

Abstract
Antisense-mediated exon skipping, which can restore the reading frame, is a most promising therapeutic approach for Duchenne muscular dystrophy. Remaining challenges include the limited applicability to patients and unclear function of truncated dystrophin proteins. Multiexon skipping targeting exons 45–55 at the mutation hotspot of the dystrophin gene could overcome both of these challenges. Previously, we described the feasibility of exons 45–55 skipping with a cocktail of Vivo-Morpholinos in vivo; however, the long-term efficacy and safety of Vivo-Morpholinos remains to be determined. In this study, we examined the efficacy and toxicity of exons 45–55 skipping by intravenous injections of 6 mg/kg 10-Vivo-Morpholino cocktail (0.6 mg/kg each vPMO) every 2 weeks for 18 weeks to dystrophic exon-52 knockout (mdx52) mice. Systemic skipping of the entire exons 45–55 region was induced, and the Western blot analysis exhibited the restoration of 5–27% of normal levels of dystrophin protein in skeletal muscles, accompanied by improvements in histopathology and muscle strength. No obvious immune response and renal and hepatic toxicity were detected at the end-point of the treatment. We demonstrate our new regimen with the 10-Vivo-Morpholino cocktail is effective and safe for long-term repeated systemic administration in the dystrophic mouse model.
Keywords: antisense therapeutics, Duchenne muscular dystrophy, dystrophin, mdx52 mouse, multiexon skipping, Vivo-Morpholinos
Introduction
Duchenne muscular dystrophy (DMD) is the most common form of muscular dystrophy, a heterogeneous group of more than 30 genetic disorders characterized by progressive muscle degeneration and weakness.1 DMD with severe muscle pathology is a fatal X-linked disorder that affects ~1 in every 3,600 live male births.2,3 Although DMD is caused by various types of mutations in the dystrophin (DMD) gene, such as deletion, duplication, and nonsense mutations, most patients with DMD have out-of-frame deletion mutations (~65%).4,5,6 Becker muscular dystrophy (BMD), a milder form of dystrophin deficiency,7 mostly results from deletion mutations (~82% of patients) that do not affect the reading frame (in-frame mutations).4,5,6 The resulting in-frame transcripts permit the expression of internally shortened but partially functional proteins.8
Currently, antisense oligonucleotide (AO)–mediated exon skipping, which can restore the disrupted reading frame by excluding the targeted exon(s), is one of the most promising approaches for the treatment of DMD.9,10,11,12,13 The aim of exon-skipping therapy for DMD is to slow the disease progression by converting severe DMD symptoms to the milder symptoms seen in patients with BMD. One major challenge to this technique is the fact that individual exons in the DMD gene need to be targeted by specifically tailored AOs to cover the majority of patients; thus, the therapy may have limited applicability.14,15,16 Another challenge is that the function and stability of each of the resulting short dystrophin proteins are unclear.17 To overcome these issues, many investigations of multiexon skipping have been conducted in preclinical trials with dystrophic animal models and DMD patient cell lines.18 In particular, multiexon skipping that targets exons 45–55 at the mutation hotspot of the DMD gene has the potential to greatly expand the applicability of exon skipping therapy for DMD patients and produce more stable/functional truncated dystrophin protein. An in-frame mutation that lacks the entire exons 45–55 region has been associated with a milder or almost asymptomatic phenotype in 95% or more of BMD patients with this type of mutation.19,20 This observation prompted us to investigate multiexon skipping of this entire region. In theory, ~63% of DMD patients with deletion mutations and 45% of all DMD patients could be rescued by skipping the exon 45–55 region according to the Universal Mutation Database (UMD)-DMD database.19
Typically, skipping multiple exons is more technically challenging and less efficient than targeting single exons. A new-generation morpholino, the Vivo-Morpholino (vPMO), which possesses a cell-penetrating octaguanidinium dendrimer, has been reported to improve the efficiency of exon skipping.21,22,23,24,25 Recently, we described a 10-vPMO cocktail (12 mg/kg every 2 weeks) that efficiently induces multiple skipping of exons 45–55 both in vitro and in vivo in dystrophic exon-52 knockout (mdx52) mice.26,27 Long-term efficacy and safety of the vPMO cocktail, including recovery of dystrophin-associated proteins (DAPs) and immune response, remain to be determined in the systemic treatment. Patients treated systemically with AO-mediated exon skipping therapy will require repeated administration of AO drugs throughout their lifetime in order to maintain the therapeutic effects. Lower concentrations of AOs that achieve sufficient exon skipping may prevent potential off-target effects associated with long-term systemic treatment.
In this study, we examined the long-term efficacy and safety of multiple exon skipping with a cocktail of vPMOs that target exons 45–51 and 53–55. We demonstrate that nine systemic injections of the cocktail every 2 weeks at a relatively low dose of 6 mg/kg induced bodywide expression of dystrophin and DAPs in the dystrophic skeletal muscles of mdx52 mice, accompanied by an improvement in pathology, functional recovery, and no detectable immune response and renal and hepatic toxicity by blood tests.
Results
Local injection with the 10-vPMO cocktail at 0.3 μg skips the entire exons 45–55 of dystrophin transcript in the tibialis anterior muscle of mdx52 mice
To validate the efficacy of exon 45–55 skipping with the vPMO cocktail at a lower dose (a total of 0.3 μg of vPMOs, 0.03 μg of each) than the previous 1.5 μg,26 we first injected the cocktail into the tibialis anterior (TA) muscles of 8-week-old mdx52 mice (Supplementary Figure S1). AO sequences used are listed in Table 1. An intended skipped mRNA band of 243 bp was detected by reverse transcriptase-PCR (RT-PCR) in the TA muscles 2 weeks after an intramuscular injection with the vPMO cocktail. Sequence analysis showed the boundary of exons 44 and 56, confirming that the entire exons 45–55 region had been removed. In addition to the intended skipped mRNA, intermediate dystrophin mRNAs in which some of the exons were removed were also observed in the nontreated and vPMO-treated samples. These intermediate transcripts were composed of both out-of-frame and in-frame transcripts. In immunohistochemistry with anti-dystrophin DYS1 and P7 antibodies, using a lower dose (0.3 μg), we induced more than 50% of dystrophin positive fibers in treated TA muscle, as well as our previous report with a vPMO cocktail at 1.5 μg.
