assay System (Promega) and normalized reporter activity was calculated by dividing Renilla activity by firefly activity of the same sample to normalize for variable transfection efficiency. For 12-point dose–response curve experiments, the siRNA series are three- or fourfold serial dilutions starting at 40 nmol/l, whereas plasmid concentration remained constant.
In vivo rat studies. SD male rats were purchased from Charles River Laboratories (Wilmington, MA). Animals had ad libitum access to food and water. All procedures were approved by the Institutional Animal Care and Research Advisory Committee at Merck.
For the LNP formulation pilot study (n = 8 per group) and the siRNA sequence qualification study (n = 3 per group), SD rats weighing ~150–170 g were utilized. Specified doses of siRNA-LNPs or vehicle control (PBS) were administered via tail vein intravenous (i.v.). Animals were euthanized on day 1, 3, and 7 postdosing (for the formulation study), or day 3 and 7 postdosing (for the sequence qualification study), and blood samples were collected for various analyses. Liver punches were collected for mRNA expression analysis immediately following euthanasia for the sequence qualification study.
For the arteriovenous shunt (AV shunt) and cuticle bleeding studies, specified dose of Klkb1-siRNAs or nontargeting (nt) siRNA control were administered via tail vein (i.v.) to SD rats weighing ~275–300 g (n = 8 per group). The AV shunt thrombosis study and cuticle bleeding study were performed at day 7 post-single dose of siRNA-LNPs based on previously described procedures.30 Briefly, an extracorporeal shunt (Tygon tubing containing a silk thread) was connected to the carotid artery and jugular vein cannulas. The blood was allowed to flow through the shunt for 15 minutes. The thrombus formed on the thread was weighted. Cuticle bleeding time was investigated by cutting a hind paw toe nail and recording the time to cessation of bleeding for up to 10 minutes. Blood and liver samples were collected following these procedures for analyses of ex vivo circulating markers and liver mRNA knockdown, respectively.
For the apixaban study in the SD rats (weighing ~350 g), apixaban (synthesized at Merck, n = 6 per group) or vehicle (35% hydroxypropyl β-cyclodextrin (HPBCD) in 10 mmol/l PBS, pH 7.0, n = 7 per group) was administered via i.v. infusion over 30 minutes at a rate of 3 ml/kg. At 15 minutes of infusion, AV shunt thrombosis procedures and cuticle bleeding procedures were carried out sequentially as described above.
In vivo rabbit studies. Male New Zealand White rabbits (14–15 weeks of age; 1.8–3.5 kg) were purchased from Covance (Denver, PA). Rabbits were allowed to acclimate for 1 week prior to study start and had ad libitum access to water and standard rabbit chow. All procedures were approved by the Institutional Animal Care and Research Advisory Committee at Merck.
Blood samples were collected via the central ear artery on day 0 for baseline measurements and on day 2, 4, and 7 after dosing with the LNPs. One hour after baseline blood samples were collected, rabbits (n = 3 per group) were dosed i.v. via the ear vein with vehicle (10 mmol/l Tris, 70 mmol/l NaCl with 5% sucrose, pH 7.5) or nontargeting control siRNA-LNP (1.0 mg/kg), or FX-siRNA-LNP (0.1 or 1.0 mg/kg) using a 23 g 3/4”needle. Dosing volume per animal was 2.7 ml/kg at a rate of 15 ml/minute. Blood samples were further processed as described below for analyses of ex vivo circulating markers.
Animals were euthanized on day 7 by lethal injection of Ketamine/Sleepaway, after which liver samples were collected for mRNA knockdown analysis.
Analysis of mRNA knockdown from liver samples. RNA isolation from the rat and rabbit liver specimens was performed using the MagMax RNA isolation method (Life Technologies). RT-PCR Taqman data analysis was performed on an ABI 7900 Real-Time PCR System, as described previously.2,3 All reactions were performed in duplicate, and data were analyzed using the ddCt method relative to Gapdh and the nontargeting (nt) control treatment.38 All Taqman probes and primers were purchased from Applied Biosystems (Foster City, CA) as prevalidated sets for rat or custom made sets for rabbit. Sequences of the probes are not shown but available upon request.
