

Abstract
GM-CSF plays a role in the nervous system, particularly in cases of injury. A therapeutic effect of GM-CSF has been reported in rat models of various central nervous system injuries. We previously showed that GM-CSF could enhance long-term recovery in a rat spinal cord injury model, inhibiting glial scar formation and increasing the integrity of axonal structure. Here, we investigated molecular the mechanism(s) by which GM-CSF suppressed glial scar formation in an in vitro system using primary astrocytes treated with TGF-β. GM-CSF repressed the expression of chondroitin sulfate proteoglycan (CSPG) core proteins in astrocytes treated with TGF-β. GM-CSF also inhibited the TGF-β-induced Rho-ROCK pathway, which is important in CSPG expression. Finally, the inhibitory effect of GM-CSF was blocked by a JAK inhibitor. These results may provide the basis for GM-CSF’s effects in glial scar inhibition and ultimately for its therapeutic effect on neural cell injuries. [BMB Reports 2014; 47(12): 679-684]
Keywords: CSPG core proteins, Glial scar, GM-CSF, Primary astrocytes, TGF-β
INTRODUCTION
Spinal cord injury (SCI) affects many people and can result in severe neurological damage. Following SCI, the central region of the spinal cord may become critically damaged by primary mechanical injury, developing hemorrhagic necrosis, which expands with time because of the activation of secondary injury processes involving apoptosis (1,2). Apoptosis plays an important role in neuronal cell death and the loss of neural function immediately after SCI. Thus, preventing apoptosis of neural cells has long been a major therapeutic principle in repairing SCI. Another problem after SCI is factors inhibiting neural regeneration. These include myelin debris-derived proteins, such as Nogo-A and myelin-associated glycoprotein (MAG), and glial scars, composed of reactive astrocytes and a network of chondroitin sulfate proteoglycans (CSPGs). In the case of glial scars, astrocytes expressing glial fibrillary acidic protein (GFAP) become hypertrophic and highly proliferative after SCI and eventually form a dense network of extracellular matrix around the cavity, thereby physically blocking axonal growth and the neural network (3,4). CSPG core proteins, such as NG2, neurocan, and phosphacan, are also upregulated and become part of the glial scar. Thus, suppression of glial scar formation and CSPG production is believed to provide a favorable environment for axonal regeneration and to be important for the repair of SCI (5).
Granulocyte macrophage colony stimulating factor (GM-CSF) is a well-known hematopoietic cytokine. It was originally identified because of its ability to stimulate the differentiation and function of hematopoietic cells. GM-CSF stimulates bone marrow stem cell proliferation and reduces leukocyte apoptosis while increasing white blood cell numbers in the peripheral blood. Due to its hematopoietic stimulating effects, GM-CSF has been used as a therapeutic cytokine in patients suffering from diseases related to bone marrow suppression. GM-CSF has also been suggested to play important roles in neural systems. GM-CSF induces microglial proliferation and activation, which can induce antigen presentation in neonates and be beneficial for axonal regeneration (6,7). GM-CSF can also function as a neurotrophic factor and induce the proliferation of neural progenitor cells (NPCs) in vitro (8,9). The therapeutic effects of GM-CSF are already well known in peripheral nervous system (PNS) injury, where it activates macrophages and Schwann cells to remove myelin debris (10-12). In central nervous system (CNS) injuries, GM-CSF has been shown to activate microglia to remove myelin debris and release brain-derived neurotrophic factor (BDNF), thereby inducing axonal regeneration and functional recovery after brain ischemic injury (13). We have also shown that GM-CSF has neuroprotective and anti-apoptotic activities in rat models of several CNS injuries including SCI, brain ischemia, and Parkinson’s disease (14-19). A study in a rat SCI model particularly showed that GM-CSF not only improved spinal cord repair, but also decreased glial scar formation, when administered by intraperitoneal injection (systemic) or local delivery using Gelfoam (20). Decreased GFAP expression and that of CSPG core proteins such as neurocan and NG2 by GM-CSF was observed clearly around the lesion area by immunohistochemistry and Western blotting analysis. This finding suggests the possibility that GM-CSF can directly inhibit the expression of these CSPG core proteins in astrocytes, thereby regulating glial scar formation after SCI.
