

Significance
Apolipoprotein (apo) A-II is the most important protein associated with senile amyloidosis. Because some variants of apoA-II protein have been found among inbred strains of mice, we hypothesized that investigating amyloidogenesis of the variants would improve our understanding of the molecular and biological mechanisms of senile amyloidosis. Here, we demonstrate that mice with type F apoA-II (APOA2F) protein were absolutely resistant to senile amyloidosis. Moreover, a selective C-terminal APOA2F peptide inhibited fibril formation of amyloidogenic apoA-II in vitro and prevented senile amyloidosis in mice. We propose an inhibitory model in which the C-terminal APOA2F peptide prevents further fibril extension by blocking the active ends of seeds. This approach could provide a novel therapeutic option for the treatment of senile amyloidosis.
Keywords: amyloid fibril formation, apolipoprotein A-II, inhibitory peptide, mouse, senile amyloidosis
Abstract
In murine senile amyloidosis, misfolded serum apolipoprotein (apo) A-II deposits as amyloid fibrils (AApoAII) in a process associated with aging. Mouse strains carrying type C apoA-II (APOA2C) protein exhibit a high incidence of severe systemic amyloidosis. Previously, we showed that N- and C-terminal sequences of apoA-II protein are critical for polymerization into amyloid fibrils in vitro. Here, we demonstrate that congenic mouse strains carrying type F apoA-II (APOA2F) protein, which contains four amino acid substitutions in the amyloidogenic regions of APOA2C, were absolutely resistant to amyloidosis, even after induction of amyloidosis by injection of AApoAII. In vitro fibril formation tests showed that N- and C-terminal APOA2F peptides did not polymerize into amyloid fibrils. Moreover, a C-terminal APOA2F peptide was a strong inhibitor of nucleation and extension of amyloid fibrils during polymerization. Importantly, after the induction of amyloidosis, we succeeded in suppressing amyloid deposition in senile amyloidosis-susceptible mice by treatment with the C-terminal APOA2F peptide. We suggest that the C-terminal APOA2F peptide might inhibit further extension of amyloid fibrils by blocking the active ends of nuclei (seeds). We present a previously unidentified model system for investigating inhibitory mechanisms against amyloidosis in vivo and in vitro and believe that this system will be useful for the development of novel therapies.
Amyloidosis refers to a group of protein structural disorders characterized by the extracellular deposits of insoluble amyloid fibrils resulting from abnormal conformational changes (1–5). Amyloid fibrils have a characteristic ultrastructural appearance and a β-pleated sheet core structure that consists of full-length proteins and/or fragments of either WT or mutant proteins found in familial diseases (2, 6–8). In humans, 28 amyloidogenic proteins have been identified. They are associated with prominent diseases such as Alzheimer’s disease, hemodialysis-associated amyloidosis, and familial amyloid polyneuropathy (2, 9, 10). To develop a therapeutic strategy for these disorders, it is essential to understand the mechanisms of amyloid fibril formation. Currently, the molecular and biological mechanisms that convert proteins into amyloid fibrils in vivo and in vitro remain largely unknown.
Apolipoprotein (apo) A-II is the second most abundant apolipoprotein in human and mouse plasma high-density lipoproteins (HDLs) (11) and the most important protein associated with murine senile amyloidosis because it is the precursor of amyloid fibrils (AApoAII) (12–15). Seven alleles of the apoA-II gene have been found among inbred strains of mice, with polymorphisms in 15 nucleotide positions comprising eight amino acid positions (16). Each inbred laboratory mouse strain has a single type apoA-II protein, and the pathological findings of senile amyloidosis in strains with type A, B, or C apoA-II (APOA2A, APOA2B, or APOA2C, respectively) have been investigated (13, 17, 18). C57BL/6J, ICR, and DBA/2 strains have APOA2A and exhibit a moderate incidence of mild amyloid deposits with aging (19, 20). BALB/c, C3H/He, N2B, 129/SV, and SAMR1 strains have APOA2B and exhibit a low incidence of slight amyloid deposits with aging. In contrast, the SAMP1 strain has APOA2C and spontaneously exhibits a high incidence of severe systemic amyloid deposits with aging (20–22). We previously reported a unique mechanism in which N- and C-terminal peptides of apoA-II protein associated into amyloid fibrils in vitro (23) according to the nucleation-dependent polymerization model, which can explain the general mechanisms of amyloid fibril formation (24–28). The 11-residue amino acid sequence from positions 6–16 in the N terminus of apoA-II protein is critical for polymerization into amyloid fibrils. The 18-residue amino acid sequence from positions 48–65 in the C terminus of apoA-II is also necessary for nucleation, but not for the extension phase. Both sequences are common, and there is no substitution among APOA2A, APOA2B, and APOA2C (Fig. 1).
Fig. 1.

Amino acid sequences of mouse apoA-II and synthetic partial peptides. For types A, B, and C apoA-II proteins (APOA2A, APOA2B, and APOA2C, respectively), the two amino acid sequences indicated in the red-colored boxes at positions 6–16 at the N terminus and 48–65 at the C terminus are the essential and common sequences required for amyloid fibril formation (23). Synthetic partial peptides were used to evaluate polymerization into amyloid fibrils in vitro and suppression against amyloid deposition in mice. The bold and blue-colored letters at positions 9, 16, 54, and 62 indicate the four variant amino acids in the core sequences for types A/B/C and F apoA-II proteins. Peptides containing orange letters represent substitutions of the a48/65(N62K) peptide.
We hypothesized that some amino acid substitutions in these N- and C-terminal amyloidogenic sequences of apoA-II might inhibit the polymerization of apoA-II into amyloid fibrils. In that regard, type F apoA-II (APOA2F) contains four substitutions in the N- and C-terminal peptides relative to APOA2C (16) (Fig. 1). In this study, we evaluated the in vivo incidence of amyloidosis in mice having APOA2F and compared it with those in mice having APOA2A or APOA2C. We also analyzed the ability of APOA2F peptides to polymerize into amyloid fibrils in vitro. In previous studies, we found that injection of a very small amount of AApoAII amyloid fibrils markedly accelerated amyloid deposition (13–15). We demonstrated that mice with APOA2F were absolutely resistant against senile amyloidosis, even after induction of amyloidosis by injection with type C AApoAII fibrils. Thus, we have succeeded in suppressing amyloid deposits in amyloidosis-susceptible mice by treatment with the C-terminal APOA2F peptide. We thus demonstrate that the C-terminal sequence of APOA2F is an important inhibitor of polymerization into amyloid fibrils in vitro and in vivo. These findings provide a previously unidentified model system for investigating inhibitory mechanisms against amyloidosis in vivo and in vitro.