Serum or plasma analyses. For determination of circulating biomarkers for liver and kidney function, whole blood was collected into serum collection tubes. Serum samples were subsequently analyzed on a Siemens Advia 1800 analyzer (Siemens Healthcare Diagnostics, Norwood, MA) for: blood urea nitrogen, creatinine (Creat), albumin, creatine kinase, ALT, and AST.
For ex vivo measurement of aPTT, PT, and circulating levels of rat plasma prekallikrein and rabbit FX, whole blood was collected into sodium citrate tubes (final concentration of citrate in blood was 11 mmol/l). Blood samples were then centrifuged at 2,500 g at 4 °C for 15 minutes to generate plasma. aPTT and PT were measured using standard procedure and reagents (TriniCLOT aPTT S (Tcoag, Bray, Ireland) and TriniCLOT PT Excel (Tcoag), respectively) on a KC4 Delta coagulation analyzer (Tcoag).
Circulating levels of rat plasma prekallikrein protein were determined by the plasma kallikrein enzyme activity assay following activation of the zymogen. Briefly, plasma samples were diluted 200-fold in 50 mmol/l Tris, pH 8, 150 mmol/l NaCl, 5 mmol/l CaCl2, 0.1% BSA, then incubated with 5 nmol/l Human FXIIa (Enzyme Research Laboratories, South Bend, IN) at 37 °C for 15 minutes, followed by addition of 1 µmol/l corn trypsin inhibitor (Haemotologic Technologies, Essex Junction, VT) to quench the activation of prekallikrein. The amount of active plasma kallikrein generated was quantified by monitoring its activity (expressed as relative fluorescent unit) on the fluorogenic substrate, HD-VLR-AFC (EMD, San Diego, CA) (Ex = 400 nm, Em = 505 nm) on the SpectraMax Plus (Molecular Devices, Sunnyvale, CA). Nonspecific background activity in the reaction system was negligible (less than 10% of the total activity in a normal rat plasma sample (data not shown)).
Circulating levels of rabbit FX protein were determined by the FXa enzyme activity assay following activation of the zymogen, using the Biophen FX Kit (Aniara, West Chester, OH) following manufacturer's instructions. The method was fully validated for use with rabbit plasma samples using purified rabbit FXa enzyme (data not shown). Briefly, plasma samples were diluted 100-fold with manufacturer-supplied buffer and mixed with Russell Viper Venom (1:1) for 3 minutes at 37 °C, to convert FX zymogen to FXa, followed by addition of 1 volume of substrate (R1). A SpectraMax Plus was used in kinetic mode set at 405 nm to determine the initial velocity of FXa conversion, which is directly proportional to the amount of FXa present in plasma as expressed as arbitrary unit of absorbance.
Rat fibrinogen total antigen was measured on the clinical analyzer described above. Rabbit fibrinogen total antigen was measured with the rabbit fibrinogen ELISA kit from Molecular Innovations (Novi, MI).
Statistical analysis. For the rat plasma kallikrein studies, two-tailed unpaired t-tests were performed for mRNA knockdown, and Mann–Whitney tests were performed for protein knockdown, efficacy and bleeding studies. For the pilot LNP formulation tolerability study, data were log transformed for analysis and presented as geometric means and geometric standard errors (GSE%); for fibrinogen unpooled variance t-tests were performed at each day; the remaining analytes were analyzed by analysis of variance with contrast t-tests performed at each day.
For the rabbit FX study, two-tailed unpaired t-tests were performed for mRNA knockdown at day 7, and two-way analysis of variance longitudinal models were fit to the day 0, 2, 4, and 7 data for PT, aPTT, and the markers for liver and renal functions. To satisfy statistical assumptions, protein knockdown was analyzed with analysis of variance separately by time point and log transforms were applied to the analytes in some cases. Comparisons of group means versus the nt control mean were conducted at each time point across the post-treatment time points. Simulation-based multiplicity adjustments were made to each set of comparisons for each analyte in the longitudinal analyses.