Table 1. Sequences of antisense oligonucleotides used for a 10-vPMO cocktail to skip exons 45–55.

Long-term systemic and repeated injections of the 10-vPMO cocktail induce exon 45–55 skipping and expression of dystrophin and its associated proteins in bodywide skeletal muscles
Next, we examined the exon skipping efficacy of systemic injections with the vPMO cocktail. We performed nine consecutive intravenous injections of the vPMO cocktail, at 6 mg/kg total per injection (0.6 mg/kg for each vPMO) at 2-week intervals, into 8-week-old mdx52 mice. The experimental term and injection frequency in the systemic treatment were almost twice as much as our preceding study, while the total dosage of 54 mg/kg remained similar to the previously tested dosage.26 Treated mice did not show any abnormal behavior after the intravenous injections. RT-PCR analysis showed exons 45–55 skipped transcripts averaged 3.5–22.7% in muscles bodywide, including the heart, at 2 weeks after the final injection (Figure 1a). The exon 44 and 56 junction of the intended band was then confirmed by direct sequencing (data not shown). Various intermediate PCR products including out-of-frame and in-frame sequences were observed in different patterns between tissue types and/or individuals after the systemic treatment as well as the intramuscular injection. We also detected dystrophin-positive fibers in all of the skeletal muscles examined by immunohistochemistry with P7 primary antibody (Figure 1b). Western blotting revealed that dystrophin expression levels in all of the tested skeletal muscles averaged 5–27% compared to normal levels in samples from wild-type (WT) mice (Figure 1c). Expression levels of induced dystrophin proteins as well as the expression pattern of the skipped mRNAs varied among different positions within a given muscle sample and among different muscle types. Unlike in the skeletal muscles, immunohistochemistry and Western blotting, respectively, revealed that there were fewer dystrophin-positive fibers and less expression of dystrophin protein (≤2.3% that of WT mice) in the heart muscle after long-term systemic vPMO treatment (Figure 1b,c). In immunohistochemistry with serial sections from biceps femoris (BF) and gastrocnemius (GC) muscles of the treated mice, we observed recovery of DAPs: α1-syntrophin, neuronal nitric oxide synthase (nNOS), α-sarcoglycan, and β-dystroglycan in the dystrophin positive fibers 2 weeks after the last administration with the vPMO cocktail (Figure 2).
Figure 1.

Long-term systemic intravenous injections of the 10-vPMO cocktail. (a) Dystrophin mRNAs with exons 45–55 skipped (detected by RT-PCR) in the various muscle types 2 weeks after the last of nine systemic intravenous (i.v.) injections of 6 mg/kg 10-vPMO cocktail (0.6 mg/kg each vPMO) at 2-week intervals. (b) Immunohistochemistry with P7 antibody against dystrophin (red) in mdx52 mice after the treatment. Representative data are shown. Scale bar, 100 μm. (c) Western blotting analysis with mouse monoclonal DYS1 antibody after repeated vPMO systemic injections into mdx52 mice. Truncated dystrophin bands at ~380 kDa (upper panel) are shown in various muscles from the vPMO cocktail–treated mdx52 mice. Positive controls, 10 and 1% protein (weight/weight percentage) of the TA muscle from WT mice; negative control, TA muscle of NT mice. Myosin heavy chain (MyHC) is shown by Coomassie Brilliant Blue staining as a loading control (lower panel). Representative data from six treated mice are shown. BB, biceps brachii; BF, biceps femoris; DIA, diaphragm; GC, gastrocnemius; HRT, heart; M, 100 bp marker; NT, nontreated mdx52-TA muscle; TA, tibialis anterior; TB, triceps brachii; QUA, quadriceps; WT, wild-type C57BL/6J.
Figure 2.

Immunohistochemistry of dystrophin-associated proteins accompanied after 9 times i.v. injections of the 10-vPMO cocktail. Dystrophin, α1-syntrophin, nNOS, α-sarcoglycan, and β-dystroglycan were stained on serial sections of the muscles form nontreated and treated mdx52 mice. Biceps femoris muscles were shown as representative data in 3 treated mice. Asterisks indicate the same muscle fiber between the images. Scale bar, 100 μm.
Systemic long-term treatment using the vPMO cocktail improves histology and muscle function in mdx52 mice without inducing detectable immunoreaction
Compared to nontreated muscles, we observed less muscle degeneration and fewer cellular infiltrations in the diaphragm (DIA), BF, quadriceps (QUA), GC, and TA muscles after long-term systemic vPMO cocktail treatment (Figure 3a). We next evaluated detailed histological changes in the muscles of treated mice compared with those in nontreated mice. The percentage of centrally nucleated fibers (CNFs), which is indicative of degeneration/regeneration cycles, was significantly reduced in the DIA, BF, QUA, GC, and TA muscle of the treated mdx52 mice compared to the nontreated mice, indicating an amelioration of muscle pathology (Figure 3b). To examine immunoglobulin (Ig) accumulation in dystrophic muscle fibers, which is a hallmark of necrotic fibers due to leaky muscle membranes in DMD patients and mdx mice,28 immunostaining with IgG antibody was performed (Figure 3c). Increased IgG signals were observed in endo- and perimysium of DIA, BF, QUA, and HRT of the nontreated and treated mice. The signal intensity and the number of IgG-positive fibers, although not statistically significant (P = 0.32 in BF and P = 0.39 in QUA, n = 4 in NT and n = 6 in treated group), were reduced in the treated mice group, indicating improved muscle pathology by the systemic treatment with the vPMO cocktail. Significant improvement in maximum hindlimb grip force was observed in treated mdx52 mice compared to nontreated mdx52 mice (Figure 3d), although the improvement in the forelimb was not significant (data not shown). To investigate an immune response to long-term administration of the vPMO cocktail, we observed changes in the number of CD3-positive T cells (Figure 4a). Although an increase of CD3-positive cells was found in DIA, BF, QUA, and HRT in both nontreated and treated mice, there was no significant difference in the number of CD3-positive cells between the treated and nontreated mice (Figure 4b).