Results
Mice Expressing APOA2F Suppress AApoAII Amyloidosis.
To determine whether amyloid fibrils would be deposited in mice expressing APOA2F, we generated B6.SPRET-Apoa2f and R1.SPRET-Apoa2f congenic mice and i.v. injected a single dose of 100 µg of sonicated AApoAII fibrils into 2-mo-old animals. Two to 9 mo later, we evaluated amyloid deposits by the presence of green birefringence in Congo red-stained tissue from whole mice and evaluated the amyloid index (AI) (13, 29, 30). In mice carrying the Apoa2a or the Apoa2c allele, AApoAII fibrils can deposit in systemic organs such as the tongue, stomach, small intestine, lung, heart, liver, spleen, and skin (but not the brain) (12, 19, 20, 30, 31). In our experiments, the organ distribution of deposited amyloid in Apoa2a mice was similar to that in Apoa2c mice (Fig. 2 and Table S1). In B6.SPRET-Apoa2f mice, amyloid deposit was detected in mice with Apoa2a alleles (Fig. 2A). The AI increased in a time-dependent manner after injection of AApoAII fibrils in Apoa2a/a mice. In contrast, in Apoa2a/f mice, the AI was approximately constant for 9 mo and was less than half that of Apoa2a/a mice. None of the Apoa2f/f mice had amyloid deposit over a 9-mo period. Representative images of early and severe amyloid deposits in the tongue, stained with Congo red or by immunohistochemical methods, are shown in Fig. 2B. Nine months after the injection of AApoAII fibrils into B6.SPRET-Apoa2f mice, amyloid deposits in the tongues of Apoa2a/a mice were readily observed in the lamina propria, and those in Apoa2a/f mice were detected in the same region. AApoAII amyloid deposits were confirmed immunohistochemically. In contrast, AApoAII deposits were not detected anywhere in the tongues of Apoa2f/f mice. In the R1.SPRET-Apoa2f congenic mice, we detected amyloid deposits in all mice with the Apoa2c allele (Fig. 2A and Table S1). In contrast, none of the Apoa2f/f mice showed amyloid deposit anywhere in the body. The AIs of Apoa2c/f mice were less than half those of Apoa2c/c mice 2 and 6 mo after induction. In addition, none of the Apoa2f/f mice showed spontaneous amyloid deposit until 13 mo of age even though Apoa2c/f mice had amyloid deposits in several organs at the same period and Apoa2c/c mice had severe amyloid deposits (Table S2).
Fig. 2.

Induction of amyloid deposits by i.v. injection of 100 µg of AApoAII amyloid fibrils. (A) The degree of amyloid deposits (amyloid index, AI) in mice at the indicated time of observation. Each bar shows the mean. Numbers in parentheses represent amyloid-positive mice/total mice examined. *P < 0.05; **P < 0.01; ***P < 0.001 (Mann–Whitney U test). (B) AApoAII deposits in the tongue of B6.SPRET-Apoa2f mice after 9 mo of the induction. (Upper) Amyloid deposits were identified by green birefringence in Congo red (CR)-stained sections using polarizing LM. According to the criteria of AApoAII amyloid deposits in mice (13, 29, 30), grades of amyloid deposit were 4, 2, and 0 in Apoa2a/a, Apoa2a/f, and Apoa2f/f, respectively. (Lower) AApoAII deposits were confirmed immunohistochemically (IHC) with anti–apoA-II antiserum. (Scale bars: 100 µm.)
Properties of Plasma HDL in Mice with APOA2F.
ApoA-II proteins in plasma are primarily distributed in HDL3 particles (15, 32). To determine the effects of the polymorphism of apoA-II protein on lipoprotein distribution, we examined the properties of HDL and levels of ApoAs and cholesterols in B6.SPRET-Apoa2f mice (Fig. 3). The contents of apoA-I and apoA-II proteins in the plasma HDL fraction in Apoa2f/f mice were similar to those in mice with the Apoa2a allele (Fig. 3A). The particle sizes of HDL3 in Apoa2f/f mice were similar to those of mice with the Apoa2a allele and were smaller than those of the amyloidosis-resistant SAMR1 mice (Apoa2b/b). The plasma levels of total and HDL cholesterols in Apoa2f/f mice were not significantly different compared with those in mice with the Apoa2a allele (Fig. 3B). In the R1.SPRET-Apoa2f mice, the plasma levels of total and HDL cholesterols in Apoa2f/f mice were not different compared with those in mice with the Apoa2c allele (Fig. S1). From these results, we conclude that the Apoa2f/f allele suppresses amyloidosis and that APOA2F affects neither the contents of apoA-I and apoA-II proteins nor the properties of plasma apoA-II–associated HDL.
Fig. 3.

Plasma concentrations of apoAs and cholesterols in B6.SPRET-Apoa2f mice at 2 mo of age. (A, Upper) ApoA-I and apoA-II in the HDL fraction were detected by CBB staining. (Lower) HDL3 particles were visualized by Sudan Black B staining. SAMR1 strain was used as a control for comparison with the Apoa2f/f mice. (B) Concentrations of plasma total and HDL cholesterols (TC and HDL-C, respectively). Each column and bar represents the mean ± SD, and numbers in parentheses represent total mice examined. There were no significant differences among three groups in various pairwise comparisons (Tukey–Kramer method for multiple comparison).
Partial Peptides of APOA2F Cannot Polymerize into Amyloid Fibrils in Vitro.