SUPPLEMENTARY MATERIAL Figure S1. In vitro Klkb1 siRNA selection. Figure S2. In vivo sequence selection study for Klkb1. Figure S3. In vitro FX siRNA selection. Table S1. Summary of in vitro screening results for the twelve most potent Klkb1 siRNAs. Table S2. Sequences of lead Klkb1 and non-targeting (nt) control siRNAs used for in vitro and in vivo experiments. Table S3. Sequences of lead FX and non-targeting (nt) control siRNAs used for in vitro and in vivo experiments.
Acknowledgments
We are grateful to Thomas F. Vogt, Martin L. Ogletree, and Patrick Andre for helpful discussions and Jeremy Caldwell for support of the work. We thank Duncan Brown for oligo design and Merck Oligo Chemistry team for synthesizing siRNA oligos. We thank Eileen Walsh, Valerie Kuzmick-Graufelds, Tayeba Khan, and Joe Davide for help with siRNA in vivo qualification studies, and Lan Ge, KehDih Lai, and Marina Ichetovkin for assistance with serum or plasma analyses. We also thank Mike Topper and Amin Rupesh for insights around siRNA-LNP tolerability findings. For declaration of conflicting interests, all authors are or were employees at Merck and may hold stock or stock options of Merck & Co. Z.C. and M.T-S. designed the overall studies, analyzed the data, and wrote the manuscript. L.L. designed and B.L., and T.L. performed the rabbit in vivo study. T.C. designed the rat in vivo studies. W.S. led the in vitro siRNA screening and lead identification. A.T., J.D., M.D., J.D., B.N., and K.L. performed experiments related to the production and in vitro characterization of siRNA oligos. Y.X., W.W., and C.J. developed methods and performed experiments related to rat plasma kallikrein and rabbit FX protein quantitation and activated partial thromboplastin time and prothrombin time measurements. N.J., L.H., and Y.Z. performed the rat in vivo studies. P.S. performed the clinical chemistry analysis. S.C. performed statistical analysis. M.G. led siRNA-LNP formulation production. B.L., T.C., Y.X., S.C., M.G., and L.L. participated in data analysis and contributed to the writing and proofreading of the manuscript. L.S-L. and D.S. coconceived the studies, proofread, and provided critical comments on the manuscript.
Supplementary Material
In vitro Klkb1 siRNA selection.
In vivo sequence selection study for Klkb1.
In vitro FX siRNA selection.
References
- Bakhtiyari S, Haghani K, Basati G, Karimfar MH. siRNA therapeutics in the treatment of diseases. Ther Deliv. 2013;4:45–57. doi: 10.4155/tde.12.136. [DOI] [PubMed] [Google Scholar]
- Tadin-Strapps M, Peterson LB, Cumiskey AM, Rosa RL, Mendoza VH, Castro-Perez J.et al. (2011siRNA-induced liver ApoB knockdown lowers serum LDL-cholesterol in a mouse model with human-like serum lipids J Lipid Res 521084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olearczyk J, Gao S, Eybye M, Yendluri S, Andrews L, Bartz S.et al. (2014Targeting of hepatic angiotensinogen using chemically modified siRNAs results in significant and sustained blood pressure lowering in a rat model of hypertension Hypertens Res 37405–412. [DOI] [PubMed] [Google Scholar]
- Safdar H, Cheung KL, Salvatori D, Versteeg HH, Laghmani el H, Wagenaar GT.et al. (2013Acute and severe coagulopathy in adult mice following silencing of hepatic antithrombin and protein C production Blood 1214413–4416. [DOI] [PubMed] [Google Scholar]
- Tep S, Mihaila R, Freeman A, Pickering V, Huynh F, Huyhn F.et al. (2012Rescue of Mtp siRNA-induced hepatic steatosis by DGAT2 siRNA silencing J Lipid Res 53859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomari Y, Zamore PD. Perspective: machines for RNAi. Genes Dev. 2005;19:517–529. doi: 10.1101/gad.1284105. [DOI] [PubMed] [Google Scholar]
