

Abstract
Billions of gallons of untreated wastewater enter the coastal ocean each year. Once sewage microorganisms are in the marine environment, they are exposed to environmental stressors, such as sunlight and predation. Previous research has investigated the fate of individual sewage microorganisms in seawater but not the entire sewage microbial community. The present study used next-generation sequencing (NGS) to examine how the microbial community in sewage-impacted seawater changes over 48 h when exposed to natural sunlight cycles and marine microbiota. We compared the results from microcosms composed of unfiltered seawater (containing naturally occurring marine microbiota) and filtered seawater (containing no marine microbiota) to investigate the effect of marine microbiota. We also compared the results from microcosms that were exposed to natural sunlight cycles with those from microcosms kept in the dark to investigate the effect of sunlight. The microbial community composition and the relative abundance of operational taxonomic units (OTUs) changed over 48 h in all microcosms. Exposure to sunlight had a significant effect on both community composition and OTU abundance. The effect of marine microbiota, however, was minimal. The proportion of sewage-derived microorganisms present in the microcosms decreased rapidly within 48 h, and the decrease was the most pronounced in the presence of both sunlight and marine microbiota, where the proportion decreased from 85% to 3% of the total microbial community. The results from this study demonstrate the strong effect that sunlight has on microbial community composition, as measured by NGS, and the importance of considering temporal effects in future applications of NGS to identify microbial pollution sources.
INTRODUCTION
Globally, swimming in and the consumption of shellfish from coastal waters polluted with sewage result in over 120 million cases of gastrointestinal (GI) illnesses, 50 million cases of respiratory illnesses, and consumption of 800 million meals of potentially contaminated shellfish each year (1). Sewage in coastal waters comes from leaking sewer lines, malfunctioning sewage treatment plants or septic systems, and overflow from combined sewer systems during heavy rainfall (2). The United States Environmental Protection Agency (USEPA) (2) estimates that there are 23,000 to 75,000 sanitary sewage overflows per year in the United States, resulting in the discharge of 3 billion to 10 billion gallons of untreated wastewater. Understanding the fate of the sewage-derived microorganisms in coastal waters is critical to identifying sewage pollution and assessing the health risks posed by sewage-polluted waters.
Throughout the world, coastal waters are monitored for fecal pollution using fecal indicator bacteria (FIB), such as Escherichia coli and enterococci (3–5). FIB enumeration methods are time-consuming (∼24 h), and the concentrations of FIB in coastal waters vary over hourly time scales, complicating their use for health risk assessment (6, 7). In addition, FIB can be found in a variety of different animal feces (8) and nonfecal environments, such as sand, sediments, and soils (9–13), as well as lacustrine and marine vegetation (14–17). Thus, FIB cannot usually be used to discern pollution sources (for example, sewage leaks or runoff from agricultural lands). The risk of GI illness associated with water impacted by nonhuman fecal pollution is believed to be less than the risk associated with human fecal pollution on the basis of quantitative microbial risk assessment (QMRA) models (18). Therefore, identification of the pollution source is important for assessing human health risks and remediating polluted waters.
Advances in technology have led to source-specific molecular methods of identifying pollution sources. Quantitative PCR (qPCR) assays that target gene sequences specific to bacteria found in the feces of different animals, including humans, gulls, and cows, have been developed (19–24). Other methods are community based and use the composition of the microbial community to identify pollution sources. Terminal restriction fragment length polymorphism (T-RFLP) analysis (25), PhyloChip analysis (26), and next-generation sequencing (NGS) (27–29) are examples of community-based methods that have been used in microbial source tracking studies. As the cost of NGS rapidly decreases, this comprehensive technique is gaining popularity as a microbial source tracking tool.
When applied to microbial source tracking, NGS is used to characterize the microbial community (based on the sequencing of a region of the 16S rRNA gene) both in contaminated water samples and in potential sources of microbial pollution. The composition of the microbial community in the water samples is compared to the composition in the potential sources to identify shared community members and deduce the pollution source (27–29). One important underlying assumption in the application of NGS to microbial source tracking is that the microbial community from a potential pollution source is temporally stable once it is released into the environment. However, this assumption has not been thoroughly tested. It is important to investigate how environmental stressors affect the temporal stability of the NGS sewage community signal in order to understand potential limitations of NGS for microbial source tracking.
Environmental stressors affecting the persistence of enteric bacteria and their DNA markers in natural waters include salinity, temperature, predation, and sunlight (30, 31). While a large amount of work has investigated the persistence of bacteria in natural waters using culture-based methods (31–36), far fewer studies have focused on the measurement of bacterial persistence by molecular methods, such as conventional PCR, qPCR, and NGS (37). Research on the persistence or decay of enteric bacteria, measured using PCR or qPCR, has shown that bacterial DNA markers persist longer at higher salinities (38, 39) and at lower water temperatures (23, 38, 40, 41). Genetic markers of enteric bacteria have also been found to persist longer when predation by natural biota is reduced or eliminated (40, 41). The findings of studies on the effect of sunlight on DNA marker persistence, however, are equivocal (33, 42). Several studies have found sunlight to have no effect on genetic marker persistence (33, 41, 43), while other studies have found that the decay rate depends on the presence of sunlight (33, 39, 41, 44). One study found that sunlight exposure affected the decay of a human-specific Bacteroidales genetic marker (a portion of the 16S rRNA gene) but not a genetic marker (a portion of the 23S rRNA gene) for Enterococcus (33). While previous studies have considered the persistence of one or several specific DNA markers, the study presented in this paper is the first to investigate the effect of environmental stressors on the entire sewage microbial community over time.
The present study focuses on the effects of natural sunlight cycles and predation by marine microbiota on microbial community diversity and composition and operational taxonomic unit (OTU) abundance. In particular, the study considers the temporal stability of the NGS signal for sewage-derived organisms in seawater. The results from this study can be used to inform the use of NGS as a microbial source tracking tool in marine waters used for recreation and shellfisheries.
MATERIALS AND METHODS
Microcosm setup.
Seawater was collected from the Fitzgerald Marine Reserve, CA (37°31′27.72″N, 122°31′3.95″W), and a portion of it was tangentially filtered through a 30-kDa (∼3-nm-pore-size) membrane (Pall Corporation, Port Washington, NY). The filtrate was used to construct filtered microcosms that contained no marine microbiota greater than approximately 3 nm in size. Unfiltered seawater was used to construct the unfiltered microcosms that contained marine microbiota. Marine microbiota may consist of bacteria, protozoa, and viruses.
Filtered seawater and unfiltered seawater were seeded with raw sewage that had been passed through a 20-μm-mesh-size sieve (nylon mesh; Nitex Bolting Cloth; WildCo Wildlife Supply Company, Yulee, FL). The sieved sewage was added to a final concentration of 15% (vol/vol). The raw sewage was collected from the Regional Water Quality Control Plant in Palo Alto, CA, on the day that the experiment was started. The plant services 220,000 residents and processes 22 million gallons of wastewater a day, according to the City of Palo Alto Regional Water Quality Control Plant website (http://www.cityofpaloalto.org/gov/depts/pwd/rwqcp/default.asp).
A volume of 600 to 700 ml of the seawater-sewage mixtures was placed in dialysis tubing (120-mm flat width) made of standard regenerated cellulose with a 6- to 8-kDa molecular weight cutoff (∼1 nm pore size; Spectra/Por 1 dialysis tubing; Spectrum Labs, Rancho Dominguez, CA) and sealed with 150-mm-width universal closures (Spectrum Labs, Rancho Dominguez, CA). The dialysis tubing allowed the exchange of solutes, including molecules with molecular masses of less than 6 to 8 kDa, between the microcosm and a seawater bath (described below) but retained particles, including bacteria, viruses, and protozoa. A total of 28 dialysis tubes were constructed; 14 contained filtered seawater plus sieved sewage and 14 contained unfiltered seawater plus sieved sewage.