We examined in vitro amyloid fibril formation by synthetic partial peptides of APOA2F using previously described methods (23). Thioflavin T (ThT)-fluorescence intensities of the APOA2A peptides (a6/16 + a48/65, TYPE A), which had common sequences of APOA2A and APOA2C proteins, were stable for up to 2 h of incubation at pH 2.5 (the nucleation phase) and then increased linearly until they reached a plateau phase after 8 h (the extension phase) (Fig. 4A). Using transmission electron microscopy (TEM) to examine reaction mixtures after 21 h of incubation, we observed abundant amyloid fibrils with characteristic structures (Fig. 4B). Moreover, apple-green birefringence was observed in the mixture stained with Congo red under polarizing light microscopy (LM). In contrast, the ThT intensities of the APOA2F peptides (f6/16 + f48/65, TYPE F) were relatively unchanged even after 21 h of incubation, and amyloid fibrils could not be detected by TEM. Moreover, the characteristic apple-green birefringence of fibrils under polarizing LM was absent from the mixture stained with Congo red dye. From these results, it was apparent that the combining of N- and C-terminal APOA2F peptides failed to form amyloid fibrils.
Fig. 4.

Synthetic partial peptides of APOA2F could not polymerize into amyloid fibrils in vitro. (A) ThT plots of synthetic partial peptides (50 µM each). Each symbol and bar represents the mean ± SD (n = 3). a.u., arbitrary units. (B) Microscopic analyses of the mixtures after 21 h of incubation. (Left) TEM Images. (Scale bars: 100 nm.) (Right) Images of Congo red-stained glass slides under bright-field or polarized LM. (Optical magnification: 100×.)
The C-Terminal APOA2F Peptide Suppressed Polymerization of Amyloid Fibrils in Vitro.
We hypothesized that the N- and/or C-terminal APOA2F peptides might inhibit polymerization into amyloid fibrils. Thus, we examined fibril formation in a reaction mixture containing both APOA2A and APOA2F peptides. First, we examined fibril formation in a mixture containing the N-terminal APOA2A peptide with the C-terminal APOA2F peptide (a6/16 + f48/65) and vice versa (f6/16 + a48/65). In each case, ThT intensity was uncharged over 21 h of incubation (Fig. 4A). Second, we examined fibril formation in reaction mixtures of a6/16 + 148/65 with APOA2F peptides at various concentrations. Reaction mixtures of TYPE A + TYPE F (1:1 mixture) showed unchanged intensities in contrast to TYPE A (Fig. 5A). N-mixture [(a6/16 + f6/16) + a48/65] showed increased ThT intensities in a fashion similar to those of TYPE A over 3 h of incubation (Fig. 5A). ThT intensities of the N-mixture increased linearly and reached a plateau phase after 6 h, but the maximal intensities were less than one-half those of TYPE A. An N-mixture 1:2 {[25 µM a6/16 + 50 µM f6/16 (1:2)] + 50 µM a48/65} showed an increasing pattern similar to the N-mixture at equal concentrations of N-terminal peptides (N-mixture 1:1) (Fig. S2). In contrast, C-mixture [a6/16 + (a48/65 + f48/65)] showed no change in ThT intensity similar to that of TYPE F (Fig. 5A). Using TEM, amyloid fibrils were observed in the N-mixture after 23 h of incubation, but characteristic fibrils were not detected in the C-mixture after the same period (Fig. 5B). We analyzed the components of the amyloid fibrils formed in the N-mixture by HPLC and liquid chromatography with tandem mass spectrometry (LC-MS/MS). After a 23-h incubation of the N-mixture, amyloid fibrils contained APOA2A peptides (a6/16, a48/65, and pyro-a48/65), but not the N-terminal APOA2F peptide (f6/16) (Fig. 5C).
Fig. 5.
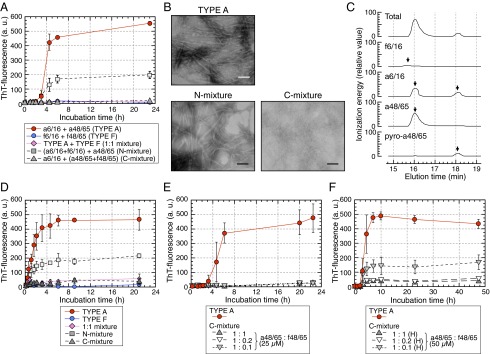
C-terminal APOA2F peptide inhibited the polymerization of APOA2A peptides in vitro. (A) ThT plots of APOA2A peptides mixed with APOA2F peptides. TYPE A, 50 µM each (a6/16 + a48/65); TYPE F, 50 µM each (f6/16 + f48/65); 1:1 mixture, 25 µM each (TYPE A + TYPE F); N-mixture, 25 µM each (a6/16 + f6/16) + 50 µM f48/65; C-mixture, 50 µM a6/16 + 25 µM each (a48/65 + f48/65). Each symbol and bar represents the mean ± SD (n = 3). a.u., arbitrary units. (B) TEM images of the mixtures after 23 h of incubation. (Scale bars: 100 nm.) (C) Representative LC-MS/MS analyses show profiles of the products of the N-mixture after 23 h of incubation. (D) Seed-dependent amyloid fibril extension of APOA2A and APOA2F peptides. In the presence of premade APOA2A fibrils, extension of TYPE A, TYPE F, or mixtures of APOA2A and APOA2F peptides were determined by ThT binding assay. Each symbol and bar represents the mean ± SD (n = 3). a.u., arbitrary units. (E and F) ThT plots of APOA2A peptides mixed with f48/65 at various concentrations. Concentrations of the a48/65 in the mixtures were 25 µM in E (n = 3) and 50 µM [high-concentration condition, (H)] in F (n = 4), and that of the a6/16 in each reaction mixture was 50 µM. Each symbol and bar represents the mean ± SD. a.u., arbitrary units.
Next, we investigated the extension of amyloid fibrils when premade fibrils were added as seeds in the reaction mixtures. With the premade fibrils in the reaction mixtures, ThT intensities of TYPE A increased rapidly from the beginning of agitation and then reached a plateau phase after ∼4 h (Fig. 5D). The lag phase shown in Fig. 5A was absent. In contrast, the ThT intensities of TYPE F did not increase. The N-mixture showed an increase resembling that of TYPE A, but the maximum intensities were less than one-half as great as TYPE A. In contrast, the ThT intensities of the reaction mixtures containing the C-terminal APOA2F peptide (C-mixture and 1:1 mixture) increased only slightly, even with premade fibrils.