- Sepp-Lorenzino L, Ruddy M. Challenges and opportunities for local and systemic delivery of siRNA and antisense oligonucleotides. Clin Pharmacol Ther. 2008;84:628–632. doi: 10.1038/clpt.2008.174. [DOI] [PubMed] [Google Scholar]
- Wooddell CI, Rozema DB, Hossbach M, John M, Hamilton HL, Chu Q.et al. (2013Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection Mol Ther 21973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furie B, Furie BC. The molecular basis of blood coagulation. Cell. 1988;53:505–518. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- Chen Z, Seiffert D, Hawes B. Inhibition of Factor XI activity as a promising antithrombotic strategy. Drug Discov Today. 2014;19:1435–1439. doi: 10.1016/j.drudis.2014.04.018. [DOI] [PubMed] [Google Scholar]
- Wong PC, Crain EJ, Watson CA, Xin B. Favorable therapeutic index of the direct factor Xa inhibitors, apixaban and rivaroxaban, compared with the thrombin inhibitor dabigatran in rabbits. J Thromb Haemost. 2009;7:1313–1320. doi: 10.1111/j.1538-7836.2009.03503.x. [DOI] [PubMed] [Google Scholar]
- Vilahur G, Padro T, Badimon L. Atherosclerosis and thrombosis: insights from large animal models. J Biomed Biotechnol. 2011;2011:907575. doi: 10.1155/2011/907575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neth P, Arnhold M, Nitschko H, Fink E. The mRNAs of prekallikrein, factors XI and XII, and kininogen, components of the contact phase cascade are differentially expressed in multiple non-hepatic human tissues. Thromb Haemost. 2001;85:1043–1047. [PubMed] [Google Scholar]
- Renné T, Gruber A. Plasma kallikrein: novel functions for an old protease. Thromb Haemost. 2012;107:1012–1013. doi: 10.1160/TH12-04-0264. [DOI] [PubMed] [Google Scholar]
- Gruber A. The role of the contact pathway in thrombus propagation. Thromb Res. 2014;133 suppl 1:S45–S47. doi: 10.1016/j.thromres.2014.03.019. [DOI] [PubMed] [Google Scholar]
- van Montfoort ML, Meijers JC. Anticoagulation beyond direct thrombin and factor Xa inhibitors: indications for targeting the intrinsic pathway. Thromb Haemost. 2013;110:223–232. doi: 10.1160/TH12-11-0803. [DOI] [PubMed] [Google Scholar]
- Bird JE, Smith PL, Wang X, Schumacher WA, Barbera F, Revelli JP.et al. (2012Effects of plasma kallikrein deficiency on haemostasis and thrombosis in mice: murine ortholog of the Fletcher trait Thromb Haemost 1071141–1150. [DOI] [PubMed] [Google Scholar]
- Revenko AS, Gao D, Crosby JR, Bhattacharjee G, Zhao C, May C.et al. (2011Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding Blood 1185302–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girolami A, Scarparo P, Candeo N, Lombardi AM. Congenital prekallikrein deficiency. Expert Rev Hematol. 2010;3:685–695. doi: 10.1586/ehm.10.69. [DOI] [PubMed] [Google Scholar]
- Wong PC, Crain EJ, Xin B, Wexler RR, Lam PY, Pinto DJ.et al. (2008Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies J Thromb Haemost 6820–829. [DOI] [PubMed] [Google Scholar]
- Lu G, DeGuzman FR, Hollenbach SJ, Karbarz MJ, Abe K, Lee G.et al. (2013A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa Nat Med 19446–451. [DOI] [PubMed] [Google Scholar]
- Yeh CH, Fredenburgh JC, Weitz JI. Oral direct factor Xa inhibitors. Circ Res. 2012;111:1069–1078. doi: 10.1161/CIRCRESAHA.112.276741. [DOI] [PubMed] [Google Scholar]