The 28 dialysis tubes were split between four seawater baths (polypropylene containers with dimensions of 75 cm by 56 cm by 18 cm containing 55 liters of water) (Fig. 1). The seawater in the bath that a dialysis tube was placed in matched the seawater in the dialysis tube. Dialysis tubes containing filtered seawater plus sieved sewage were split between 2 water baths containing filtered seawater. Dialysis tubes containing unfiltered seawater plus sieved sewage were split between 2 seawater baths containing unfiltered seawater. One of each set of seawater baths was placed in the dark in a room with a constant temperature (15°C), and the other was placed on the rooftop of the laboratory to receive natural sunlight. The microcosms were kept at ∼15°C to mimic the ocean temperatures along the central California coast. The seawater baths exposed to natural sunlight were maintained at 15°C ± 5°C by running freshwater chilled by a Neslab RTE-7 Digital One refrigerated bath (Thermo Fisher Scientific, Waltham, MA) through 0.5-in. vinyl tubing along the bottom of the baths. The temperature of the seawater baths was monitored throughout the day using a handheld YSI-30 system (YSI, Yellow Springs, OH), and adjustments to the chilled water were made to maintain their temperatures.
FIG 1.
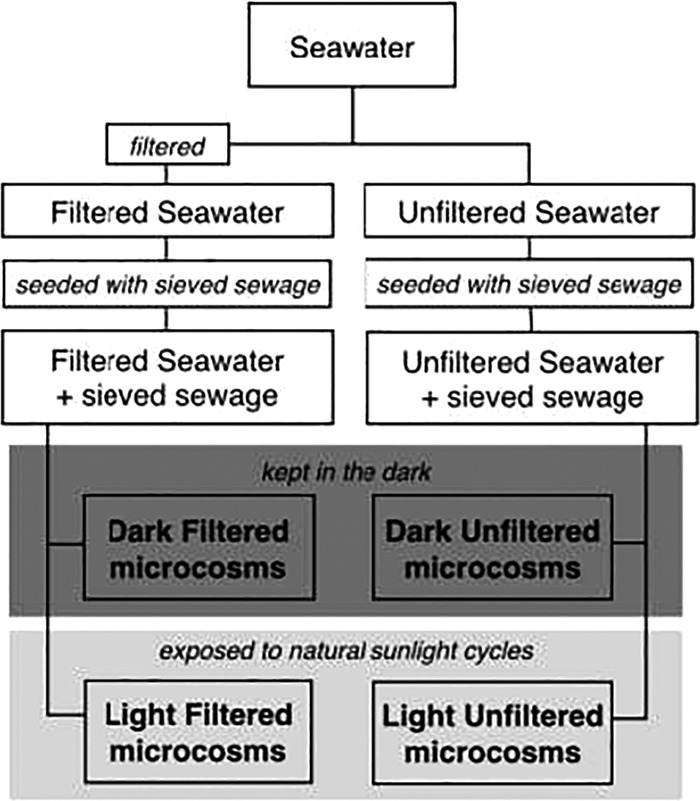
Experimental design. Seawater was collected at a site along the California coast. Half of the seawater was filtered through a tangential membrane filter (molecular weight cutoff, 30 kDa). Filtered and unfiltered seawater was seeded with sieved (pore size, 20 μm) sewage to make microcosms. Filtered and unfiltered microcosms were then divided between water baths and either kept in the dark or exposed to 2 natural sunlight cycles on the laboratory rooftop.
To determine if the dialysis tubing blocked any wavelengths of sunlight, we measured the spectra of artificial sunlight in a solar simulator (Atlas Suntest CPS+; Linsengericht-Altenhaßlau, Germany) with an International Light ILT950 UV-visible-near infrared spectroradiometer (International Light, Peabody, MA) and compared the spectra obtained when the simulator was uncovered with those obtained when it was covered with dialysis tubing (data not shown). The dialysis tubing was found not to block any wavelengths of sunlight.
Sampling procedure.
Experiments were conducted from midnight on 26 April 2013 to midnight on 28 April 2013. We used destructive sampling; at each time point, one dialysis tube from each seawater bath was removed. At the start of the experiment (time [t] = 0 h), one dialysis tube was processed for the filtered microcosm (the initial mixture of filtered seawater plus sieved sewage) and one was processed for the unfiltered microcosm (the initial mixture of unfiltered seawater plus sieved sewage). Subsequently, four dialysis tubes (one from each experimental bath) were collected at 12, 24, 36, and 48 h after the start of the experiment and are referred to here as dialysis tubes or microcosms collected at time points of 12 h, 24 h, 36 h, and 48 h. Dialysis tubes collected at the 12-h and 36-h time points were collected during the day when sunlight was incident on the sunlit treatments. Dialysis tubes collected at the 0-h, 24-h, and 48-h time points were collected when the sun was set.
A fraction of the bath seawater was cycled at every time point; 3 liters of filtered or unfiltered seawater from the bath was removed, and 3 liters of new filtered or unfiltered seawater that was being stored at 4°C was added to the bath to replenish the seawater. This was done to refresh dissolved constituents, such as nutrients, that might have been taken up by the organisms in the dialysis tubes.
The solar radiation on the roof was measured using a Sunshine Pyranometer SPN1 instrument (Delta-T Devices Ltd., Cambridge, United Kingdom). Solar radiation ranged from 0 to 240 W/m2 on the first day of the experiment and from 0 to 239 W/m2 on the second day (the solar irradiance measured every 30 min is provided in Fig. S1 in the supplemental material).
DNA extraction.
One hundred milliliters of fluid from each dialysis tube was filtered through 0.22-μm-pore-size, 47-mm-diameter Durapore filters (Millipore, Billerica, MA) to collect microorganisms and stored at −80°C for analysis. DNA was extracted from frozen filters using a MO BIO PowerWater DNA isolation kit following the manufacturer's instructions (MO BIO Laboratories, Inc., Carlsbad, CA). DNA was eluted in 100 μl warmed solution PW6 (MO BIO Laboratories, Inc., Carlsbad, CA) and stored at −20°C.
16S rRNA PCR amplification and 454 pyrosequencing.
The bacterial community in the samples was characterized using 454 pyrosequencing. Indexed amplicon libraries were constructed using fusion PCR primers targeting the V6-V4 hypervariable domains of the bacterial 16S rRNA. One-hundred-microliter reaction mixtures were prepared, and 25 μl was aliquoted into triplicate reaction mixtures with the template and a no-template control (NTC). The reaction mixtures consisted of 1× Platinum PCR SuperMix High Fidelity master mix (Invitrogen, Life Technologies, Carlsbad, CA), 50 nM forward and reverse primers, and 2 μl of undiluted DNA template. The fusion primers contained A (forward) and B (reverse) 454 titanium adapters. In each forward primer, a unique 11-nucleotide multiplex identifier (MID) was included between the adapter sequence and the 16S rRNA-specific sequence. The 16S rRNA sequences used to target the V6-V4 region were 518F (CCAGCAGCYGCGGTAAN) and 1064R (CGACRRCCATGCANCACCT) (27, 46). PCR cycling parameters included an initial denaturation step at 94°C for 3 min, followed by 30 cycles of 94°C for 30 s, 57°C for 45 s, and 72°C for 1 min and a final extension at 72°C for 2 min. Triplicate reaction mixtures were pooled, and 5 μl was run on a 1.5% agarose gel for 60 min at 100 V. Five microliters of NTC was also included in the gel. After confirmation of the target amplicon in the gel, the pooled amplicons were processed with a QIAquick cleanup kit (Qiagen, Valencia, CA). Samples were pooled in equal molar concentrations for sequencing. Pyrosequencing was performed at the Genome Sequencing and Analysis Core Resource at Duke University in half of a picotiter plate on a Roche 454 GS-FLX sequencing instrument.