To investigate the suppressive effects of the C-terminal APOA2F peptide on polymerization into amyloid fibrils, we analyzed the polymerization of the C-mixtures at several ratios of f48/65 to APOA2A peptides. The f48/65 at concentrations of one-fifth and one-tenth of 25 µM a48/65 peptide (1:0.2 and 1:0.1 ratios) completely inhibited the polymerization of APOA2A peptides into fibrils, results similar to those seen at a 1:1 ratio (Fig. 5E). The inhibition of polymerization continued over a 15-d period (Fig. S3). We also examined polymerization of other C-mixtures at higher concentrations of f48/65. The f48/65 at a concentration of 10 µM with 50 µM a48/65 [1:0.2(H)] (H, high-concentration condition), which mimicked the native situation of N- and C-terminal sequences of apoA-II, inhibited the polymerization of APOA2A peptides (Fig. 5F). In contrast, 5 µM f48/65 plus 50 µM a48/65 [1:0.1(H)] was less effective at inhibition than 1:0.2(H). Thus, the f48/65 inhibited the polymerization of APOA2A peptides into amyloid fibrils in a dose-dependent manner.
Next, we analyzed the polymerization of APOA2A peptides when there was a single amino acid substitution at positions 54 or 62 of APOA2A as shown in Fig. 1. The ThT intensities of a6/16 + a48/65(R54K) stimulated polymerization into fibrils (Fig. 6A). Using TEM, the amyloid fibrils were observed in the reaction mixture of a6/16 + a48/65(R54K), but, in addition, aggregated amorphous bodies were also detected in abundance (Fig. 6D). An amino acid substitution at position 62 (N62K) of APOA2A completely inhibited not only the polymerization of a6/16 + a48/65(N62K) into amyloid fibrils (Fig. 6A), but also the polymerization of the amyloidogenic APOA2A peptides (Fig. 6B). Moreover, the inhibitory ability of a48/65(N62K) tended to be stronger than that of the f48/65 (Figs. 5F and 6B and Fig. S4). We performed the polymerization using other variants [a48/65(N62R) and a48/65(N62A)] (Fig. 6 C and E). Similar to the abilities of the a48/65(N62K), a48/65(N62R) almost completely inhibited the polymerization into fibrils with the a6/16 and polymerization of the amyloidogenic APOA2A peptides. However, the inhibitory ability of a48/65(N62A) was weaker than that of a48/65(N62K), and only a few characteristic forms similar to fibrils derived from TYPE A were detected in the concentrated mixture of a6/16 + a48/65(N62A) (Fig. 6D).
Fig. 6.
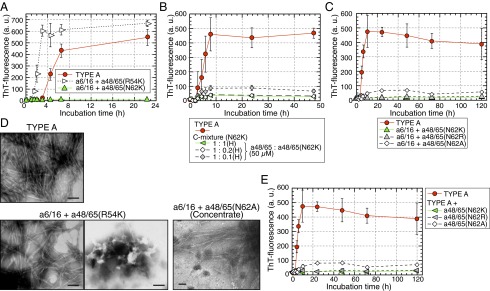
Single amino acid substitutions in the C-terminal sequence at position 62 of APOA2A inhibited the polymerization of amyloidogenic peptides. (A and C) ThT plots of the N-terminal APOA2A peptide plus a single amino acid substitution. Each concentration of peptides in the mixture was 50 µM. Each symbol and bar represents the mean ± SD (n = 3). a.u., arbitrary units. (B) ThT plots of APOA2A peptides mixed with a48/65(N62K) at various concentrations. Concentration of the a48/65 in the reaction mixtures was 50 µM similar to the experimental condition shown in Fig. 5F. Each symbol and bar represents the mean ± SD [n = 3 (TYPE A) and n = 4 (others)]. a.u., arbitrary units; H, high-concentration conditions. (E) ThT plots of APOA2A peptides mixed with a single amino acid substitution. The plot of TYPE A represents the same as in C because the experiments in C and E were performed at the same time. Each symbol and bar represents the mean ± SD (n = 3). a.u., arbitrary units. In C and E, the ThT intensities of the mixture using a48/65(N62A), but not a48/65(N62R), were higher compared with those of a48/65(N62K) after 10 h of incubation (P < 0.05, Student’s t test). (D) TEM images of the mixtures after 22.5 h of incubation shown in A. Because no characteristic form of amyloid fibrils was detected in the mixture of a6/16 + a48/65(N62A) after 120 h of incubation shown in C, the mixtures were concentrated ∼18-fold according to the method used to collect seeds. Only a few characteristic forms similar to fibrils derived from TYPE A in the concentrate were detected [a48/65(N62A) (Concentrate)]. (Scale bars: 100 nm.)
Testing the Hypothesis That the C-Terminal Sequence of APOA2F Might Block Further Amyloid Fibril Formation by Inhibiting Seeding Activity of Amyloid Fibrils.
We conducted experiments to elucidate whether C-terminal sequences of APOA2F might have other inhibitory effects on amyloid fibril formation. First, we performed polymerization of APOA2A peptides mixed with a48/65(N62K) for various incubation times (Fig. 7A). The ThT plot of APOA2A peptides plus the a45/65(N62K) at the initial phase increased only slightly and resembled that of the C-mixture 1:1(H) in Fig. 6B. Mixing APOA2A peptides with a48/65(N62K) at the plateau phase of polymerization yielded a plot that was similar to that obtained with the TYPE A over a 48-h incubation period. Interestingly, the plot of the mixture in the case of the addition of a45/65(N62K) at linear phase increased to 8 h and then decreased to less than half maximal intensity.
Fig. 7.