In the present study, we investigated whether GM-CSF can inhibit the expression of CSPG core proteins in rat primary astrocytes treated with TGF-β to mimic glial scar formation in vitro. Signal pathways involved in GM-CSF’s effect were also investigated to understand its molecular mechanism of action.
RESULTS
GM-CSF repressed expression of CSPG core proteins induced by TGF-β3 in primary astrocytes
TGF-β family proteins are involved in the glial scar formation and have been used to induce the expression of scar genes including CSPG core proteins in an in vitro astrocyte model (21, 22). Primary astrocytes isolated from rats were treated with TGF-β3 for 6 h, and the expression of CSPG core proteins was examined. As shown in Fig. 1, TGF-β3 increased the expression of CSPG core proteins, including NG2, neurocan, and phosphacan, indicating that the astrocyte model of glial scar formation was established. Then, we examined whether GM-CSF could inhibit glial scar formation in the astrocyte model. As shown in Fig. 1A, GM-CSF repressed the TGF-β3-mediated induction of CSPG core proteins in a dose-dependent manner, and GM-CSF receptor antibody abrogated the effects of GM-CSF while G-CSF receptor antibody had no effect. However, G-CSF did increase the TGF-β3-mediated induction of CSPG core proteins, and G-CSF receptor antibody prevented the effects of G-CSF while GM-CSF receptor antibody did not (Fig. 1B). Furthermore, GM-CSF inhibited the TGF-β3-mediated induction of xylosyltransferase (xylT) 1 and 2, which are important in the biosynthesis of CSPG core proteins, but G-CSF had little effect (Fig. 1C). Additionally, G-CSF increased the expression of CSPG core proteins without TGF-β3 treatment, as it did in the TGF-β3-treated astrocytes (Fig. 1B), but GM-CSF did not affect their expression when astrocytes were not treated with TGF-β3 (Fig. 2). Together, these results indicated that GM-CSF can inhibit the TGF-β3-mediated induction of CSPG core proteins through receptor-mediated signal transduction in primary astrocytes, and suggested that GM-CSF may suppress glial scar formation through regulating expression of CSPG core proteins.
Fig. 1. Effects of GM-CSF and G-CSF on the expression of CSPG core proteins in the astrocyte model of glial scar formation. (A, B) Primary astrocytes were treated with TGF-β3 (10 ng/ml) for 24 h with or without pretreatment of GM-CSF and G-CSF for 6 h as indicated. The cells were also treated with antibody against GMRα (GMRα Ab) or G-CSF receptor (GCSF-R Ab) prior to GM-CSF and G-CSF. Then, the expression of neurocan, phosphacan, and NG2 was analyzed. β-actin was used as an internal control. (C) Primary astrocytes were treated with TGF-β3 (10 ng/ml) for 24 h with or without pretreatment with GM-CSF (10 ng/ml) and G-CSF (10 ng/ml) for 6 h. Then, cell lysates were prepared, and subjected to Western blot analysis. β-actin was used as an internal control. Values below each panel indicate fold normalized expression ratio of each protein to β-actin relative to that of no treatment, taken as 1.
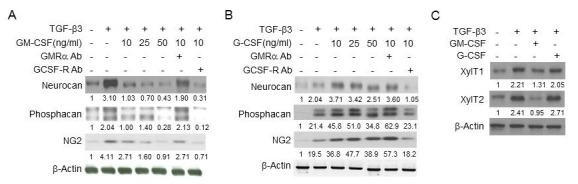
Fig. 2. Effects of GM-CSF and G-CSF on the expression of glial CSPG core proteins. Primary astrocytes were treated with GM-CSF (A) or G-CSF (B) for 24 h as indicated. Cell lysates were prepared, and then subjected to Western blot analysis using neurocan, phosphacan, and NG2 antibodies. β-actin was used as an internal control.