Sequence processing and statistical analyses.
We processed the raw sequences using Quantitative Insights into Microbial Ecology (MacQIIME) (47). Reads were assigned to samples using the MID attached to the forward primers. Sequences were retained if they passed the following quality controls: quality scores greater than 25, more than 300 but less than 1,000 bases, no primer mismatches, and no more than 5 homopolymers. Sequences were assigned OTUs by de novo picking and were clustered on the basis of 97% sequence similarity using the UCLUST program (48). A representative sequence was chosen for each OTU, aligned using the PyNAST tool with default parameters, and assigned a taxonomic identity using the RDP Classifier program (49, 50). Greengenes (GG; version 13_5) was used as the reference database (51–53). Only OTUs assigned a taxonomic designation in the Greengenes database were used for downstream analysis (54). To account for unequal sequencing depths, OTU counts were rarefied to the minimum number of reads in a sample (4,635 sequences) (55, 56). The 20 OTUs that had the highest relative abundances (using the rarefied data for all time points, all the microcosm treatments, and the unfiltered seawater before it was seeded with sewage) are referred to here as the “20 most abundant OTUs.”
All statistical analyses were conducted at the OTU level with R Bioconductor software (57) using the phyloseq package (58), the vegan package (59), the Empirical Analysis of Digital Gene Expression in R (edgeR) package (60), and the SourceTracker package (61). Statistical analyses are described further in the supplemental material. We loaded the data from QIIME into R, calculated the observed richness and the Shannon-Weaver diversity index, and rarefied the data using the phyloseq package (62, 63). Rarefied OTU data were used for all subsequent analyses except those conducted using edgeR (those analyses are described in the supplemental material). Similarities in microbial community composition were investigated using the vegan package (59). Rarefied OTU counts were square root transformed, and Bray-Curtis dissimilarities between samples were calculated. Similarities were visualized using a nonmetric multidimensional scaling (nMDS) ordination plot. Mantel tests were used to investigate the relationship between community composition and time. Analysis of similarity (ANOSIM) was used to test if there was a significant difference in community composition at the OTU level on the basis of the experimental treatments. The SourceTracker package (61) was used to investigate how the sewage microbial community changed with time in each treatment (described in the supplemental material). For SourceTracker, the unfiltered seawater (before seeding with sieved sewage) and the filtered seawater seeded with sieved sewage at the initial time point were considered sources. All other samples were considered “sinks.”
Changes in the relative OTU abundance at each time point due to the different treatments were investigated using the edgeR package (60). We investigated four hypotheses using the edgeR package: (i) OTU abundance is lower in the dark unfiltered relative to the dark filtered microcosms in the absence of sunlight due to the effect of the microbiota, (ii) OTU abundance is lower in the light unfiltered relative to the light filtered microcosm in the presence of sunlight due to the effect of the microbiota, (iii) OTU abundance is lower in the light filtered relative to the dark filtered microcosms due to the effect of sunlight in the absence of microbiota, and (iv) OTU abundance is lower in the light unfiltered relative to the dark unfiltered microcosms due to the effect of sunlight in the presence of microbiota. The data were filtered in edgeR to remove OTUs with very low raw counts (OTUs that had a count of 2 or more in at least one sample were retained by the program). The filtered data set contained 5,338 unique OTUs. Raw OTU counts (not rarefied) were normalized to pseudocounts using a model-based method that applies a correction factor to scale all the library sizes to one effective library size. Since we did not have biological replicates to calculate the biological coefficient of variation (BCV) (1, 2, 4–6), we based the dispersion estimates (used to calculate the BCV) on the samples from the dark filtered microcosm at 0 h and 12 h (further described in the supplemental material). To account for type 1 errors, all P values were adjusted to false discovery rates (FDRs) (64) and were considered statistically significant if the FDR was less than 10% (FDR < 0.1).
Nucleotide sequence accession number.
Raw sequence data were submitted to NCBI's Sequence Read Archive (SRA) under study accession number SRP042614.
RESULTS
NGS data.
After quality control in QIIME, samples had between 4,635 reads (dark filtered microcosm at 48 h) and 41,597 reads (light unfiltered microcosm at 36 h), with the average being 15,519 reads (see Table S1 in the supplemental material). The concatenated, unrarified data set contained 9,723 unique OTUs. Although the PCR primers were designed to target bacteria, the concatenated, unrarified data set contained 7 archaeal OTUs (0.07%). The rarefied data contained 4,810 unique OTUs (3 of which were archaeal).
Microbial community characterization.
We defined seawater OTUs to be OTUs with a relative abundance of 1.0% or greater in the unfiltered seawater (before being seeded with sewage). Sewage OTUs were defined to be OTUs with a relative abundance of 1.0% or greater in the initial filtered seawater plus sieved sewage mixture (containing only bacteria from sewage). The lowest taxonomic designations provided by QIIME using the Greengenes database for the seawater OTUs and sewage OTUs and their relative abundances are shown in Tables 1 and 2, respectively.
TABLE 1.
Seawater OTUs
| Seawater OTUa | Relative abundance (%) |
|---|---|
| Octadecabacter (genus) | 8.4 |
| Pelagibacteraceae (family) | 5.4 |
| Tenacibaculum (genus) | 5.0 |
| Candidatus Portiera (genus) | 3.3 |
| Flavobacteriaceae I (family) | 3.1 |
| Flavobacteriaceae II (family) | 2.1 |
| Haptophyceae (order) | 2.0 |
| Olleya (genus) | 2.0 |
| Polaribacter (genus) | 1.9 |
| Mamiellaceae (family) | 1.9 |
| Flavobacteriaceae III (family) | 1.9 |
| Cryomorphaceae (family) | 1.8 |
| Bacteroidetes (phylum) | 1.6 |
| Stramenopiles (order) | 1.3 |
| Flavobacteriaceae IV (family) | 1.2 |
| Flavobacteriaceae V (family) | 1.1 |
| Flavobacteriaceae VI (family) | 1.1 |
The lowest taxonomic designation (and its level) assigned to the seawater OTUs through QIIME using the Greengenes database. Six unique seawater OTUs were assigned to the Flavobacteriaceae (denoted by roman numerals).
TABLE 2.
Sewage OTUs
| Sewage OTUa | Relative abundance (%) |
|---|---|
| Prevotella copri (species) | 6.0 |
| Acinetobacter johnsonii (species) | 4.1 |
| Faecalibacterium prausnitzii (species) | 3.6 |
| Roseburia (genus) | 3.3 |
| Cloacibacterium (genus) | 2.5 |
| Blautia (genus) | 2.4 |
| Comamonadaceae (genus) | 2.3 |
| Bacteroides (genus) | 1.7 |
| Bifidobacterium adolescentis (species) | 1.6 |
| Lachnospiraceae (genus) | 1.5 |
| Bacteroides uniformis (species) | 1.4 |
| Lactobacillus (genus) | 1.4 |
| Prevotella (genus) | 1.4 |
| Bacteroides ovatus (species) | 1.1 |
| Coprococcus (genus) | 1.1 |
| Enhydrobacter (genus) | 1.0 |
The lowest taxonomic designation (and its level) assigned to the sewage OTUs through QIIME using the Greengenes database.