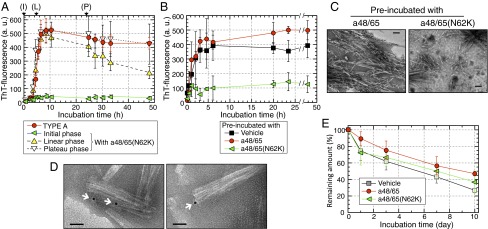
The a48/65(N62K) blocked amyloid fibril formation of amyloidogenic APOA2A peptides by inhibition of seeding activities. Amyloid fibril formation by the APOA2A peptides (a6/16 + a48/65) mixed with a48/65(N62K) at various times (A), with or without preincubated seeds (B), and the remaining amounts of premade fibrils after treatments with C-terminal peptides (E). Each symbol and bar represents the mean ± SD (n = 3 in A and B, and 5 in E). a.u., arbitrary units. (A) ThT plots of APOA2A peptides mixed with a48/65(N62K) from the beginning of agitation [initial phase (I)], after 4.5 h [linear phase (L)] and 24 h [plateau phase (P)] of incubation. Each concentration of peptides in the mixture was 50 µM. (B) ThT plots of the APOA2A peptides in the presence of preincubated seeds. (C) TEM images of the mixtures with preincubated seeds after 48 h of incubation in B. (Scale bars: 100 nm.) (D) TEM images of the preincubated seeds with biotin-labeled a48/65(N62K) using immunoelectron microscopy. Colloidal gold with streptavidin was detected close to the head of fibrils (arrows). (Scale bars: 50 nm.) (E) Remaining amounts of premade fibrils after incubation with C-terminal peptides. ThT intensities are expressed as a percentage of the level observed at the beginning of agitation. There was no significant difference among the curves of ThT intensities (one-way ANOVA).
Next, we examined extension of APOA2A peptides in the presence of preincubated seeds with C-terminal peptides. In the presence of preincubated seeds with a48/65, the ThT intensities increased from the beginning of agitation and reached a plateau phase after ∼4 h (Fig. 7B). The increase was similar to that of the APOA2A peptides mixture containing preincubated seeds with vehicle. The characteristic forms of amyloid fibrils were readily detected in the mixture containing preincubated seeds with a48/65 after 48 h incubation (Fig. 7C). In contrast, ThT intensities of the mixture in the presence of preincubated seeds with a48/65(N62K) increased moderately and reached a plateau phase at approximately one-quarter that of preincubated seeds with a48/65 or vehicle after 48 h incubation (Fig. 7B). In the mixtures with preincubated seeds with a48/65(N62K), shorter formed fibrils were detected compared with those of the mixtures with preincubated seeds with a48/65 (Fig. 7C).
We investigated the binding site of a48/65(N62K) on amyloid fibrils. First, we confirmed that the biotin-labeled a48/65(N62K) had an inhibitory effect on polymerization of APOA2A peptides almost equal to that of a48/65(N62K) (Fig. S5). Using the interaction of biotin and streptavidin, we observed that biotin-labeled a48/65(N62K) existed near amyloid fibrils. Under TEM, colloidal gold with streptavidin was detected close to the head, but not the body, of fibrils preincubated with the biotin-labeled a48/65(N62K) (Fig. 7D and Fig. S6).
We also analyzed the degradation of amyloid fibrils in the presence of C-terminal peptide. Premade seeds gradually decreased regardless of the C-terminal peptides added (Fig. 7E).
The C-Terminal Sequence of APOA2F Suppressed AApoAII Amyloidosis in Vivo.
We investigated whether the C-terminal sequence of APOA2F had inhibitory effects on AApoAII amyloidosis in vivo. All mice implanted with the a48/65(N62K) pump showed the onset of amyloidosis, but their amyloid deposits were lower than those in mice implanted with the vehicle or a48/65 pumps (Fig. 8A and Table S3). The severe amyloid deposits were observed abundantly in the tongues of mice implanted with the vehicle or a48/65 pumps (Fig. 8B). In contrast, in mice implanted with the a48/65(N62K) pump, amyloid deposits were comparatively moderate.
Fig. 8.

Suppression of amyloid deposits in vivo by treatment with a48/65(N62K). A single administration of 5 µg of AApoAII fibrils was given to 2-mo-old amyloidosis-susceptible mice using osmotic pumps implanted in their abdominal cavities. After 27 d, amyloid deposits were detected. (A) The AI in mice induced by AApoAII fibrils. Each bar and dot shows the mean of the group and individual grade of amyloid deposit. P value, Mann–Whitney U test. (B) LM images of amyloid deposits in the tongues by Congo red staining. (Left) Under normal light. (Right) Under polarized light. Grades of amyloid deposits in the tongues were 3, 3, and 2 in mice with vehicle, a48/65, and a48/65(N62K) pumps, respectively. (Scale bars: 100 µm.)
Discussion
We reported previously that the onset and frequency of senile amyloidosis was related to the type of apoA-II protein in mice (17, 19, 29, 33). As shown in Fig. 1, there are only two instances in which proline (Pro, P) was replaced by glutamine (Gln, Q) at position 5 and alanine (Ala, A) was replaced by valine (Val, V) at position 38 in a comparison between amyloidosis-low–sensitive APOA2B and amyloidosis-susceptible APOA2C mice (17, 19, 33). We previously demonstrated in vitro that the N-terminal peptides of both APOA2B and APOA2C (positions 1–16 with substitution of Gln for Pro at position 5) showed the same properties in the nucleation and extension phases of amyloid fibril formation in combination with the C-terminal peptide (positions 48–65) (23). Moreover, the N-terminal sequence from positions 1–5 was not critical for polymerization into fibrils. Although the C-terminal region was necessary for nucleation, its sequence was common between APOA2B and APOA2C. We could not explain the mechanisms underlying the reduced amyloidogenicity of APOA2B from the in vivo polymerization analysis. Genetic analyses suggested that higher levels of cholesterol and larger particles of HDL were present in the plasma of mice with APOA2B compared with mice with APOA2C (15, 34) and that it was due to an Ala-to-Val substitution at position 38 (35). In fact, we found that i.v. injection of AApoAII fibrils derived from APOA2B induced severe amyloidosis in SAMR1 mice with APOA2B (13). Thus, we expect that APOA2B might differ from APOA2C in binding affinity for HDL and in metabolic processes and that amyloidosis could be easily induced in mice with APOA2B by changing the microenvironment into a pathogenic state.