GM-CSF inhibited the TGF-β3-induced Rho-ROCK pathway in primary astrocytes
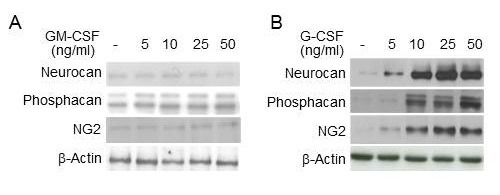
The Rho-ROCK signal pathway is known to mediate the inhibitory effect of CSPG on neuronal regeneration (23). It is also known to be activated by TGF-β in other cell types (24) but the role of the Rho-ROCK pathway in the TGF-β-induced CSPG expression in astrocytes is not well understood. In this study, both Rho and ROCK inhibitors (statin and Y27632) suppressed the TGF-β3-mediated induction of CSPG core proteins in primary astrocytes (Fig. 3A, B) indicating that the Rho-ROCK pathway is involved in TGF-β’s effects. TGF-β3 actually induced phosphorylation of Rho and ROCK signals and also myosin light chain (MLC), a downstream molecule in the Rho-ROCK pathway, which was inhibited effectively by GM-CSF but not by G-CSF (Fig. 3C). We also observed that a ROCK inhibitor suppressed the TGF-β3-induced phosphorylation of MLC (data not shown). These results suggest that GM-CSF repressed TGF-β-induced CSPG core protein expression via blocking the Rho-ROCK signal pathway.
Fig. 3. Effects of GM-CSF and G-CSF on the TGF-β3-induced Rho-ROCK signaling pathway. (A, B) Primary astrocytes were treated with TGF-β3 (10 ng/ml) for 24 h with pre-treatment of Rho inhibitor (10 or 25 μM) or ROCK inhibitor (Y-27632: 10 or 25 μM) for 1 h. Then, the expression of neurocan, phosphacan and NG2 was analyzed. β-actin was used as an internal control. Values below each panel indicate fold normalized expression ratio of each protein to β-actin relative to that of no treatment, taken as 1. (C) Primary astrocytes were treated with TGF-β3 (10 ng/ml) for 24 h with or without pre-treatment of GM-CSF or G-CSF for 6 h, as indicated. Then, the phosphorylation levels of RhoA, ROCK, and MLC were analyzed by Western blotting. Values below each panel indicate fold normalized phosphorylation ratio of each protein to unphosphorylated level of the protein relative to that of no treatment, taken as 1.
The JAK pathway is responsible for the GM-CSF inhibition of TGF-β signals
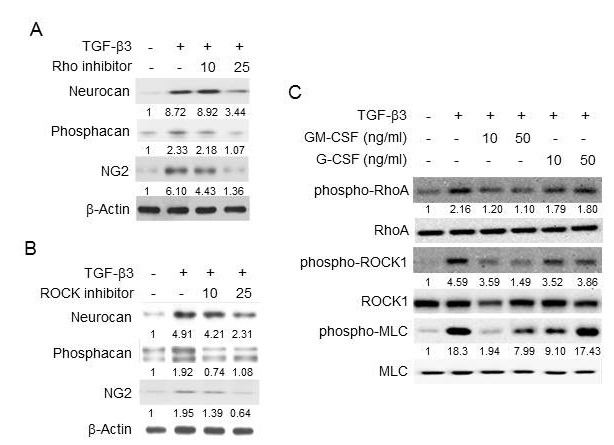
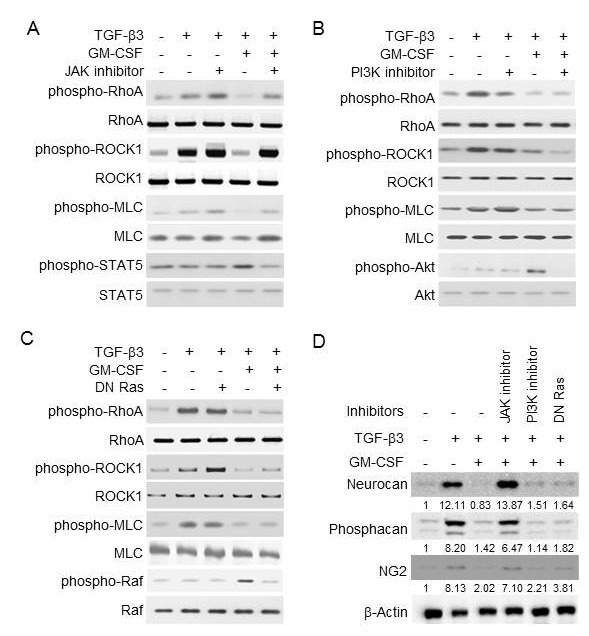
To find the link between the GM-CSF receptor and the Rho-ROCK pathway of TGF-β, we next examined the effects of GM-CSF signal inhibitors in inhibiting TGF-β function. GM-CSF is known to primarily activate the JAK-STAT, PI3K-Akt, and Ras-Raf pathways in hematopoietic cells and similarly in astrocyte cells (25, 26). To block JAK and PI3K, primary astrocytes were treated with JAK inhibitor I and LY294002, respectively. To inhibit Ras, cells were transfected with a plasmid expressing the dominant negative mutant of Ras (DN Ras). JAK inhibitor I completely recovered the GM-CSF-mediated