Sewage OTUs dominated the initial unfiltered seawater plus sieved sewage mixture (at 0 h), even though this mixture also contained bacteria from seawater. All the sewage OTUs and none of the seawater OTUs had a relative abundance of 1.0% or more in the unfiltered seawater plus sieved sewage mixture at 0 h.
All samples, including the unfiltered seawater (before being seeded with sewage), were used to identify the 20 most abundant OTUs in the data set. Of the 20 most abundant OTUs, 14 OTUs were sewage OTUs and 0 were seawater OTUs, based on the definitions above. The relative abundance of the 20 most abundant OTUs for each microcosm treatment at each time point is shown in Fig. S2 in the supplemental material (see also Table S2).
The effect of sunlight, marine microbiota, and aging on community diversity and composition.
The observed richness and alpha diversity, quantified using the Shannon-Weaver diversity index (62), were calculated for every sample to investigate changes at the community level over time within each treatment. Observed richness varied between 240 and 1,246, while the Shannon-Weaver index varied from 1.7 to 5.5 (see Table S1 in the supplemental material). Both observed richness and the Shannon-Weaver index remained relatively constant in both the dark filtered and unfiltered microcosms and decreased with time in the light filtered and unfiltered microcosms (P < 0.001 and Pearson's r = −0.9 for both light filtered and unfiltered microcosms). While a decrease in observed richness and alpha diversity was observed in the light filtered and unfiltered microcosms, the total number of bacterial 16S rRNA gene copies in the sunlit microcosms remained relatively stable over time (see Fig. S3 in the supplemental material).
A Mantel test investigated whether a relationship between community composition and time existed within and across microcosm treatments. For each of the four microcosm treatments, the community composition was significantly more similar (as determined by the Bray-Curtis similarity coefficient) the closer the samples were collected in time (Mantel test for each microcosm treatment, 0.71 < r < 0.94 and P < 0.05). We also grouped the data from all the microcosms and found a significant relationship between community composition and time across all microcosm treatments (Mantel test, r = 0.50 and P = 0.001). Results indicate that as the microbial community in seawater ages, the community composition changes significantly both in the presence and in the absence of sunlight and marine microbiota.
Similarities in community composition between samples were visualized using a nonmetric multidimensional scaling (nMDS) ordination plot (see Fig. S4 in the supplemental material). Samples from the dark filtered and dark unfiltered microcosms formed a distinct cluster in the ordination plot, while samples from the light filtered and light unfiltered microcosms formed another distinct cluster. Also, dark filtered and unfiltered microcosms collected close together in time clustered, and light filtered and unfiltered microcosms collected close together in time clustered. These results support the Mantel test results and indicate that while aging changed the community composition, it did so in different ways in the dark microcosms and the light microcosms. The community compositions of samples exposed to sunlight cycles were significantly more similar to each other than to the community compositions of samples kept in the dark (ANOSIM, global R = 0.33 and P = 0.009). The presence of marine microbiota, however, did not significantly affect the similarity of the community composition between samples (ANOSIM, global R = −0.013 and P = 0.46).
To better understand the observed changes in community composition, we examined temporal trends in the relative abundance of the 20 most abundant OTUs for each microcosm treatment. The relative abundance of each of the 20 most abundant OTUs was normalized by the maximum relative abundance of that OTU in a given treatment, here referred to as the normalized relative abundance (37). Normalized relative abundances ranged from 0 to 1. Dark filtered microcosms showed trends similar to those for the dark unfiltered microcosms. The majority of OTUs showed a decrease in normalized relative abundance over time for each treatment (from a normalized relative abundance of 1 to 0) (Fig. 2). The normalized relative abundance of 4 (out of 20) OTUs, however, increased over the course of the experiment in both microcosms (from 0 to 1). The light microcosms showed patterns similar to those shown by the dark microcosms, except that the changes between time points were more pronounced. Because there were changes in OTU relative abundances in all microcosms, changes in the abundances of particular OTUs due to a particular treatment (sunlight or the presence of microbiota) were assessed by comparing treatments to an appropriate reference, as described below.
FIG 2.

Changes in normalized relative abundance for the 20 most abundant OTUs in the NGS data set. The size of the symbol represents the relative abundance of the OTU normalized by the highest relative abundance for that OTU in that microcosm type (dark filtered, dark unfiltered, light filtered, light unfiltered). The largest shape is equal to 1, while the smallest is equal to 0. *, an OTU that was also classified as a sewage OTU, defined in the Materials and Methods section to be an OTU with a relative abundance of 1.0% or greater in the initial filtered seawater plus sieved sewage mixture.
Significant differences in OTU abundance between treatments.
Whether or not there was a significant difference in the relative abundance of each OTU between treatments was evaluated at 0 h, 12 h, 24 h, 36 h, and 48 h using the edgeR package (60, 65). The FDR cutoff was set at 0.1 (10%). Four comparisons were made to test four specific hypotheses about the effects of the microcosm treatments (described in Materials and Methods).
The first comparison tested whether OTU abundance was lower due to the presence of marine microbiota in the absence of sunlight by comparing OTU abundance in the dark unfiltered and the dark filtered microcosms. No OTUs were significantly less abundant in the dark unfiltered than the dark filtered microcosms at 0 h, 12 h, 36 h, and 48 h (Table 3). At 24 h, 6 OTUs were significantly less abundant in the dark unfiltered relative to the dark filtered microcosms (FDR < 0.1) (see Table S3 in the supplemental material). The lowest taxonomic designation of these 6 OTUs was Acinetobacter (2/6 OTUs), Pseudoalteromonas (2/6), Cloacibacterium (1/6), and Gammaproteobacteria (1/6).
TABLE 3.
Number of OTUs with significantly reduced abundances (FDR < 10%) due to sunlight and marine microbiotaa
| Time (h) | OTU reduced by sunlight, −biota (LF vs DF) | OTU reduced by sunlight, +biota (LU vs DU) | OTU reduced by biota, −sunlight (DU vs DF) | OTU reduced by biota, +sunlight (LU vs LF) |
|---|---|---|---|---|
| 0 | — | — | 0 | 0 |
| 12 | 0 | 0 | 0 | 0 |
| 24 | 22 | 7 | 6 | 2 |
| 36 | 19 | 28 | 0 | 0 |
| 48 | 31 | 22 | 0 | 0 |
—, no data were available for the comparisons between sunlit and dark microcosms at 0 h because the microcosms had not yet been exposed to the sunlight treatment. −biota, absence of biota; +biota, presence of biota; LF, light filtered; DF, dark filtered; LU, light unfiltered; DU, dark unfiltered.
The second comparison tested whether OTU abundance was lower due to the presence of marine microbiota in the presence of sunlight by comparing OTU abundance in the light unfiltered and the light filtered microcosms. No OTUs were significantly less abundant in the light unfiltered relative to the light filtered microcosms at 0 h, 12 h, 36 h, and 48 h (Table 3). At 24 h, 2 OTUs were significantly less abundant in the light unfiltered relative to the filtered microcosms (FDR < 0.1). The lowest taxonomic designations of the 2 OTUs were Paludibacter and Streptococcus minor (see Table S4 in the supplemental material).