In the present study, using congenic mouse strains carrying APOA2F, we demonstrated that homozygous Apoa2f/f mice were “absolutely” resistant to amyloidosis (Fig. 2 and Tables S1 and S2). We also demonstrated that none of the plasma biochemical parameters of Apoa2f/f mice were significantly different from those of Apoa2a/a and Apoa2c/c mice (Fig. 2 and Fig. S1). In addition, the particle sizes of plasma HDL3 of homozygous Apoa2f/f mice were the same as those of Apoa2a/a (Fig. 3). From in vitro analyses, we demonstrated that the APOA2F peptides alone and also N- or C-terminal APOA2F peptide with C- or N-terminal APOA2A peptide did not polymerize into amyloid fibrils (Figs. 4 and 5). Those findings suggest that the mechanism underlying the resistance of APOA2F against amyloidosis might be different from the mechanism discussed for APOA2B in our previous reports (17, 29, 36) and that it might be based on the nonamyloidogenicity of APOA2F protein. However, our congenic mice were generated by backcrossing only six generations, and it is possible that other genes could play a major role in amyloidogenicity. Further genetic investigations of those mice will need to be conducted.
Most importantly, we found that APOA2F had an inhibitory effect on amyloid fibril formation by APOA2A. APOA2F had protective effects in vivo as seen in amyloidosis-induced Apoa2a/f mice (Fig. 2A) and in the spontaneous amyloidosis in Apoa2c/f mice (Table S2). In vitro, the C-terminal APOA2F peptide (f48/65) inhibited not only the polymerization into fibrils but also the extension phases of the amyloidogenic APOA2A peptides (Figs. 5 and 6 and Fig. S3). In particular, the a48/65(N62K) was a stronger inhibitor than the f48/65 (Fig. S4). We also showed that the a48/65(N62K) suppressed amyloid deposits in vivo (Fig. 8). Furthermore, we obtained findings supporting our hypothesis that the C-terminal APOA2F peptide might inhibit the extension by capping the active ends of the seeds or new amyloid fibrils made by fragmentation (Fig. 7 A–D and Fig. S6). The premade fibrils decreased gradually over a 10-d period regardless of the C-terminal peptides added (Fig. 7E). In contrast, when both N- and C-terminal APOA2A peptides were present in the reaction mixtures, the fibrils made from APOA2A peptides after 24 h of incubation were almost constant over a 15-d period (Fig. S3). The constant amount of amyloid fibrils over 24 h of incubation as TYPE A (Fig. S3) might be due to chemical equilibrium between the combination of peptides and fibrils. If this interpretation is correct, these results could suggest that the stability of amyloid fibrils derived from apoA-II peptides might require a supply of both N- and C-terminal peptides.
Based on our in vitro studies, we have proposed models describing the inhibition of amyloid fibril formation by apoA-II protein via a selective C-terminal APOA2F peptide (Fig. 9). At present, we do not know whether C-terminal APOA2F peptides directly contact APOA2A peptides and inhibit the nucleation phase of fibril formation. The C-terminal APOA2F peptide could inhibit fibril formation from the initial phase of polymerization (Figs. 5F and 6B). Thus, we believe that the C-terminal APOA2F peptide’s inhibition of polymerization at the extension phase cannot be explained without interaction between APOA2A and APOA2F. Further studies are required to investigate the inhibitory effect of the C-terminal APOA2F peptide on polymerization of other amyloid fibrils and/or amyloidogenic proteins.
Fig. 9.

Models showing amyloid fibril formation by apoA-II protein and the selective inhibition by the C-terminal APOA2F peptide. (A) Amyloid fibril formation of apoA-II peptides in vitro (23). N- and C-terminal APOA2A peptides change their secondary structures under acidic conditions (pH 2.5) with amyloidogenic conformation (intermediate) and polymerize into a nucleus (seed) (step 1). Once a nucleus is formed, or new nuclei (seeds) are made by fragmentation of amyloid fibrils, further extension of amyloidogenic peptides occurs (steps 2 and 3). Amyloid fibrils are degraded in the course of time, except for the supply of combination of N- and C-terminal amyloidogenic peptides (step 4). (B) Hypothetical mechanism by which the C-terminal APOA2F peptide inhibits amyloid fibril formation. The C-terminal APOA2F peptide strongly inhibits the extension phase of amyloidogenic peptides by capping possible active ends of seeds (step 5). As a result, amyloidogenic APOA2A peptides cannot polymerize into amyloid fibrils (step 2).
The Asp76Asn variant of β2-microglobulin (D76N β2M) is notable because the substituted amino acid changed the electric charge from negative to neutral at the single amino acid residue within the region promoting polymerization of WT β2M (37). The change resulted in accelerated polymerization of β2M into amyloid fibrils. We speculated that the Asn76 amide might establish a new hydrogen bond with Tyr78, as a result of which the equivalent region of D76N β2M would become more rigid than WT β2M. The a48/65(N62K) changed the electric charge to positive and eliminated the amide residue at position 62 of APOA2A. Its capacity for amyloidogenesis was the opposite of the D76N β2M. We speculate that the lack of an amide at position 62 of APOA2A, like a48/65(N62K), resulted in decreased amyloidogenicity. This hypothesis is supported by findings using another variant [a48/65(N62R)]. It has a positive electric charge but lacks an amide similar to the a48/65(N62K), and it almost completely inhibited the polymerization into amyloid fibrils (Fig. 6 C and E). On the other hand, another variant a48/65(N62A), without an amide residue at position 62 of APOA2A (but having the same electric charge as the C-terminal APOA2A peptide), was hard to polymerize into fibrils with the N-terminal APOA2A peptide, but its inhibitory ability was weaker than that of a48/65(N62K) (Fig. 6 C–E). We speculate that the inhibitory mechanism of the C-terminal APOA2F peptide requires both the absence of amide as well as a change of electric charge at position 62 of APOA2A.