suppression of the Rho-ROCK pathway of TGF-β3, while LY294002 and DN Ras had little effect (Fig. 4A-C). Additionally, JAK inhibitor I completely blocked the GM-CSF-mediated repression of CSPG core proteins, including neurcan, phosphacan, and NG2, induced by TGF-β3, while LY294002 and DN Ras did not (Fig. 4D). Each inhibitor was also confirmed to downregulate GM-CSF-induced phosphorylation of STAT5, Akt and Raf, respectively (Fig. 4A-C). The inhibitors by themselves did not block the TGF-β3 signals, and TGF-β3 alone did not induce the GM-CSF signals. These results suggest that JAK played a critical role in the GM-CSF-mediated suppression of Rho-ROCK activation and CSPG core protein expression by TGF-β.
Fig. 4. Signal pathways of GM-CSF responsible for the suppression of Rho-ROCK activation by TGF-β3. Primary astrocytes were pretreated with 10 μM of JAK inhibitor I or LY294002 (PI3K inhibitor) for 1 h, or were transfected with DN Ras. Then, the cells were treated with TGF-β3 (10 ng/ml) and GM-CSF (10 ng/ml), as indicated. (A-C) The phosphorylation levels of RhoA, ROCK, MLC, STAT5, Akt, and Raf were analyzed by Western blotting as indicated. (D) The expression levels of neurocan, phosphacan and NG2 were analyzed by Western blotting. β-actin was used as an internal control. Values below each panel indicate fold normalized expression ratio of each protein to β-actin relative to that of no treatment, taken as 1.
DISCUSSION
We demonstrated that GM-CSF could inhibit expression of CSPG core proteins, such as NG2, neurocan, and phosphacan, induced by TGF-β in primary astrocytes. Additionally, the expression of CSPG core proteins by TGF-β was mediated via activation of the RhoA-ROCK pathway, which was also inhibited by GM-CSF. These results support that the anti-scar forming activity of GM-CSF observed previously in a rat SCI model was mediated by a specific biological action of GM-CSF but not by its neuroprotective effects. Glial scars hinder the exchange of small molecules, cytokines, and neurotrophic factors between lesion areas and adjacent intact neural tissues, which, in turn, inhibits the repair and regeneration of damaged neural tissues (27-29). The scar is composed mainly of various CSPGs secreted by astrocytes, oligodendrocyte precursor cells (OPCs), and meningeal cells, and by GFAP-positive astrocytes during the late stage after injury. Many studies have suggested that TGF-β plays an important role in the expression of CSPGs and glial scar formation (22, 30, 31) and a strategy to inhibit TGF-β signaling may be a potential therapy to repair CNS injuries (32). Thus, the specific action of GM-CSF in inhibiting glial scar formation could be a promising therapeutic tool for CNS injuries, including SCI.