The third comparison tested whether OTU abundance was lower due to the presence of sunlight in the absence of marine microbiota by comparing OTU abundance in the light filtered and dark filtered microcosms. OTU abundances in the light filtered and dark filtered microcosms at 12 h were not significantly different. At 24 h, 36 h, and 48 h there were 22, 19, and 31 OTUs, respectively, that were significantly less abundant in the light filtered microcosm relative to the dark filtered microcosm (FDR < 0.1) (Table 3). The OTUs that were significantly less abundant represented 47 unique OTUs. The family-level taxonomic assignments of the 47 OTUs were Colwelliaceae (8/47 OTUs less abundant in the light filtered microcosm than the dark filtered microcosm), Pseudoalteromonadaceae (7/47), Oceanospirillaceae (6/47), Moraxellaceae (4/47), Flavobacteriaceae (3/47), Psychromonadaceae (2/47), Ruminococcaceae (2/47), Bacteroidaceae (2/47), and Vibrionaceae (2/47) (the remaining 11 families with only one OTU assignment are not listed here but can be found in Table S5 in the supplemental material). One of the 47 OTUs was a sewage OTU (the lowest taxonomic designation of Flavobacteriaceae, Olleya), and 2 OTUs were among the 20 most abundant OTUs overall (lowest taxonomic designations of Colwelliaceae and Pseudoalteromonas).
The fourth comparison tested whether OTU abundance was lower due to the presence of sunlight in the presence of marine microbiota by comparing OTU abundance in the light unfiltered and dark unfiltered microcosms. No OTUs were significantly less abundant in the light unfiltered relative to the dark unfiltered microcosms at 12 h. At 24 h, 36 h, and 48 h there were 7, 28, and 22 OTUs, respectively, that were significantly less abundant (FDR < 0.1) (Table 3). The OTUs that were significantly less abundant represented 37 unique OTUs. The family-level taxonomic assignments of the 37 OTUs (grouped at the family level) were Oceanospirillaceae (10/37 OTUs), Colwelliaceae (8/37), Flavobacteriaceae (3/37), Lachnospiraceae (2/37), Pseudoalteromonadaceae (2/37), Alteromonadaceae (1/37), Lactobacillales (1/37), Moraxellaceae (1/37), Psychromonadaceae (1/37), Vibrionaceae (1/37), Weeksellaceae (1/37), and 6 OTUs whose lowest taxonomic assignment was only at the class or order level (see Table S6 in the supplemental material). Two of the 37 less abundant OTUs were sewage OTUs (lowest taxonomic designations of Lactobacillus and Cloacibacterium), and 1 OTU was among the 20 most abundant OTUs overall (lowest taxonomic designation of Colwelliaceae).
Decline of the sewage microbial community over time.
SourceTracker was used to estimate the proportion of all OTUs in each sample that originated from the sieved sewage. The proportion of OTUs from sewage decreased in all the microcosms with time (Fig. 3). The change was the most pronounced in the microcosms exposed to sunlight, decreasing from approximately 100% to 80% in the first 24 h and then to 9% at 48 h in the sunlit microcosm with no microbiota (light filtered microcosm) and from 85% to 24% in the first 24 h and then to 3% at 48 h in the sunlit microcosm with microbiota (light unfiltered microcosm). In the dark filtered and dark unfiltered microcosms, the proportion of OTUs from sewage decreased from approximately 100% to 33% at 48 h and from 85% to 33% at 48 h, respectively.
FIG 3.
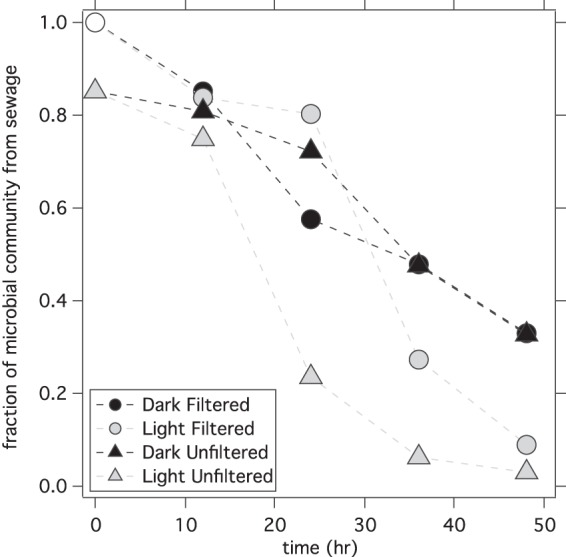
Changes in the proportion of the sewage microbial community with time. The open circle at 1.0 represents the assumed fraction of the microbial community from sewage in the filtered-seawater-plus-sewage mixture at 0 h. The fraction of the microbial community from sewage in the unfiltered-seawater-plus-sewage mixture at 0 h is less than 1 because the community contains microorganisms from both sewage and seawater. Time since the start of the experiment is shown on the x axis.
DISCUSSION
While previous research has shown that exposure to environmental stressors affects enteric bacterial concentrations when the effects are measured by culture-based and molecular methods (31, 38, 39, 66), no work has explored how environmental stressors impact the entire community of sewage-contaminated waters. The results presented here illustrate the effects of natural sunlight cycles and marine microbiota on the microbial community of sewage-impacted seawater over 48 h and provide insights into the use of NGS for microbial source tracking.
Sunlight affects microbial community composition and OTU abundance.
While community composition and OTU abundance changed with time in all microcosm treatments, the changes in the sunlit microcosms were more pronounced than those in the dark microcosms. The number and evenness of OTUs present in the sunlit microcosms decreased with time but did not decrease in the dark microcosms. The microbial communities in the sunlit microcosms tended to be more similar to each other than those in the dark microcosms, and a number of OTUs were significantly less abundant in the sunlit than the dark microcosms. The taxonomic designations of the sunlight-sensitive OTUs in treatments with and without marine microbiota were very similar. However, a common trait was not inferred from their designations. They included aerobes and anaerobes, Gram-negative and Gram-positive bacteria, and bacteria found in environments ranging from the human gut to soils, plants, marine waters, and freshwaters (67). It is important to note that these microcosms mimic conditions in clear, shallow seawater, where all wavelengths of sunlight may be received. This would mimic conditions that occur when freshwater effluent from a broken sewer pipe or sewer overflow floats at the surface of the coastal ocean or conditions that occur when a contaminated water column is particularly clear (68). It is possible that the effect of sunlight on the microbial community would be diminished in colored or very deep water if UVB and UVA wavelengths are filtered from the light as it passes through the water column (69).
Marine microbiota minimally affect microbial community composition and OTU abundance.
While the community composition and OTU abundance changed with time in all microcosm treatments, the effect of marine microbiota in particular (including viruses, bacteria, and protozoa of seawater origin) on the community composition and OTU abundance was minimal. Microbial community composition was not significantly different between microcosms with and without microbiota, holding sunlight exposure status constant. Very few OTUs (8 out of 5,338) were significantly less abundant in microcosms with microbiota relative to in microcosms without microbiota. This finding suggests that the marine microbiota are not particularly important in shaping the sewage microbial community over the time scale of our experiment. However, this conclusion relies on the assumption that destruction of a microbe by marine microbiota results in the destruction of the 16S rRNA gene segment targeted by NGS. In fact, it is possible that the segment remains intact and trapped inside the dialysis membrane. It is also important to note that our experiments were designed to test only for the effects of marine microbiota and not those of sewage-derived microorganisms of less than 20 μm that might contribute to NGS target degradation.
Previous research showed that indigenous biota can have a negative impact on fecal indicator bacterial concentrations due to predation and competition (36, 40, 70–75). Declines in the concentration of culturable Escherichia coli and Enterococcus faecalis have been observed when indigenous microbiota were present in both freshwater and marine water (72, 73). Few studies have investigated the effect of indigenous biota on the decay of nucleic acid targets. A recent study comparing the effects of sunlight and indigenous microbiota on the decay of nucleic acid targets (Enterococcus 23S rRNA gene, human-specific Bacteroidales 16S rRNA gene, and general Bacteroidales 16S rRNA gene) showed that sunlight had a significant effect at between 0 and 120 hours and indigenous microbiota had a significant effect after 120 hours (44). This is consistent with the results presented herein and suggests that we might have observed an effect attributable to marine microbiota had our experiments continued past 48 h.