Other reports showed that the hydrophobic core regions in α-synuclein and prion protein (associated with Parkinson's and prion diseases, respectively) polymerized into amyloid fibrils (38, 39). Based on those findings, we calculated the physicochemical properties of a48/65 and f48/65 in silico using Hydrophilicity Plot (Innovagen AB). The averaged hydrophilicity of the f48/65 was larger than that of the a48/65 (−0.4 and −0.6, respectively). The increased hydrophilicity of the f48/65 might be important in part for inhibiting polymerization under the conditions described here.
In light of those suggestions, we speculate that the mechanism involves a change of electric charge and the absence of the amide group and possibly the increased hydrophilicity at the amyloidogenic amino acid residue of APOA2A. One plausible explanation for this hypothesis could be the ability of N62 to engage in an amide zipper to stabilize an intermolecular β-sheet in which it is found. Other reports suggest that the amide in the amyloidogenic regions of proteins such as β2M and amylin are associated with amyloidogenicity (37, 40, 41). However, the 3D structure of mouse apoA-II is unknown, and its role in polymerization is unclear. Further studies are required to investigate the intermolecular mechanisms at residue 62 of apoA-II in amyloidogenicity.
We also calculated the amyloidogenic properties of APOA2C and APOA2F proteins in silico using the Zyggregator algorithm (University of Cambridge). This software predicts the intrinsic aggregation propensities of amino acid sequences by considering the physicochemical properties of amino acids (42–45). In the case of APOA2C, our selected N- and C-terminal peptides almost matched the in silico amyloidogenic sequences of apoA-II protein, and our in vitro strategy was supported (Fig. S7). At neutral pH, APOA2C tends to be amyloidogenic, similar to that under acidic conditions. The calculations suggest that mice having APOA2C are amyloidosis-susceptible. In contrast, the amyloidogenic properties of APOA2F were lower than those of APOA2C in the oligomeric and the amyloid states. Those calculations suggested that APOA2F might resist amyloid fibril formation compared with APOA2C and strongly support our in vivo and in vitro findings.
Our in vitro studies were performed under optimal acidic conditions to polymerize the fibrils in the absence of biomolecules such as lipids, proteoglycans, sialoglycolipids, and extracellular matrices present in the microenvironment of amyloid fibril formation in mice. We suspect that in vitro studies might exaggerate the inhibitory effect against the polymerization of amyloidosis-susceptible proteins, but we anticipate that our strategy could lead to new approaches to the inhibition of amyloid fibril formation and treatments that suppress amyloidosis in vivo.
Materials and Methods
Animals.
B6.SPRET-Apoa2f congenic mice were generated on a C57BL/6J genetic background using standard procedures. Mus spretus (SPRET/Ei) mice with the type F apoA-II allele (Apoa2f/f) were obtained from National Bio-Resource Project (National Institute of Genetics, Mishima, Japan). C57BL/6J mice with the type A apoA-II allele (Apoa2a/a) were purchased from Japan SLC, Inc. SPRET/Ei mice were backcrossed to C57BL/6J mice for six generations, after which heterozygous littermates (Apoa2a/f) were interbred and progeny (Apoa2a/a, Apoa2a/f and Apoa2f/f) were used for the indicated experiments. The apoA-II allele was determined by use of the PCR using specific primers for either Apoa2a or Apoa2f (Table S4). To confirm the strain-specific character of mice with APOA2F, we generated R1.SPRET-Apoa2f congenic mice on the SAMR1 genetic background. SPRET/Ei mice were backcrossed to R1.P1-Apoa2c (Apoa2c/c) for six generations. Due to fighting among the males, only female mice were used for induction of AApoAII amyloidosis. The R1.P1-Apoa2c congenic strain was developed in our laboratory and had the amyloidogenic Apoa2c/c allele from the SAMP1 strain on the genetic background of the SAMR1 strain (29). An amyloidosis-susceptible transgenic mouse strain (Apoa2c-Tg) that systemically overexpressed APOA2C was established in our laboratory as described previously (15). We used transgene-heterozygous (Tg+/−) mice to investigate the suppression of amyloid deposit by C-terminal peptides of apoA-II.
Mice were raised in the Division of Laboratory Animal Research, Research Center for Human and Environmental Sciences, Shinshu University, under specific pathogen-free conditions at 24 ± 2 °C with a light-controlled regimen (12 h light/dark cycle). A commercial diet (MF; Oriental Yeast) and tap water were available ad libitum.
Ethics Statement.
All experimental procedures conducted with mice were carried out in accordance with the Regulations for Animal Experimentation of Shinshu University.
Induction of AApoAII Amyloidosis.
AApoAII fibrils were obtained from the liver of an R1.P1-Apoa2c mouse that had severe amyloid deposits induced by an i.v. injection of AApoAII fibrils (14, 29, 46). Fibrils were isolated as a distilled water suspension using Pras’s method (47) and further purified by ultracentrifugation. The purified AApoAII fibrils were stored in a –80 °C freezer until used for induction of amyloidosis in mice. For induction of AApoAII amyloidosis, AApoAII fibrils were resuspended in sterile distilled water and sonicated according to our previous method (14). Two-month-old B6.SPRET-Apoa2f and R1.SPRET-Apoa2f mice were injected i.v. with a single dose of 100 µg of fibrils, after 2–9 mo, and were killed by cardiac puncture under deep diethyl ether anesthesia. Several organs were collected from these congenic animals and used to detect amyloid deposits.
Detection of Amyloid Deposits in Mice.
Organs were fixed in 10% (vol/vol) neutral buffered formalin, embedded in paraffin, and cut into 4-µm sections using standard procedures. Amyloid deposits were identified with polarized LM (Axioskop 2; Carl Zeiss Japan) to detect apple-green color birefringence in tissue sections stained with 1% (wt/vol) Congo red (19, 48, 49). The intensity of deposit was quantitated using the AI, which was the average of grades 0–4 in the following seven organs: tongue, stomach, small intestine, liver, spleen, heart, and skin (13, 29, 30). AApoAII fibrils were also identified immunohistochemically using the horseradish peroxidase-labeled streptavidin-biotin method (DAKO) with specific antiserum against mouse apoA-II. This anti–apoA-II antiserum was prepared in our laboratory (30). It reacts specifically with plasma APOA2A, APOA2B, and APOA2C on Western blotting and with AApoAII fibrils derived from three types of apoA-II proteins in both Western blotting and immunohistochemical analyses (13, 15, 31).