The Rho-ROCK signaling pathway is one of the downstream signals of TGF-β (24). Previously, interleukin-1β (IL-1β) was shown to suppress astroglial activation after CNS injury via inhibiting the Rho-ROCK signal pathway (33). Statin, a Rho inhibitor, was shown to inhibit CSPGs expression and astrocyte activation, but Y27362, a ROCK inhibitor, increased their expression (34, 35). Thus, the roles of Rho and ROCK in CSPG expression in astrocytes are not yet clearly understood, particularly when cells are treated with TGF-β in vitro. This study first showed that inhibition of Rho and ROCK signals of TGF-β using specific inhibitors or GM-CSF decreased CSPGs expression in cultured astrocytes significantly. The exact mechanism(s) of GM-CSF’s effect remain(s) unclear but seem to involve the JAK signal pathway and act on Rho or its upstream pathway of TGF-β signals in astrocytes.
The Rho-ROCK signaling pathway is also implicated in the CSPG inhibition of axon growth and neuronal regeneration (23). Several ROCK inhibitors, such as dimethylfasudil, fasudil, and Y27632, were shown to increase neurite outgrowth in neuronal cells in vitro and axonal regeneration of rat optic nerve in vivo (36, 37). Bacterial C3 exoenzyme, a Rho inhibitor and dominant negative mutant of Rho were also reported to alleviate CSPG-mediated inhibition of neurite outgrowth and to promote neuronal regeneration after injury (38, 39). Having similar inhibitory effects on both Rho and ROCK signals, GM-CSF could inhibit CSPG production in astrocytes and their inhibitory effect on axonal regeneration in neurons.
MATERIALS AND METHODS
Treatment of cytokines and inhibitors
Primary astrocytes were treated with GM-CSF or G-CSF (Chemicon Inc.) for 24 h, alone or in combination with TGF-β3 (Chemicon Inc.). For combinatorial treatments, cells were pretreated with GM-CSF at 6 h before TGF-β3 treatment. The inhibitors used were LY294002 (PI3K inhibitor; Merck KGaA), statin (Rho inhibitor), Y27632 (ROCK inhibitor; Sigma Chemical Co.), JAK inhibitor I, GM-CSF receptor α antibody, and G-CSF receptor antibody (Santa Cruz Biotech.). The plasmid expressing DN-Ras was kindly provided by Professors Gautam Sethi (National University of Singapore) and Kwang Seok Ahn (Kyung Hee University).
Isolation and culture of primary astrocytes
All animal experiments were performed in the authorized animal care facility and approved by the ‘Committee for the Care and Use of Laboratory Animals’ at Inha University College of Medicine. Primary astrocytes were prepared from cerebral cortices of 1-day old neonatal Sprague Dawley rats. Briefly, dissociated cortical cells were suspended in DMEM (Gibco-BRL) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate and 10% FBS, and plated on uncoated T75 flasks at a density of 6 × 105 cells/cm2. Monolayer of type 1 astrocytes was obtained 12-14 days after plating. Non-astrocytic cells such as microglia and neurons were detached from the flasks by shaking and removed by changing the medium.
Western blotting
Whole cell lysates were prepared, and Western blotting was performed as described (17). The antibodies were purchased as follows: neurocan and phosphacan from Chemicon Inc., NG2 from Upstate USA Inc., phospho-ROCK from Bioss, horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody, RhoA, phospho-RhoA, ROCK, xylT1, xylT2, Raf, and phospho-Raf from Santa Cruz Biotech., MLC, phospho-MLC, Akt, phospho-Akt, STAT5 and phospho-STAT5 from Cell Signaling Technology Inc., and β-actin from Sigma Chemical Co..
Acknowledgments
We thank Professors Kwang Seok Ahn (Kyung Hee University, Republic of Korea) and Gautam Sethi (National University of Singapore, Singapore) for the gift of DN-Ras. This work was supported by research grants from the National Research Foundation of Korea (2007-0054931 to SGL and 2009-0079196 to BHC).