The microbial community in sewage changes as it ages in seawater.
Aging of the microbial community in sewage-impacted seawater for 48 hours resulted in changes to both the OTU abundance and the community composition in the presence and absence of sunlight and marine microbiota. However, aging had an uneven effect on OTU abundance (in all microcosm treatments), as illustrated by changes in the 20 most abundant OTUs. The relative abundances of the 20 most abundant OTUs both decreased and increased with time, depending on the OTU. OTUs that increased (designated the Colwelliaceae, Flavobacteriaceae, and Pseudoalteromonas) were not sewage OTUs. OTUs that decreased in relative abundance included many sewage OTUs (designated Roseburia, Prevotella, Prevotella copri, Enhydrobacter, Coprococcus, Comamonadaceae, Cloacibacterium, Blautia, Bifidobacterium adolescentis, Bacteroides uniformis, Bacteroides ovatus, Bacteroides, Acinetobacter johnsonii). It is not surprising that sewage OTUs decreased in relative abundance, given that many feces-associated bacteria are anaerobes and unlikely to survive in oxic seawater (76); however, the time scale of the decrease is of particular interest for the use of NGS for microbial source tracking applications.
Implications for the use of NGS to identify sources of microbial pollution.
Changes to community composition over time in all microcosm treatments indicate that the longer that the sewage was aged, the more dissimilar it became from fresh sewage (defined as sewage collected directly from the wastewater treatment plant). After 48 hours, only between 3% and 33% (depending on the microcosm treatment) of the OTUs were identified by SourceTracker as being from the fresh sewage community, despite the fact that 100% and 85% of the bacteria present in the filtered and unfiltered microcosms, respectively, originated from sewage. While caution should be taken when extrapolating the microcosm results to the natural environment, this result suggests that the sewage signal detected by NGS from a sewage spill event might be difficult to detect after 48 hours. This would make it difficult to confidently link an aged sewage microbial community in coastal waters to its source using NGS. It is also important to note that NGS gives relative abundance and not concentrations. Thus, while the sewage signal detected by NGS decreases in the first 48 hours, the concentration of viable sewage bacteria and, potentially, pathogens after 48 hours is unknown. We measured the concentration of culturable enterococci in the microcosms and found that they declined rapidly in samples receiving the sunlight treatment but did not decline in samples kept in the dark (see Fig. S3 in the supplemental material). Thus, the fate of enterococci mirrored the decline in the sewage bacterial community observed in the sunlit microcosms using SourceTracker but not the decline observed in the dark microcosms.
One limitation of the present study is the lack of biological replicates. Lacking biological replicates, we used the dark filtered microcosms at 0 h and 12 h to estimate the biological coefficient of variation used in edgeR (see the explanation in the supplemental material). These samples were not exact replicates, so the estimate of biological variability used in our study may be an overestimate. An overestimate of biological variability could have resulted in the identification of fewer OTUs being identified as having significantly different abundances in the compared treatments. Unlike edgeR, SourceTracker does not require biological replicates; however, there is some error associated with the proportion calculation that replicates would likely help to minimize (Dan Knights, personal communication).
This study investigated the temporal stability of sewage microbial communities in seawater by NGS. Temporal stability is not typically considered in the development of microbial source tracking tools. However, it is extremely important to understand the relative persistence of the targets used to identify microbial pollution sources and the targets used to indicate the presence of microbial pollution. A mismatch between persistence could be problematic and lead to the misidentification of pollution sources. Future work should consider the relative persistence of microbial source tracking targets, fecal indicator bacteria, and pathogens. This metric should be considered just as important as sensitivity and specificity (24) in evaluating the performance of microbial source tracking tools. Microbial source tracking models that incorporate pollutant aging (77) would further our ability to identify and remediate microbial pollution in coastal waters.
Supplementary Material
ACKNOWLEDGMENTS
We thank Allison Pieja and Raylan Willis for their assistance with the experiments.
This research was funded by the National Science Foundation (NSF OCE-1129270 and NSF OCE-0910491).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03950-14.
REFERENCES
- 1.Shuval H. 2003. Estimating the global burden of thalassogenic diseases: human infectious diseases caused by wastewater pollution in the environment. J Water Health 1:53–64. [PubMed] [Google Scholar]
- 2.U.S. Environmental Protection Agency. 2004. Office of Water report to Congress: impacts and control of CSOs and SSOs. U.S. Environmental Protection Agency, Washington, DC. [Google Scholar]
- 3.Ashbolt NJ, Grabow WOK, Snozzi M. 2001. Indicators of microbial water quality. In Fewtrell L, Bartram J (ed), World Health Organization (WHO) water quality: guidelines, standards and health. IWA Publishing, London, United Kingdom. [Google Scholar]
- 4.U.S. Environmental Protection Agency. 2006. Method 1600: enterococci in water by membrane filtration using membrane-Enterococcus indoxyl-β-d-glucoside agar (mEI). Report EPA-821-R-06-009. U.S. Environmental Protection Agency, Washington, DC. [Google Scholar]
- 5.U.S. Environmental Protection Agency. 2002. Method 1603: Escherichia coli (E. coli) in water by membrane filtration using modified membrane-thermotolerant Escherichia coli agar (modified mTEC). Report EPA 821-R-02-023. U.S. Environmental Protection Agency, Washington, DC. [Google Scholar]
- 6.Boehm AB. 2007. Enterococci concentrations in diverse coastal environments exhibit extreme variability. Environ Sci Technol 41:8227–8232. doi: 10.1021/es071807v. [DOI] [PubMed] [Google Scholar]
- 7.Boehm AB, Yamahara KM, Love DC, Peterson BM, McNeill K, Nelson KL. 2009. Covariation and photoinactivation of traditional and novel indicator organisms and human viruses at a sewage-impacted marine beach. Environ Sci Technol 43:8046–8052. doi: 10.1021/es9015124. [DOI] [PubMed] [Google Scholar]
- 8.Ervin JS, Russell TL, Layton BA, Yamahara KM, Wang D, Sassoubre LM, Cao Y, Kelty CA, Sivaganesan M, Boehm AB, Holden PA, Weisberg SB, Shanks OC. 2013. Characterization of fecal concentrations in human and other animal sources by physical, culture-based, and quantitative real-time PCR methods. Water Res 47:6873–6882. doi: 10.1016/j.watres.2013.02.060. [DOI] [PubMed] [Google Scholar]
- 9.Yamahara KM, Layton BA, Santoro AE, Boehm AB. 2007. Beach sands along the California coast are diffuse sources of fecal bacteria to coastal waters. Environ Sci Technol 41:4515–4521. doi: 10.1021/es062822n. [DOI] [PubMed] [Google Scholar]