- Novobrantseva TI, Borodovsky A, Wong J, Klebanov B, Zafari M, Yucius K.et al. (2012Systemic RNAi-mediated Gene Silencing in Nonhuman Primate and Rodent Myeloid Cells Mol Ther Nucleic Acids 1e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B, Keough E, Matter A, Leander K, Young S, Carlini E.et al. (2011Biodistribution of small interfering RNA at the organ and cellular levels after lipid nanoparticle-mediated delivery J Histochem Cytochem 59727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannen BH, Robotham JL. The acute-phase response. New Horiz. 1995;3:183–197. [PubMed] [Google Scholar]
- Giovannini I, Chiarla C, Giuliante F, Vellone M, Nuzzo G. Modulation of plasma fibrinogen levels in acute-phase response after hepatectomy. Clin Chem Lab Med. 2004;42:261–265. doi: 10.1515/CCLM.2004.048. [DOI] [PubMed] [Google Scholar]
- Walker B, Lynas JF. Strategies for the inhibition of serine proteases. Cell Mol Life Sci. 2001;58:596–624. doi: 10.1007/PL00000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry BL, Desai UR. Discovery methodology for the development of direct factor VIIa inhibitors. Expert Opin Drug Discov. 2014;9:859–872. doi: 10.1517/17460441.2014.923398. [DOI] [PubMed] [Google Scholar]
- Beaubien G, Rosinski-Chupin I, Mattei MG, Mbikay M, Chrétien M, Seidah NG. Gene structure and chromosomal localization of plasma kallikrein. Biochemistry. 1991;30:1628–1635. doi: 10.1021/bi00220a027. [DOI] [PubMed] [Google Scholar]
- Xu Y, Cai TQ, Castriota G, Zhou Y, Hoos L, Jochnowitz N.et al. (2014Factor XIIa inhibition by Infestin-4: in vitro mode of action and in vivo antithrombotic benefit Thromb Haemost 111694–704. [DOI] [PubMed] [Google Scholar]
- Liu J, Gao BB, Clermont AC, Blair P, Chilcote TJ, Sinha S.et al. (2011Hyperglycemia-induced cerebral hematoma expansion is mediated by plasma kallikrein Nat Med 17206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost C, Wang J, Nepal S, Schuster A, Barrett YC, Mosqueda-Garcia R.et al. (2013Apixaban, an oral, direct factor Xa inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects Br J Clin Pharmacol 75476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewerchin M, Liang Z, Moons L, Carmeliet P, Castellino FJ, Collen D.et al. (2000Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice Thromb Haemost 83185–190. [PubMed] [Google Scholar]
- Lee G, Duan-Porter W, Metjian AD. Acquired, non-amyloid related factor X deficiency: review of the literature. Haemophilia. 2012;18:655–663. doi: 10.1111/j.1365-2516.2012.02773.x. [DOI] [PubMed] [Google Scholar]
- Strapps WR, Pickering V, Muiru GT, Rice J, Orsborn S, Polisky BA.et al. (2010The siRNA sequence and guide strand overhangs are determinants of in vivo duration of silencing Nucleic Acids Res 384788–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gindy ME, Leone AM, Cunningham JJ. Challenges in the pharmaceutical development of lipid-based short interfering ribonucleic acid therapeutics. Expert Opin Drug Deliv. 2012;9:171–182. doi: 10.1517/17425247.2012.642363. [DOI] [PubMed] [Google Scholar]
- Gindy ME, Feuston B, Glass A, Arrington L, Haas RM, Schariter J.et al. (2014Stabilization of Ostwald ripening in low molecular weight amino lipid nanoparticles for systemic delivery of siRNA therapeutics Mol Pharm 114143–4153. [DOI] [PubMed] [Google Scholar]
- Ariani F, Mari F, Pescucci C, Longo I, Bruttini M, Meloni I.et al. (2004Real-time quantitative PCR as a routine method for screening large rearrangements in Rett syndrome: Report of one case of MECP2 deletion and one case of MECP2 duplication Hum Mutat 24172–177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In vitro Klkb1 siRNA selection.
In vivo sequence selection study for Klkb1.
In vitro FX siRNA selection.