References
- 1.Amar A. P., Levy M. L. Pathogenesis and pharmacological strategies for mitigating secondary damage in acute spinal cord injury. Neurosurgery. (1999);44:1027–1039. doi: 10.1097/00006123-199905000-00052. discussion 1039-1040. [DOI] [PubMed] [Google Scholar]
- 2.Crowe M. J., Bresnahan J. C., Shuman S. L., Masters J. N., Beattie M. S. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat. Med. (1997);3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- 3.Sandvig A., Berry M., Barrett L. B., Butt A., Logan A. Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: expression, receptor signaling, and correlation with axon regeneration. Glia. (2004);46:225–251. doi: 10.1002/glia.10315. [DOI] [PubMed] [Google Scholar]
- 4.Jones L. L., Margolis R. U., Tuszynski M. H. The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp. Neurol. (2003);182:399–411. doi: 10.1016/S0014-4886(03)00087-6. [DOI] [PubMed] [Google Scholar]
- 5.Silver J., Miller J. H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. (2004);5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 6.David S., Ousman S. S. Recruiting the immune response to promote axon regeneration in the injured spinal cord. Neuroscientist. (2002);8:33–41. doi: 10.1177/107385840200800108. [DOI] [PubMed] [Google Scholar]
- 7.Schermer C., Humpel C. Granulocyte macrophage-colony stimulating factor activates microglia in rat cortex organotypic brain slices. Neurosci. Lett. (2002);328:180–184. doi: 10.1016/S0304-3940(02)00496-2. [DOI] [PubMed] [Google Scholar]
- 8.Kim J. K., Choi B. H., Park H. C., Park S. R., Kim Y. S., Yoon S. H., Park H. S., Kim E. Y., Ha Y. Effects of GM-CSF on the neural progenitor cells. Neuroreport. (2004);15:2161–2165. doi: 10.1097/00001756-200410050-00003. [DOI] [PubMed] [Google Scholar]
- 9.Kannan Y., Moriyama M., Sugano T., Yamate J., Kuwamura M., Kagaya A., Kiso Y. Neurotrophic action of interleukin 3 and granulocyte-macrophage colony-stimulating factor on murine sympathetic neurons. Neuroimmunomodulation. (2000);8:132–141. doi: 10.1159/000054273. [DOI] [PubMed] [Google Scholar]
- 10.Franzen R., Bouhy D., Schoenen J. Nervous system injury: focus on the inflammatory cytokine 'granulocyte-macrophage colony stimulating factor'. Neurosci. Lett. (2004);361:76–78. doi: 10.1016/j.neulet.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 11.Saada A., Reichert F., Rotshenker S. Granulocyte macrophage colony stimulating factor produced in lesioned peripheral nerves induces the up-regulation of cell surface expression of MAC-2 by macrophages and Schwann cells. J. Cell Biol. (1996);133:159–167. doi: 10.1083/jcb.133.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ousman S. S., David S. MIP-1alpha, MCP-1, GM-CSF, and TNF-alpha control the immune cell response that mediates rapid phagocytosis of myelin from the adult mouse spinal cord. J. Neurosci. (2001);21:4649–4656. doi: 10.1523/JNEUROSCI.21-13-04649.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bouhy D., Malgrange B., Multon S., Poirrier A. L., Scholtes F., Schoenen J., Franzen R. Delayed GM-CSF treatment stimulates axonal regeneration and functional recovery in paraplegic rats via an increased BDNF expression by endogenous macrophages. FASEB J. (2006);20:1239–1241. doi: 10.1096/fj.05-4382fje. [DOI] [PubMed] [Google Scholar]
- 14.Ha Y., Kim Y. S., Cho J. M., Yoon S. H., Park S. R., Yoon D. H., Kim E. Y., Park H. C. Role of granulocyte-macrophage colony-stimulating factor in preventing apoptosis and improving functional outcome in experimental spinal cord contusion injury. J. Neurosurg. (2005);2:55–61. doi: 10.3171/spi.2005.2.1.0055. [DOI] [PubMed] [Google Scholar]