Figure 3.

Histopathology and grip strength after systemic injections of the 10-vPMO cocktail. (a) Hematoxylin and eosin staining in the diaphragm (DIA), biceps femoris (BF), quadriceps (QUA), gastrocnemius (GC), and tibialis anterior (TA) 2 weeks after the last of nine systemic injections of 6 mg/kg 10-vPMO cocktail at 2-week intervals (9× i.v. vPMOs) compared with wild-type (WT) and nontreated (NT) muscles. Scale bar, 100 μm. (b) Significant reduction in percentage of centrally nucleated fibers in skeletal muscles after long-term systemic treatment (n = 4 in nontreated and n = 6–7 in treated group). (c) Detection of IgG (green) in DIA, BF, QUA and HRT muscles of WT, nontreated and treated mdx52 mice 2 weeks after the final injection. (d) Increased grip power (kilogram force/kg) of hind limb in mdx52 mice after eight administrations of the cocktail. Data (n = 5 in nontreated and n = 3 in treated group) are presented as mean ± SD. * P < 0.05 and ** P < 0.01.
Figure 4.

Immune responses against the 10-vPMO cocktail after the long-term systemic treatment.(a) Immunostaining against CD3 antigen (red) of a pan T cell marker in diaphragm (DIA), biceps femoris (BF), quadriceps (QUA) and heart (HRT) muscles from wild-type at 6-month-old, nontreated and treated mdx52 mice 2 weeks after the final i.v. injection of the vPMO cocktail. Nuclei (blue) were counterstained with DAPI. Scale bar, 100 μm. (b) Counting CD3-positive cells on the muscle sections. The number of sporadic CD3-positive cells was counted on 10 section areas at random through a 20× objective lens. Data (n = 4 in nontreated and n = 6 in treated group) are presented as mean ± SD.
Serum biochemical parameters for renal and liver functions are not significantly altered by long-term systemic treatment with the 10-vPMO cocktail
To investigate the potential toxicity of long-term systemic injections of the 10-vPMO cocktail, serum biochemical parameters were tested and statistically analyzed among the groups at the end-point, which was 2 weeks after the final injection (Figure 5). A reduction in serum creatine kinase (CK) level, an indicator of muscle damage, was accompanied by histological and functional recovery, but this result did not reach the level of significance. The level of blood urea nitrogen (BUN) in the treated mice was significantly reduced to a level equivalent to that in WT mice. Levels of creatine (Cre), total bilirubin (T-bil), and γ-glutamyl transpeptidase (γ-GTP), a more specific indicator of liver lesions than aspartate aminotransferase (AST) and alanine aminotransferase (ALT) affected by muscle lesions,26,29,30 were not significantly changed by the long-term treatment. Thus, renal and hepatic toxicity were not detected in the mdx52 mice after nine consecutive injections of the 10-vPMO cocktail at 2-week intervals.
Figure 5.
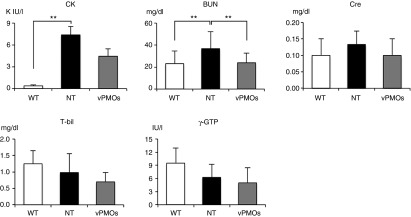
Evaluation of serum creatine kinase level and renal and hepatic toxicity in the blood test at the end-point of 2 weeks after the last of nine systemic injections of 6 mg/kg 10-vPMO cocktail at 2-week intervals.Blood biochemical tests were performed for serum creatine kinase (CK), blood urea nitrogen (BUN), creatinine (Cre), total bilirubin (T-bil), and γ-glutamyl transpeptidase (γ-GTP) in wild-type (WT), nontreated (NT), and treated mdx52 mice (n = 4 in WT, n = 6 in NT and n = 4 in treated group). Data are presented as mean ± SE.