- 10.Solo-Gabrielle HM, Wolfert MA, Desmarais TR, Palmer CJ. 2000. Sources of Escherichia coli in a coastal subtropical environment. Appl Environ Microbiol 66:230–237. doi: 10.1128/AEM.66.1.230-237.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desmarais TR, Solo-Gabriele HM, Palmer CJ. 2002. Influence of soil on fecal indicator organisms in a tidally influenced subtropical environment. Appl Environ Microbiol 68:1165–1172. doi: 10.1128/AEM.68.3.1165-1172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halliday E, Gast RJ. 2011. Bacteria in beach sands: an emerging challenge in protecting coastal water quality and bather health. Environ Sci Technol 45:370–379. doi: 10.1021/es102747s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitman RL, Nevers MB. 2003. Foreshore sand as a source of Escherichia coli in nearshore water of a Lake Michigan beach. Appl Environ Microbiol 69:5555–5562. doi: 10.1128/AEM.69.9.5555-5562.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imamura G, Thompson RM, Boehm AB, Jay JA. 2011. Beach wrack is a reservoir for faecal indicator bacteria along the California coast. FEMS Microbiol Ecol 77:40–49. doi: 10.1111/j.1574-6941.2011.01082.x. [DOI] [PubMed] [Google Scholar]
- 15.Whitman RL, Shively DA, Pawlik H, Nevers MB, Byappanahalli MN. 2003. Occurrence of Escherichia coli and enterococci in Cladophora (Chlorophyta) in nearshore water and beach sand of Lake Michigan. Appl Environ Microbiol 69:4714–4719. doi: 10.1128/AEM.69.8.4714-4719.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byappanahalli MN, Shively DA, Nevers MB, Sadowsky MJ, Whitman RL. 2003. Growth and survival of Escherichia coli and enterococci populations in the macro-alga Cladophora (Chlorophyta). FEMS Microbiol Ecol 46:203–211. doi: 10.1016/S0168-6496(03)00214-9. [DOI] [PubMed] [Google Scholar]
- 17.Badgley BD, Thomas FI, Harwood VJ. 2011. Quantifying environmental reservoirs of fecal indicator bacteria associated with sediment and submerged aquatic vegetation. Environ Microbiol 13:932–942. doi: 10.1111/j.1462-2920.2010.02397.x. [DOI] [PubMed] [Google Scholar]
- 18.Soller JA, Schoen ME, Bartrand T, Ravenscroft JE, Ashbolt NJ. 2010. Estimated human health risks from exposure to recreational waters impacted by human and non-human sources of faecal contamination. Water Res 44:4674–4691. doi: 10.1016/j.watres.2010.06.049. [DOI] [PubMed] [Google Scholar]
- 19.Haugland RA, Siefring SC, Wymer LJ, Brenner KP, Dufour AP. 2005. Comparison of Enterococcus measurements in freshwater at two recreational beaches by quantitative polymerase chain reaction and membrane filter culture analysis. Water Res 39:559–568. doi: 10.1016/j.watres.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 20.Wade TJ, Calderon RL, Sams E, Beach M, Brenner KP, Williams AH, Dufour AP. 2006. Rapidly measured indicators of recreational water quality are predictive of swimming-associated gastrointestinal illness. Environ Health Perspect 114:24–28. doi: 10.1289/ehp.8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.U.S. Environmental Protection Agency. 2012. Method 1611: enterococci in water by TaqMan® quantitative polymerase chain reaction (qPCR) assay. U.S. Environmental Protection Agency, Washington, DC. [Google Scholar]
- 22.Bernhard AE, Field KG. 2000. Identification of nonpoint sources of fecal pollution in coastal waters by using host-specific 16S ribosomal DNA genetic markers from fecal anaerobes. Appl Environ Microbiol 66:1587–1594. doi: 10.1128/AEM.66.4.1587-1594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seurinck S, Defoirdt T, Verstraete W, Siciliano SD. 2005. Detection and quantification of the human-specific HF183 Bacteroides 16S rRNA genetic marker with real-time PCR for assessment of human faecal pollution in freshwater. Environ Microbiol 7:249–259. doi: 10.1111/j.1462-2920.2004.00702.x. [DOI] [PubMed] [Google Scholar]
- 24.Boehm AB, Van de Werfhorst LC, Griffith JF, Holden PA, Jay JA, Shanks OC, Wang D, Weisberg SB. 2013. Performance of forty-one microbial source tracking methods: a twenty-seven lab evaluation study. Water Res 47:6812–6828. doi: 10.1016/j.watres.2012.12.046. [DOI] [PubMed] [Google Scholar]
- 25.Cao Y, Van De Werfhorst LC, Scott EA, Raith MR, Holden PA, Griffith JF. 2013. Bacteroidales terminal restriction fragment length polymorphism (TRFLP) for fecal source differentiation in comparison to and in combination with universal bacteria TRFLP. Water Res 47:6944–6955. doi: 10.1016/j.watres.2013.03.060. [DOI] [PubMed] [Google Scholar]
- 26.Dubinsky EA, Esmaili L, Hulls JR, Cao Y, Griffith JF, Andersen GL. 2012. Application of phylogenetic microarray analysis to discriminate sources of fecal pollution. Environ Sci Technol 46:4340–4347. doi: 10.1021/es2040366. [DOI] [PubMed] [Google Scholar]
- 27.Newton RJ, Bootsma MJ, Morrison HG, Sogin ML, McLellan SL. 2013. A microbial signature approach to identify fecal pollution in the waters off an urbanized coast of Lake Michigan. Microb Ecol 65:1011–1023. doi: 10.1007/s00248-013-0200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Unno T, Jang J, Han D, Kim JH, Sadowsky MJ, Kim OS, Chun J, Hur HG. 2010. Use of barcoded pyrosequencing and shared OTUs to determine sources of fecal bacteria in watersheds. Environ Sci Technol 44:7777–7782. doi: 10.1021/es101500z. [DOI] [PubMed] [Google Scholar]
- 29.Cao Y, Van De Werfhorst LC, Dubinsky EA, Badgley BD, Sadowsky MJ, Andersen GL, Griffith JF, Holden PA. 2013. Evaluation of molecular community analysis methods for discerning fecal sources and human waste. Water Res 47:6862–6872. doi: 10.1016/j.watres.2013.02.061. [DOI] [PubMed] [Google Scholar]
- 30.Barnes MA, Turner CR, Jerde CL, Renshaw MA, Chadderton WL, Lodge DM. 2014. Environmental conditions influence eDNA persistence in aquatic systems. Environ Sci Technol 48:1819–1827. doi: 10.1021/es404734p. [DOI] [PubMed] [Google Scholar]
- 31.Davies-Colley RJ, Donnison AM, Speed DJ, Ross CM, Nagels JW. 1999. Inactivation of feacal indicator microorganisms in waste stabilization ponds: interactions of environmental factors with sunlight. Water Res 33:1220–1230. doi: 10.1016/S0043-1354(98)00321-2. [DOI] [Google Scholar]
- 32.Anderson KL, Whitlock JE, Harwood VJ. 2005. Persistence and differential survival of fecal indicator bacteria in subtropical waters and sediments. Appl Environ Microbiol 71:3041–3048. doi: 10.1128/AEM.71.6.3041-3048.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walters SP, Yamahara KM, Boehm AB. 2009. Persistence of nucleic acid markers of health-relevant organisms in seawater microcosms: implications for their use in assessing risk in recreational waters. Water Res 43:4929–4939. doi: 10.1016/j.watres.2009.05.047. [DOI] [PubMed] [Google Scholar]
- 34.Sinton L, Hall C, Braithwaite R. 2007. Sunlight inactivation of Campylobacter jejuni and Salmonella enterica, compared with Escherichia coli, in seawater and river water. J Water Health 5:357–365. doi: 10.2166/wh.2007.031. [DOI] [PubMed] [Google Scholar]
- 35.Sinton LW, Hall CH, Lynch PA, Davies-Colley RJ. 2002. Sunlight inactivation of fecal indicator bacteria and bacteriophages from waste stabilization pond effluent in fresh and saline waters. Appl Environ Microbiol 68:1122–1131. doi: 10.1128/AEM.68.3.1122-1131.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korajkic A, McMinn BR, Harwood VJ, Shanks OC, Fout GS, Ashbolt NJ. 2013. Differential decay of enterococci and Escherichia coli originating from two fecal pollution sources. Appl Environ Microbiol 79:2488–2492. doi: 10.1128/AEM.03781-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiao TH, Clancy TM, Pinto A, Xi C, Raskin L. 2014. Differential resistance of drinking water bacterial populations to monochloramine disinfection. Environ Sci Technol 48:4038–4047. doi: 10.1021/es4055725. [DOI] [PubMed] [Google Scholar]