- 15.Park H. C., Shim Y. S., Ha Y., Yoon S. H., Park S. R., Choi B. H., Park H. S. Treatment of complete spinal cord injury patients by autologous bone marrow cell transplantation and administration of granulocyte-macrophage colony stimulating factor. Tissue Engineering. (2005);11:913–922. doi: 10.1089/ten.2005.11.913. [DOI] [PubMed] [Google Scholar]
- 16.Yoon S. H., Shim Y. S., Park Y. H., Chung J. K., Nam J. H., Kim M. O., Park H. C., Park S. R., Min B. H., Kim E. Y., Choi B. H., Park H., Ha Y. Complete spinal cord injury treatment using autologous bone marrow cell transplantation and bone marrow stimulation with granulocyte macrophage-colony stimulating factor: Phase I/II clinical trial. Stem Cells. (2007);25:2066–2073. doi: 10.1634/stemcells.2006-0807. [DOI] [PubMed] [Google Scholar]
- 17.Huang X., Choi J. K., Park S. R., Ha Y., Park H., Yoon S. H., Park H. C., Park J. O., Choi B. H. GM-CSF inhibits apoptosis of neural cells via regulating the expression of apoptosis-related proteins. Neurosci. Res. (2007);58:50–57. doi: 10.1016/j.neures.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 18.Kim N. K., Choi B. H., Huang X., Snyder B. J., Bukhari S., Kong T. H., Park H., Park H. C., Park S. R., Ha Y. Granulocyte-macrophage colony-stimulating factor promotes survival of dopaminergic neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced murine Parkinson's disease model. European J. Neurosci. (2009);29:891–900. doi: 10.1111/j.1460-9568.2009.06653.x. [DOI] [PubMed] [Google Scholar]
- 19.Kong T., Choi J. K., Park H., Choi B. H., Snyder B. J., Bukhari S., Kim N. K., Huang X., Park S. R., Park H. C., Ha Y. Reduction in programmed cell death and improvement in functional outcome of transient focal cerebral ischemia after administration of granulocyte-macrophage colony-stimulating factor in rats. Laboratory investigation. J. Neurosurg. (2009);111:155–163. doi: 10.3171/2008.12.JNS08172. [DOI] [PubMed] [Google Scholar]
- 20.Huang X., Kim J. M., Kong T. H., Park S. R., Ha Y., Kim M. H., Park H., Yoon S. H., Park H. C., Park J. O., Min B. H., Choi B. H. GM-CSF inhibits glial scar formation and shows long-term protective effect after spinal cord injury. J. Neurol. Sci. (2009);277:87–97. doi: 10.1016/j.jns.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 21.Asher R. A., Morgenstern D. A., Fidler P. S., Adcock K. H., Oohira A., Braistead J. E., Levine J. M., Margolis R. U., Rogers J. H., Fawcett J. W. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J. Neurosci. (2000);20:2427–2438. doi: 10.1523/JNEUROSCI.20-07-02427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith G. M., Strunz C. Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia. (2005);52:209–218. doi: 10.1002/glia.20236. [DOI] [PubMed] [Google Scholar]
- 23.Monnier P. P., Sierra A., Schwab J. M., Henke-Fahle S., Mueller B. K. The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol. Cell Neurosci. (2003);22:319–330. doi: 10.1016/S1044-7431(02)00035-0. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y. E. Non-Smad pathways in TGF-beta signaling. Cell Res. (2009);19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guthridge M. A., Stomski F. C., Thomas D., Woodcock J. M., Bagley C. J., Berndt M. C., Lopez A. F. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells. (1998);16:301–313. doi: 10.1002/stem.160301. [DOI] [PubMed] [Google Scholar]
- 26.Choi J. K., Choi B. H., Ha Y., Park H., Yoon S. H., Park H. C., Park S. R. Signal transduction pathways of GM-CSF in neural cell lines. Neurosci. Lett. (2007);420:217–222. doi: 10.1016/j.neulet.2007.03.065. [DOI] [PubMed] [Google Scholar]