Discussion
In this study, we demonstrated the feasibility of skipping exons 45–55 in their entirety, which is the mutation hotspot region in the dystrophin gene, and rescued expression of dystrophin and DAPs over 18 weeks without any detectable immune response or nephro- and hepatotoxicity in the blood test after nine intravenous injections of the 10-vPMO cocktail at a low concentration of 6 mg/kg. These results indicate that our new regimen for long-term treatment with the vPMO cocktail at 6 mg/kg can safely achieve therapeutic effects similar to our previous study with a dose of 12 mg/kg.26
Antisense-mediated exon skipping targeting a single exon is currently close to clinical application for the treatment of DMD, but the multiexon skipping strategy is still far from the clinical trial stage, due to the limited amount of information yielded thus far by preclinical trials.18 To date, several clinical trials of exon skipping using unmodified morpholinos and 2'O-methylated phosphorothioates (2'OMePSs) are ongoing for DMD. A phase 1/2 clinical trial of exon 44 skipping has been completed and skipping of exons 45 and 53 is being investigated in a phase 1/2a clinical trial conducted by Prosensa (http://www.prosensa.eu/). The exon-skipping phase 3 trial, which targets exon 51 with a 2'OMePS AO, is ongoing.13 Sarepta Therapeutics is currently conducting a phase 2b clinical trial of exon 51 skipping with a morpholino,12 while Nippon Shinyaku has started a phase 1 trial of exon 53 skipping (UMIN Clinical Trials Registry number UMIN000010964). Although these targeted exons comprise portions of the hotspot region, skipping of exons 44, 45, 51, and 53 would be applicable to only 6.2% (8.8%), 8.2% (11.8%), 13.0% (19.1%), and 7.7% (11.4%) of all DMD patients (patients with deletion mutations), respectively.16 In addition to the limitations of therapeutic coverage, another concern is that single exon skipping therapy produces distinct truncated dystrophin proteins depending on patient-specific mutation patterns; this raises a concern that ongoing skipping strategies may not always induce stable/functional dystrophin proteins. By contrast, more than 95% of patients in whom exons 45–55 are deleted in their entirety are reported to have milder BMD symptoms or are asymptomatic.19,20 BMD is caused by a reduction in the amount functional dystrophin protein.31 This fact suggests that truncated proteins that lack this specific region are more stable and functionally able to protect muscle tissue from deterioration. It is worth noting that multiexon skipping targeting exons 45–55 would cover ~63% of DMD patients with deletion mutations, according to the UMD-DMD database.19 Aartsma-Rus's group first tested the concept of skipping of the hotspot region with 2'OMePS cocktails in DMD patient cell lines.32 Unfortunately, this trial did not achieve sufficient skipping of the intended exons and dystrophin expression. Recently, we demonstrated proof of concept for skipping exons 45–55 with a cocktail of vPMOs; effective expression of dystrophin protein was induced after five consecutive systemic injections at 12 mg/kg every 2 weeks.26 Positively charged vPMOs have higher cell membrane permeability and improved delivery into muscle fibers. One concern is that vPMOs might have a narrower therapeutic window due to their chemical feature, compared to uncharged morpholinos. Long-term low-dose therapy could be an excellent solution to overcome this limitation. Thus, this therapeutic strategy has potential for the treatment of ~30 and 45% of all DMD patients based on the LOVD and the UMD-DMD database, respectively.19,33
New-generation morpholinos, such as cell-penetrating peptide-conjugated PMOs (PPMOs) and vPMOs, are reported to enable high-efficiency exon skipping at lower doses than unmodified morpholinos and 2'OMePSs.22,24,27,34 In a previous study, five administrations of a vPMO at a low dose of 6 mg/kg were more effective than a single bolus administration at 30 mg/kg in the mdx mouse model.24 Moreover, intravenous injections every 2 weeks of a PPMO at a dose of 6 mg/kg for 1 year achieved successful dystrophin expression in body-wide skeletal muscles in the mdx mouse.35 In this study, we employed a relatively low dose of 0.6 mg/kg of each vPMO (6 mg/kg in total) and found that long-term treatment with nine injections of the 10-vPMO cocktail at 2-week intervals successfully induced expression of mRNA in which exons 45–55 had been skipped; the result was recovery of dystrophin and DAPs body-wide in mdx52 mice. Although there were variations in the distribution and expression levels of the skipped dystrophin mRNA/proteins among the different muscle types and/or within positions of a given muscle, and no significant reduction in creatine kinase levels in the treated-mouse group, the amelioration of skeletal muscle pathology and functional recovery that we observed provide evidence of the therapeutic effects of the regimen.
Another concern was that very little dystrophin protein was detected in the heart muscle (≤1% that of WT mice), even though exons 45–55 skipped mRNA was detected by RT-PCR in heart tissue from treated mice. The lower expression of dystrophin protein may be attributed to differences in the posttranscriptional processing for dystrophin generated in skeletal muscle compared to heart muscle. This is supported by a recent study that dystrophin expression level in the heart is higher than in the skeletal muscles and that mutated dystrophin transcripts are less stable than normal transcripts.36 This could include the rate of decay for the AO-induced dystrophin mRNA and protein, because effective exon skipping in the heart muscle of transgenic mice, which bear target human β-globin intron, has been reported with four intravenous injections of a vPMO at 12.5 mg/kg/day for 4 days as well as in other tissues, including skeletal muscle.21 Neither bolus nor repeated intravenous/intraperitoneal administration of a vPMO at doses from 15 mg/kg to 30 mg/kg resulted in similar levels of dystrophin mRNA and protein produced in heart muscle compared to skeletal muscles.24,25 A similar tendency was reported in studies in which 2'OMePSs, morpholinos, and PPMOs were administered to mdx mice;30,35,37,38,39 moreover, this tendency was also seen in mdx mice subjected to adenoassociated virus-mediated exon skipping.40 In addition, distinct cellular trafficking systems for AOs between skeletal muscle cells and cardiac cells may affect the efficiency of AO-mediated exon skipping in these tissues.41 Although the processing mechanism of dystrophin transcripts and AO delivery system will need to be more thoroughly characterized in order to increase protein expression, it is encouraging to note that as little as 1–2% dystrophin protein expression in the heart muscle improves cardiac function and pathology in mdx mice.30,42
A remaining challenge facing the new-generation morpholinos is that there is still insufficient information available about their toxicity. As described above, thus far, frequent systemic intravenous injections at a dose of 6 mg/kg of a single vPMO and PPMO for long-term therapy seem to be an effective regimen with no potential adverse effects in mdx mice, which is a rationale for the evaluation of efficacy and safety of the 10-vPMO cocktail at 6 mg/kg in this study.24,35 Lethal toxicity emerges following single intravenous administration at a dose of 60 mg/kg PPMO in the mdx mouse, and 30 mg/kg PPMO systemic injections also cause transient activity reduction in treated mice, indicating potential off-target effects.35 In nonhuman primates treated by systemic injections with PPMO at 9 mg/kg for 4 weeks, mild tubular degeneration in the kidney has been reported.34 Although it is speculated that acute lethality/abnormal behavior after an intravenous injection with vPMOs may be caused by multimers clustered by hybridization between their sequences,43 no overt toxicity due to the chemical components of vPMO was recorded at up to 15–20 mg/kg in mice.25,44 However, the presence of dose-dependent and delayed side effects by long-term treatment must be determined to use this chemistry as a clinical grade. Neither the current 18-week regimen of 6 mg/kg vPMOs (0.6 mg/kg each vPMO) every 2 weeks, nor the previous study using the vPMO cocktail at a higher dose of 12 mg/kg, lead to any deterioration of the serum parameters that were tested.26 Notably, we found a significant reduction in BUN, an indicator of progression of protein catabolism, to the normal range observed in WT mice. Mussini et al. also have shown that catabolism in DMD muscles is increased.45 A relatively higher level of BUN is reported in mdx mice compared with WT C57BL/6 mice.46 The reduced BUN level with no significant change in the creatinine level may result from amelioration of muscle degeneration by the treatment. In addition, we confirmed that our vPMO cocktail does not induce a detectable immune response of T cells. Although pathological and immunological side effects for long-term treatment with vPMOs need to be more closely examined in various tissue types of animal models, these results indicate that this regimen may have clinical potential for DMD patients who require repeated administration over the course of their lifetime.