- 38.Okabe S, Shimazu Y. 2007. Persistence of host-specific Bacteroides-Prevotella 16S rRNA genetic markers in environmental waters: effects of temperature and salinity. Appl Microbiol Biotechnol 76:935–944. doi: 10.1007/s00253-007-1048-z. [DOI] [PubMed] [Google Scholar]
- 39.Green HC, Shanks OC, Sivaganesan M, Haugland RA, Field KG. 2011. Differential decay of human faecal Bacteroides in marine and freshwater. Environ Microbiol 13:3235–3249. doi: 10.1111/j.1462-2920.2011.02549.x. [DOI] [PubMed] [Google Scholar]
- 40.Kreader CA. 1998. Persistence of PCR-detectable Bacteroides distasonis from human feces in river water. Appl Environ Microbiol 64:4103–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dick LK, Stelzer EA, Bertke EE, Fong DL, Stoeckel DM. 2010. Relative decay of Bacteroidales microbial source tracking markers and cultivated Escherichia coli in freshwater microcosms. Appl Environ Microbiol 76:3255–3262. doi: 10.1128/AEM.02636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogers SW, Donnelly M, Peed L, Kelty CA, Mondal S, Zhong Z, Shanks OC. 2011. Decay of bacterial pathogens, fecal indicators, and real-time quantitative PCR genetic markers in manure-amended soils. Appl Environ Microbiol 77:4839–4848. doi: 10.1128/AEM.02427-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bae S, Wuertz S. 2009. Rapid decay of host-specific fecal Bacteroidales cells in seawater as measured by quantitative PCR with propidium monoazide. Water Res 43:4850–4859. doi: 10.1016/j.watres.2009.06.053. [DOI] [PubMed] [Google Scholar]
- 44.Korajkic A, McMinn BR, Shanks OC, Sivaganesan M, Fout GS, Ashbolt NJ. 2014. Biotic interactions and sunlight affect persistence of fecal indicator bacteria and microbial source tracking genetic markers in the upper Mississippi River. Appl Environ Microbiol 80:3952–3961. doi: 10.1128/AEM.00388-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reference deleted.
- 46.Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc Natl Acad Sci U S A 103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 49.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG, Angenent LT, Knight R, Ley RE. 2012. Impact of training sets on classification of high-throughput bacterial 16S rRNA gene surveys. ISME J 6:94–103. doi: 10.1038/ismej.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hewitt KM, Mannino FL, Gonzalez A, Chase JH, Caporaso JG, Knight R, Kelley ST. 2013. Bacterial diversity in two neonatal intensive care units (NICUs). PLoS One 8:e54703. doi: 10.1371/journal.pone.0054703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bryant JA, Stewart FJ, Eppley JM, DeLong EF. 2012. Microbial community phylogenetic and trait diversity declines with depth in a marine oxygen minimum zone. Ecology 93:1659–1673. doi: 10.1890/11-1204.1. [DOI] [PubMed] [Google Scholar]
- 56.Flores GE, Bates ST, Knights D, Lauber CL, Stombaugh J, Knight R, Fierer N. 2011. Microbial biogeography of public restroom surfaces. PLoS One 6:e28132. doi: 10.1371/journal.pone.0028132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McMurdie PJ, Holmes S. 2012. Phyloseq: a bioconductor package for handling and analysis of high-throughput phylogenetic sequence data. Pac Symp Biocomput 2012:235–246. doi: 10.1142/9789814366496_0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Wagner H. 2013. vegan: community ecology package. R package version 2.2-0. http://CRAN.R-project.org/package=vegan. [Google Scholar]
- 60.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, Bushman FD, Knight R, Kelley ST. 2011. Bayesian community-wide culture-independent microbial source tracking. Nat Methods 8:761–763. doi: 10.1038/nmeth.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shannon CE, Weaver W. 1949. The mathematical theory of communication. University of Illinois Press, Urbana, IL. [Google Scholar]
- 63.Hill TC, Walsh KA, Harris JA, Moffett BF. 2003. Using ecological diversity measures with bacterial communities. FEMS Microbiol Ecol 43:1–11. doi: 10.1111/j.1574-6941.2003.tb01040.x. [DOI] [PubMed] [Google Scholar]
- 64.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57:289–300. [Google Scholar]
- 65.McCarthy DJ, Chen Y, Smyth GK. 2012. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Curtis TP, Mara DD, Silva SA. 1992. Influence of pH, oxygen, and humic substances on ability of sunlight to damage fecal coliforms in waste stabilization pond water. Appl Environ Microbiol 58:1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenberg E. 2013. The prokaryotes, 4th ed. Springer, New York, NY. [Google Scholar]
- 68.Boehm AB, Fuhrman JA, Mrse RD, Grant SB. 2003. A tiered approach for the identification of a human fecal pollution source at a recreational beach: Case study at Avalon Bay, Catalina Island, California. Environ Sci Technol 37:673–680. doi: 10.1021/es025934x. [DOI] [PubMed] [Google Scholar]
- 69.Jerlov NG. 1950. Ultra-violet radiation in the sea. Nature 166:111–112. doi: 10.1038/166111a0. [DOI] [PubMed] [Google Scholar]
- 70.Jannasch HW. 1968. Competitive elimination of Enterobacteriaceae from seawater. Appl Microbiol 16:1616–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feng F, Goto D, Yan T. 2010. Effects of autochthonous microbial community on the die-off of fecal indicators in tropical beach sand. FEMS Microbiol Ecol 74:214–225. doi: 10.1111/j.1574-6941.2010.00921.x. [DOI] [PubMed] [Google Scholar]
- 72.Korajkic A, Wanjugi P, Harwood VJ. 2013. Indigenous microbiota and habitat influence Escherichia coli survival more than sunlight in simulated aquatic environments. Appl Environ Microbiol 79:5329–5337. doi: 10.1128/AEM.01362-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wanjugi P, Harwood VJ. 2013. The influence of predation and competition on the survival of commensal and pathogenic fecal bacteria in aquatic habitats. Environ Microbiol 15:517–526. doi: 10.1111/j.1462-2920.2012.02877.x. [DOI] [PubMed] [Google Scholar]
- 74.Harwood VJ, Staley C, Badgley BD, Borges K, Korajkic A. 2014. Microbial source tracking markers for detection of fecal contamination in environmental waters: relationships between pathogens and human health outcomes. FEMS Microbiol Rev 38:1–40. doi: 10.1111/1574-6976.12031. [DOI] [PubMed] [Google Scholar]
- 75.Boehm AB, Keymer DP, Shellenbarger GG. 2005. An analytical model of enterococci inactivation, grazing, and transport in the surf zone of a marine beach. Water Res 39:3565–3578. doi: 10.1016/j.watres.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 76.Kovatcheva-Datchary P, Tremaroli V, Backhed F. 2013. The gut microbiota. In Rosenberg E. (ed), The prokaryotes—human microbiology. Springer, Berlin, Germany. [Google Scholar]
- 77.Wang D, Farnleitner AH, Field KG, Green HC, Shanks OC, Boehm AB. 2013. Enterococcus and Escherichia coli fecal source apportionment with microbial source tracking genetic markers—is it feasible? Water Res 47:6849–6861. doi: 10.1016/j.watres.2013.02.058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.