- 27.Iannotti C., Zhang Y. P., Shields L. B., Han Y., Burke D. A., Xu X. M., Shields C. B. Dural repair reduces connective tissue scar invasion and cystic cavity formation after acute spinal cord laceration injury in adult rats. J. Neurotrauma. (2006);23:853–865. doi: 10.1089/neu.2006.23.853. [DOI] [PubMed] [Google Scholar]
- 28.Roitbak T., Sykova E. Diffusion barriers evoked in the rat cortex by reactive astrogliosis. Glia. (1999);28:40–48. doi: 10.1002/(sici)1098-1136(199910)28:1<40::aid-glia5>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 29.Hynds D. L., Snow D. M. Neurite outgrowth inhibition by chondroitin sulfate proteoglycan: stalling/stopping exceeds turning in human neuroblastoma growth cones. Exp. Neurol. (1999);160:244–255. doi: 10.1006/exnr.1999.7212. [DOI] [PubMed] [Google Scholar]
- 30.Logan A., Berry M., Gonzalez A. M., Frautschy S. A., Sporn M. B., Baird A. Effects of transforming growth factor beta 1 on scar production in the injured central nervous system of the rat. Eur. J. Neurosci. (1994);6:355–363. doi: 10.1111/j.1460-9568.1994.tb00278.x. [DOI] [PubMed] [Google Scholar]
- 31.Schwab J. M., Beschorner R., Nguyen T. D., Meyermann R., Schluesener H. J. Differential cellular accumulation of connective tissue growth factor defines a subset of reactive astrocytes, invading fibroblasts, and endothelial cells following central nervous system injury in rats and humans. J. Neurotrauma. (2001);18:377–388. doi: 10.1089/089771501750170930. [DOI] [PubMed] [Google Scholar]
- 32.Logan A., Green J., Hunter A., Jackson R., Berry M. Inhibition of glial scarring in the injured rat brain by a recombinant human monoclonal antibody to transforming growth factor-beta2. Eur. J. Neurosci. (1999);11:2367–2374. doi: 10.1046/j.1460-9568.1999.00654.x. [DOI] [PubMed] [Google Scholar]
- 33.John G. R., Chen L., Rivieccio M. A., Melendez-Vasquez C. V., Hartley A., Brosnan C. F. Interleukin-1beta induces a reactive astroglial phenotype via deactivation of the Rho GTPase-Rock axis. J. Neurosci. (2004);24:2837–2845. doi: 10.1523/JNEUROSCI.4789-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmberg E., Zhang S. X., Sarmiere P. D., Kluge B. R., White J. T., Doolen S. Statins decrease chondroitin sulfate proteoglycan expression and acute astrocyte activation in central nervous system injury. Exp. Neurol. (2008);214:78–86. doi: 10.1016/j.expneurol.2008.07.020. [DOI] [PubMed] [Google Scholar]
- 35.Chan C. C., Wong A. K., Liu J., Steeves J. D., Tetzlaff W. ROCK inhibition with Y27632 activates astrocytes and increases their expression of neurite growth-inhibitory chondroitin sulfate proteoglycans. Glia. (2007);55:369–384. doi: 10.1002/glia.20466. [DOI] [PubMed] [Google Scholar]
- 36.Gopalakrishnan S. M., Teusch N., Imhof C., Bakker M. H., Schurdak M., Burns D. J., Warrior U. Role of Rho kinase pathway in chondroitin sulfate proteoglycan-mediated inhibition of neurite outgrowth in PC12 cells. J. Neurosci. Res. (2008);86:2214–2226. doi: 10.1002/jnr.21671. [DOI] [PubMed] [Google Scholar]
- 37.Lingor P., Teusch N., Schwarz K., Mueller R., Mack H., Bahr M., Mueller B. K. Inhibition of Rho kinase (ROCK) increases neurite outgrowth on chondroitin sulphate proteoglycan in vitro and axonal regeneration in the adult optic nerve in vivo. J. Neurochem. (2007);103:181–189. doi: 10.1111/j.1471-4159.2007.04756.x. [DOI] [PubMed] [Google Scholar]
- 38.Jain A., Brady-Kalnay S. M., Bellamkonda R. V. Modulation of Rho GTPase activity alleviates chondroitin sulfate proteoglycan-dependent inhibition of neurite extension. J. Neurosci. Res. (2004);77:299–307. doi: 10.1002/jnr.20161. [DOI] [PubMed] [Google Scholar]
- 39.Just I., Rohrbeck A., Huelsenbeck S. C., Hoeltje M. Therapeutic effects of Clostridium botulinum C3 exoenzyme. Naunyn. Schmiedebergs. Arch. Pharmacol. (2011);383:247–252. doi: 10.1007/s00210-010-0589-3. [DOI] [PubMed] [Google Scholar]