In summary, we demonstrated that long-term repeated intravenous injections of the 10-vPMO cocktail at a lower dose than described previously was both effective and safe in the DMD animal model. However, further investigations of vPMO cocktails will need to be conducted at the preclinical stage, such as dose-escalation and -reduction studies, as well as acute and chronic toxicity assessments, since multiexon skipping has more potential for off-target effects due to the repeated administration of many different AOs. Intermediate products induced by AO cocktails also should be considered a potential side effect, as shown in this study. Multiexon skipping with a number of AOs is likely to generate a variety of dystrophin mRNA/protein types including in-frame and out-of-frame transcripts due to the unequal skipping efficiency of individual AOs.26 Reducing the number of AOs used in a cocktail—depending on mutation patterns in the patients—or developing a new strategy with fewer AOs to induce skipping of the entire exons 45–55 region, regardless of the mutation patterns, may prevent unexpected effects and lead to formation of intended dystrophin proteins more effectively. Such advancements would aid in forwarding this strategy and other multiple exon skipping strategies into clinical application.33 In addition, sequence-specific AO cocktails optimized for the human DMD gene will need to be tested with human DMD patient skeletal muscle cell lines. Currently, the drug regulatory authorities consider individual AOs targeting different sequences to be separate drugs. This stance may need to be changed for developing cocktail drugs. Nevertheless, the results of this study should contribute not only to the clinical development of an exon 45–55 skipping therapy for DMD, but should also open up the possibility of using antisense drug cocktails for other genetic diseases in which long-term administration is required.
Material and methods
Animals. Eight-week-old male exon 52–deficient mdx52 mice were used in this study. As a control in the systemic treatment, male WT C57BL/6J mice at 6 months old were used for comparing to treated mdx52 at the end-point of the treatment. All mice were housed in an individually ventilated cage system with a 12-hour light–dark cycle; they received standard mouse chow (Harlan Teklad, Madison, WI) and water ad libitum. All mice were allowed to rest for at least 7 days in the facility before acclimatizing them on the instruments and taking baseline readings for behavioral assays. All mice were handled according to local Institutional Animal Care and Use Committee (IACUC) guidelines (University of Alberta, Edmonton, Canada and Children's National Medical Center, Worthington, DC).
Antisense oligonucleotides. Ten AOs targeted to exons 45–51 and 53–55 in mouse dystrophin gene were designed using ESEfinder software to anneal to the exon-splicing enhancer sites of each exon or exon/intron boundary, as previously described (Table 1).26,27 Specificity of the designed AOs was confirmed by NCBI blast software (https://www.ncbi.nlm.nih.gov/), which shows that our AO sequences theoretically do not bind any untargeted RNA sequences in 100% identity. All sequences were synthesized using vPMO backbones (Gene-Tools, LLC, Philomath, OR).21
Vivo-Morpholino injections. Animals were anesthetized by inhalation of isoflurane (Baxter, Deerfield, IL) for injections. A total of 0.3 μg of vPMOs targeting exons 45–55 in a total volume of 36 μl of saline were injected into the TA muscle of mdx52 mice. Muscle samples were obtained 2 weeks after the intramuscular injection. For long-term systemic treatment, a total of 6 mg/kg per injection of 10-vPMOs (0.6 mg/kg for each) in 150 μl of saline was injected into the tail vein of mdx52 mice, nine times at 2-week intervals. The mice were examined 2 weeks after the final injection. Muscle samples were obtained immediately after the mice were killed. The samples were snap frozen in liquid nitrogen–cooled isopentane and stored at −80 °C before use.
RT-PCR. Total RNA from muscle sections of WT, nontreated mdx52, and treated mdx52 mice were extracted as previously described.47 Total RNA template (200 ng) was used for a 25 μl RT-PCR with the SuperScript III One-Step RT-PCR System (Invitrogen, Carlsbad, CA) and 0.2 μmol/l of each primer, in accordance with the manufacturer's instructions. Primer sequences for the PCR were designed with Primer3Plus software: Exon 44_F, CAGTTGAAAAATGGCGACAC and Exon 56_R, GTAACAGGGGTGCTTCATCC. The cycling conditions were as follows: 55 °C for 30 minutes; 94 °C for 2 minutes; 35 cycles at 94 °C for 15 seconds, 60 °C for 30 seconds, and 68 °C for 1.2 minutes; and 68 °C for 5 minutes. PCR products were separated on a 2% agarose gel and then visualized by SYBR Safe DNA Gel Stain (Invitrogen). Skipping percentage was calculated as 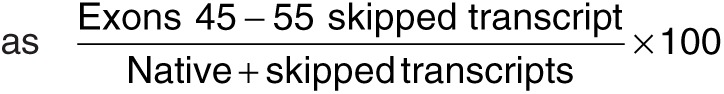 using ImageJ software (NIH). Bands of the expected size for the transcript were extracted with a gel extraction kit (Promega, Madison, WI). The sequencing reactions were performed with Big Dye Terminator v3.1 (Applied Biosystems, Foster City, CA).
using ImageJ software (NIH). Bands of the expected size for the transcript were extracted with a gel extraction kit (Promega, Madison, WI). The sequencing reactions were performed with Big Dye Terminator v3.1 (Applied Biosystems, Foster City, CA).
Immunohistochemistry. Sections (7 μm thickness) of the TA muscle after single intramuscular injection were incubated with two anti-dystrophin antibodies: mouse monoclonal DYS1 against peptides encoded by exons 26–30 (1:200; Novocastra Laboratories, Newcastle upon Tyne, UK) and rabbit polyclonal P7 against peptides encoded by exon 57 (1:200; Fairway BioTech, Shrewsbury, UK). For quantification of the number of dystrophin-positive fibers, we made several tissue sections and selected representative sections of TA muscles, then stained these sections with DYS1 antibody (n = 3). The number of dystrophin-positive fibers were counted in sections having at least 200 total muscle fibers using a BZ-9000 fluorescence microscope (Keyence, Osaka, Japan), as previously described.48 In systemic treatment with the 10-vPMO cocktail, the diaphragm, biceps femoris, quadriceps, gastrocnemius, tibialis anterior, biceps brachii, triceps brachii, and heart muscles were examined 2 weeks after the final injection using anti-dystrophin (P7) antibody and antibodies against dystrophin-associated proteins: anti-α1-syntrophin rabbit polyclonal antibody (1:200, Abcam, Cambridge, UK), anti-nNOS rabbit polyclonal antibody (1:100, Invitrogen), anti-α-sarcoglycan mouse monoclonal antibody (1:10, Novocastra Laboratories), and anti-β-dystroglycan mouse monoclonal antibody (1:5, Novocastra Laboratories). To detect the primary antibodies, Alexa Fluor 594–conjugated goat antimouse or rabbit IgG (1:2,000; Invitrogen) were used as secondary antibodies. To examine IgG accumulation in muscle fibers and immune response to the vPMO cocktail, anti-mouse IgG F(ab')2 (1:750, Invitrogen) and anti-CD3 rabbit monoclonal (1:25, Abcam) antibodies were used on nonfixed and 4% PFA-fixed sections (7 μm thickness), respectively. The number of IgG-positive fibers and sporadic CD3-positive cells as pan T cells were counted in 10 section areas randomly selected through a 20× objective lens. In immunostaining against CD3 antigen, cells were regarded as positive when more than half the membrane circumference was stained on cross-sections.
Hematoxylin and eosin staining. For counting CNFs, muscle sections (7 μm thickness) were stained with Mayer's hematoxylin and eosin (H&E) solutions and images were taken with a DMR microscope (Leica Micro-systems, Newcastle upon Tyne, UK) with a 20× objective lens, as previously described.49 The percentage of CNFs was calculated in 400–1,100 fibers in DIA, BF, QUA, GC, and TA muscles of nontreated (n = 4) and treated mdx52 mice (n = 6–7).
Western blotting. Protein extraction from frozen muscle sections and Western blot analysis were performed as previously described.47 In brief, 10 and 1% (4 and 0.4 μg of protein, respectively) of the TA muscle from WT mice were used as a positive control, 40 μg of protein of the TA muscle from nontreated mdx52 mice were used as a negative control, and 40 μg of protein from the indicated muscles of treated mdx52 mice were loaded onto a NuPAGE Novex 3–8% Tris-Acetate Midi Gel (Invitrogen) and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis at 150 V for 75 minutes. The proteins were transferred onto an Immobilon PVDF membrane (Millipore, Billerica, MA) by semidry blotting at 20 V of constant voltage for 70 minutes. The membrane was blocked with phosphate-buffered saline containing 0.05% Tween 20, 0.1% casein, and 0.1% gelatin, then incubated with the DYS1 monoclonal antibody (1:400 in blocking solution) at 4 °C overnight. Using ImageJ software, the intensities of the bands were compared with those from WT muscles, as previously described.48 Myosin heavy chain (MyHC) stained by Coomassie Brilliant Blue was used as a loading control.
Muscle function test. The grip strength test for hind and forelimbs of the mice was performed 2 weeks after the eighth of the every 2 weeks intravenous injections, as previously described.50
Biochemical blood test. Serum samples were collected from WT mice, nontreated and treated mdx52 mice 2 weeks after the final injection of vPMO cocktail. Serum biochemical parameters of creatine kinase, blood urea nitrogen, creatinine, total bilirubin, and γ-GTP were assayed with the Fuji Drychem system (Fuji Film Medical, Tokyo, Japan).
Statistical analysis. For analysis of dystrophin-positive fibers in the locally injected TA muscle and CNFs in the systemic treatment, statistical differences were assessed by F test and Student's t-test. Mann–Whitney U-test was performed to analyze the number of IgG-positive fibers and CD3-positive cells. One-way analysis of variance with a Tukey–Kramer multiple-comparison test was performed for statistical analysis of the serum biochemical test. Data are reported as mean values ± SD or ± SE. The level of significance was set at P < 0.05.
SUPPLEMENTARY MATERIAL Figure S1. Intramuscular (i.m.) injection of the 10-vPMO cocktail at 0.3 μg (0.03 μg of each vPMO).
Acknowledgments
This work was supported by the Foundation to Eradicate Duchenne, the University of Alberta Faculty of Medicine and Dentistry, Parent Project Muscular Dystrophy (USA), the Canadian Institutes of Health Research (CIHR), the Friends of Garrett Cumming Research Funds, HM Toupin Neurological Science Research Funds, Muscular Dystrophy Canada, the Canada Foundation for Innovation, Alberta Enterprise and Advanced Education, and the Women and Children's Health Research Institute (WCHRI). Y.A. is supported by MRC Confidence in Concept award. K.N. is supported by NIH grants (1K26RR032082, 1P50AR060836-01, 1U54HD071601, and 2R24HD050846-06), DOD grants (W81XWH-11-1-0330, W81XWH-11-1-0782, W81XWH-10-1-0659, W81XWH-11-1-0809, and W81XWH-09-1-0599), a translational research grant from the MDA, and a pilot grant from PPMD.
Supplementary Material
Intramuscular (i.m.) injection of the 10-vPMO cocktail at 0.3 μg (0.03 μg of each vPMO).
References
- Mercuri E, Muntoni F. Muscular dystrophies. Lancet. 2013;381:845–860. doi: 10.1016/S0140-6736(12)61897-2. [DOI] [PubMed] [Google Scholar]
- Zellweger H, Antonik A. Newborn screening for Duchenne muscular dystrophy. Pediatrics. 1975;55:30–34. [PubMed] [Google Scholar]
- Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 2014;24:482–491. doi: 10.1016/j.nmd.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Takeshima Y, Yagi M, Okizuka Y, Awano H, Zhang Z, Yamauchi Y.et al. (2010Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center J Hum Genet 55379–388. [DOI] [PubMed] [Google Scholar]
- Magri F, Govoni A, D'Angelo MG, Del Bo R, Ghezzi S, Sandra G.et al. (2011Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up J Neurol 2581610–1623. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Kimura E, Mori-Yoshimura M, Komaki H, Matsuda Y, Goto K.et al. (2013Characteristics of Japanese Duchenne and Becker muscular dystrophy patients in a novel Japanese national registry of muscular dystrophy (Remudy) Orphanet J Rare Dis 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelly J, Gilgenkrantz H, Lambert M, Hamard G, Chafey P, Récan D.et al. (1990Effect of dystrophin gene deletions on mRNA levels and processing in Duchenne and Becker muscular dystrophies Cell 631239–1248. [DOI] [PubMed] [Google Scholar]
- Kunkel LM, Beggs AH, Hoffman EP. Molecular genetics of Duchenne and Becker muscular dystrophy: emphasis on improved diagnosis. Clin Chem. 1989;35 7 Suppl:B21–B24. [PubMed] [Google Scholar]
- Lu QL, Yokota T, Takeda S, Garcia L, Muntoni F, Partridge T. The status of exon skipping as a therapeutic approach to duchenne muscular dystrophy. Mol Ther. 2011;19:9–15. doi: 10.1038/mt.2010.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Yokota T. Antisense therapy in neurology. J Pers Med. 2013;3:144–176. doi: 10.3390/jpm3030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. Make or break for first splice-modulating agents. Nat Rev Drug Discov. 2013;12:813–815. doi: 10.1038/nrd4151. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP.et al.; Eteplirsen Study Group 2013Eteplirsen for the treatment of Duchenne muscular dystrophy Ann Neurol 74637–647. [DOI] [PubMed] [Google Scholar]
- Voit T, Topaloglu H, Straub V, Muntoni F, Deconinck N, Campion G.et al. (2014Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study Lancet Neurol 13987–996. [DOI] [PubMed] [Google Scholar]
- Yokota T, Pistilli E, Duddy W, Nagaraju K. Potential of oligonucleotide-mediated exon-skipping therapy for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2007;7:831–842. doi: 10.1517/14712598.7.6.831. [DOI] [PubMed] [Google Scholar]
- Yokota T, Takeda S, Lu QL, Partridge TA, Nakamura A, Hoffman EP. A renaissance for antisense oligonucleotide drugs in neurology: exon skipping breaks new ground. Arch Neurol. 2009;66:32–38. doi: 10.1001/archneurol.2008.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen GJ.et al. (2009Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations Hum Mutat 30293–299. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Bronson A, Levin AA, Takeda S, Yokota T, Baudy AR.et al. (2011Restoring dystrophin expression in duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through Am J Pathol 17912–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echigoya Y, Yokota T. Skipping multiple exons of dystrophin transcripts using cocktail antisense oligonucleotides. Nucleic Acid Ther. 2014;24:57–68. doi: 10.1089/nat.2013.0451. [DOI] [PubMed] [Google Scholar]
- Béroud C, Tuffery-Giraud S, Matsuo M, Hamroun D, Humbertclaude V, Monnier N.et al. (2007Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy Hum Mutat 28196–202. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Yoshida K, Fukushima K, Ueda H, Urasawa N, Koyama J.et al. (2008Follow-up of three patients with a large in-frame deletion of exons 45-55 in the Duchenne muscular dystrophy (DMD) gene J Clin Neurosci 15757–763. [DOI] [PubMed] [Google Scholar]
- Morcos PA, Li Y, Jiang S. Vivo-Morpholinos: a non-peptide transporter delivers Morpholinos into a wide array of mouse tissues. BioTechniques. 2008;45:613–4, 616, 618 passim. doi: 10.2144/000113005. [DOI] [PubMed] [Google Scholar]
- Yokota T, Nakamura A, Nagata T, Saito T, Kobayashi M, Aoki Y.et al. (2012Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs Nucleic Acid Ther 22306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Nagata T, Yokota T, Nakamura A, Wood MJ, Partridge T.et al. (2013Highly efficient in vivo delivery of PMO into regenerating myotubes and rescue in laminin-a2 chain-null congenital muscular dystrophy mice Hum Mol Genet 224914–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Li Y, Morcos PA, Doran TJ, Lu P, Lu QL. Octa-guanidine morpholino restores dystrophin expression in cardiac and skeletal muscles and ameliorates pathology in dystrophic mdx mice. Mol Ther. 2009;17:864–871. doi: 10.1038/mt.2009.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Benrashid E, Lu P, Cloer C, Zillmer A, Shaban M.et al. (2011Targeted skipping of human dystrophin exons in transgenic mouse model systemically for antisense drug development PLoS ONE 6e19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Yokota T, Nagata T, Nakamura A, Tanihata J, Saito T.et al. (2012Bodywide skipping of exons 45-55 in dystrophic mdx52 mice by systemic antisense delivery Proc Natl Acad Sci USA 10913763–13768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Yokota T, Wood MJ. Development of multiexon skipping antisense oligonucleotide therapy for Duchenne muscular dystrophy. Biomed Res Int. 2013;2013:402369. doi: 10.1155/2013/402369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R, Harris DC, Whitington PF. Relative elevations of serum alanine and aspartate aminotransferase in muscular dystrophy. J Pediatr Gastroenterol Nutr. 2005;41:121–124. doi: 10.1097/01.wno.0000161657.98895.97. [DOI] [PubMed] [Google Scholar]
- Wu B, Xiao B, Cloer C, Shaban M, Sali A, Lu P.et al. (2011One-year treatment of morpholino antisense oligomer improves skeletal and cardiac muscle functions in dystrophic mdx mice Mol Ther 19576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bergen JC, Wokke BH, Janson AA, van Duinen SG, Hulsker MA, Ginjaar HB.et al. (2014Dystrophin levels and clinical severity in Becker muscular dystrophy patients J Neurol Neurosurg Psychiatr 85747–753. [DOI] [PubMed] [Google Scholar]
- van Vliet L, de Winter CL, van Deutekom JC, van Ommen GJ, Aartsma-Rus A. Assessment of the feasibility of exon 45-55 multiexon skipping for Duchenne muscular dystrophy. BMC Med Genet. 2008;9:105. doi: 10.1186/1471-2350-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A, Kaman WE, Weij R, den Dunnen JT, van Ommen GJ, van Deutekom JC. Exploring the frontiers of therapeutic exon skipping for Duchenne muscular dystrophy by double targeting within one or multiple exons. Mol Ther. 2006;14:401–407. doi: 10.1016/j.ymthe.2006.02.022. [DOI] [PubMed] [Google Scholar]
- Moulton HM, Moulton JD. Morpholinos and their peptide conjugates: therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim Biophys Acta. 2010;1798:2296–2303. doi: 10.1016/j.bbamem.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Wu B, Lu P, Cloer C, Shaban M, Grewal S, Milazi S.et al. (2012Long-term rescue of dystrophin expression and improvement in muscle pathology and function in dystrophic mdx mice by peptide-conjugated morpholino Am J Pathol 181392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitali P, van den Bergen JC, Verhaart IE, Wokke B, Janson AA, van den Eijnde R.et al. (2013DMD transcript imbalance determines dystrophin levels FASEB J 274909–4916. [DOI] [PubMed] [Google Scholar]
- Lu QL, Rabinowitz A, Chen YC, Yokota T, Yin H, Alter J.et al. (2005Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles Proc Natl Acad Sci USA 102198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Lu P, Benrashid E, Malik S, Ashar J, Doran TJ.et al. (2010Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino Gene Ther 17132–140. [DOI] [PubMed] [Google Scholar]
- Tanganyika-de Winter CL, Heemskerk H, Karnaoukh TG, van Putten M, de Kimpe SJ, van Deutekom J, et al. Long-term exon skipping studies with 2'-o-methyl phosphorothioate antisense oligonucleotides in dystrophic mouse models. Mol Ther Nucleic Acids. 2012;1:e44. doi: 10.1038/mtna.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denti MA, Incitti T, Sthandier O, Nicoletti C, De Angelis FG, Rizzuto E.et al. (2008Long-term benefit of adeno-associated virus/antisense-mediated exon skipping in dystrophic mice Hum Gene Ther 19601–608. [DOI] [PubMed] [Google Scholar]
- Lehto T, Castillo Alvarez A, Gauck S, Gait MJ, Coursindel T, Wood MJ.et al. (2014Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells Nucleic Acids Res 423207–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jearawiriyapaisarn N, Moulton HM, Sazani P, Kole R, Willis MS. Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc Res. 2010;85:444–453. doi: 10.1093/cvr/cvp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson DP, Dangott LJ, Lightfoot JT. Lessons learned from vivo-morpholinos: How to avoid vivo-morpholino toxicity. BioTechniques. 2014;56:251–256. doi: 10.2144/000114167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widrick JJ, Jiang S, Choi SJ, Knuth ST, Morcos PA. An octaguanidine-morpholino oligo conjugate improves muscle function of mdx mice. Muscle Nerve. 2011;44:563–570. doi: 10.1002/mus.22126. [DOI] [PubMed] [Google Scholar]
- Mussini E, Cornelio F, Colombo L, De Ponte G, Giudici G, Cotellessa L.et al. (1984Increased myofibrillar protein catabolism in duchenne muscular dystrophy measured by 3-methylhistidine excretion in the urine Muscle Nerve 7388–391. [DOI] [PubMed] [Google Scholar]
- Sazani P, Ness KP, Weller DL, Poage D, Nelson K, Shrewsbury AS. Chemical and mechanistic toxicology evaluation of exon skipping phosphorodiamidate morpholino oligomers in mdx mice. Int J Toxicol. 2011;30:322–333. doi: 10.1177/1091581811403504. [DOI] [PubMed] [Google Scholar]
- Yokota T, Hoffman E, Takeda S. Antisense oligo-mediated multiple exon skipping in a dog model of duchenne muscular dystrophy. Methods Mol Biol. 2011;709:299–312. doi: 10.1007/978-1-61737-982-6_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S.et al. (2009Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs Ann Neurol 65667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echigoya Y, Lee J, Rodrigues M, Nagata T, Tanihata J, Nozohourmehrabad A.et al. (2013Mutation types and aging differently affect revertant fiber expansion in dystrophic mdx and mdx52 mice PLoS ONE 8e69194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Nakamura A, Yokota T, Saito T, Okazawa H, Nagata T.et al. (2010In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse Mol Ther 181995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Intramuscular (i.m.) injection of the 10-vPMO cocktail at 0.3 μg (0.03 μg of each vPMO).