Levels of apoA-I and apoA-II in HDL, HDL Particle Size, and Plasma Cholesterols.
Plasma was collected from 2-mo-old B6.SPRET-Apoa2f mice that had fasted for 12–16 h and was stored in a −20 °C freezer until used for analysis. Aliquots were thawed on ice. To detect the levels of apoA-I and apoA-II in HDL, we collected the HDL fraction from plasma using the standard method of density gradient ultracentrifugation (15, 32, 34). HDL samples were loaded on a Tris-Tricine SDS polyacrylamide gel [16.5% (wt/vol) acrylamide] for electrophoresis (SDS/PAGE). After electrophoresis, the gel was stained with 0.2% Coomassie Brilliant Blue (CBB) dye (R-250; MP Biomedicals). ApoA-I and apoA-II were identified by the markers of molecular size (Kaleidoscope Polypeptide Standards 95790; Bio-Rad), which were loaded at the same time. To determine the HDL particle size, plasma was prestained by Sudan Black B dye and followed by electrophoresis on a nondenaturing PAGE gel containing a 5–15% (wt/vol) linear polyacrylamide gradient. Plasma total and HDL cholesterol levels in 2-mo-old mice were determined using quantitative assay kits based on an enzymatic procedure or by a modified heparin-manganese precipitation procedure, respectively (15, 32).
Polymerization of Synthetic Partial Peptides into Amyloid Fibrils.
Synthetic partial peptides [>95.0% purity; biotin-labeled a48/65(N62K), 90.3% purity] of mouse apoA-II protein were obtained from Sigma-Genosys. We used the two distinct domains of apoA-II: The N-terminal peptide (6/16) from positions 6–16 and the C-terminal peptide (48/65) from positions 48–65 were critical for polymerization into amyloid fibrils (23). We prepared solutions of each peptide in 100% dimethyl sulfoxide (DMSO) using methods described previously (23). The peptide contents of these solutions were determined using a BCA Protein Assay Reagent Kit (Pierce). Aliquots of these peptide solutions were stored in a refrigerator (4 °C) and thawed at 20 °C with shaking at 300 rpm for 1 h and used immediately in each experiment.
Polymerization of synthetic peptides was performed using a method described previously (23). Briefly, reaction mixtures in prechilled tubes (Safe-lock tube; Eppendorf AG) contained synthetic peptides, 50 mM sodium citrate buffer (pH 2.5), 100 mM NaCl, and 5% (vol/vol) DMSO at final concentrations. After brief blending, the reaction mixtures were incubated with agitation at 300 rpm at 37 °C in a shaker (Thermomixer Comfort; Eppendorf AG), and aliquots were used for ThT binding assays at arbitrary intervals. These mixtures were also used for detecting characteristic structures and properties of amyloid fibrils using TEM and LM. The components of amyloid fibrils were analyzed by LC-MS/MS.
Extension of Amyloid Fibrils by Synthetic Partial Peptides.
Extension of amyloid fibrils by synthetic peptides was also performed using methods described previously (23). To generate “seeds” for extension reactions of amyloid fibril formation, we performed polymerization of APOA2A peptides (a6/16 + a48/65) for 3 d of incubation. “Premade fibrils” were collected by centrifugation at 1.61 × 104 × g at 4 °C for 3 h using a high speed refrigerated microcentrifuge (5415R; Eppendorf) and were stored in a –80 °C freezer until used.
To examine in detail the inhibitory effects of the C-terminal APOA2F peptide on the extension phase of amyloid fibril formation, 25 µg/mL premade fibrils were resuspended in the reaction solution containing 50 µM a48/65 or a48/65(N62K). The mixtures were incubated with agitation at 300 rpm at 37 °C for 30 min and were recentrifuged. Pellets were carefully collected as “preincubated seeds.” We performed extension of the APOA2A peptides with these preincubated seeds. We also prepared preincubated seeds with biotin-labeled a48/65(N62K) and used them immediately for immunoelectron microscopy.
Degradation of Amyloid Fibrils Derived from Synthetic Partial Peptides.
Twenty-five micrograms per milliliter premade fibrils and 50 µM a48/65 or a48/65(N62K) were mixed in the reaction solution. After brief blending, the mixtures were incubated with agitation at 300 rpm at 37 °C for 0–10 d, and aliquots were used for ThT binding assays at arbitrary intervals.
ThT Binding Assay.
The amount of amyloid fibrils derived from synthetic peptides was determined with a fluorometric assay with ThT (23, 24, 31, 50). Measurements were made in a total volume of 1 mL, containing 2.5- or 5-µL aliquots of the reaction mixtures, 250 nM ThT, and 50 mM glycine-NaOH buffer (pH 9.0). After briefly mixing the solutions at room temperature, the ThT-fluorescence intensities were measured using a fluorescence spectrophotometer (RF-5300PC; Shimadzu Corporation) at the excitation and emission wavelengths of 450 nm and 482 nm, respectively. Each reaction mixture was assayed in triplicate, and the average was calculated.
Microscopic Analyses of the Experiments Using Synthetic Peptides.
For TEM experiments, we prepared the sample according to methods described previously (23). The reaction mixtures were spread on carbon-coated grids (400 mesh; NEM) and were negatively stained with 1% (wt/vol) phosphotungstic acid (electron microscopy grade; TAAB Laboratories Equipment Ltd.)-NaOH (pH 7.0). These samples were examined under an electron microscope (1200EX or 1400EX; JEOL) with an acceleration voltage of 80 kV. Immunoelectron microscopy was performed using a modified method in parts described previously (51). The preincubated seeds with biotin-labeled a48/65(N62K) were resuspended in the reaction solution and recentrifuged. Pellets were resuspended again and loaded immediately onto carbon-coated grids at room temperature (25 °C) for 15 min. The grids were immersed in 1:4 streptavidin-conjugated 10 nm colloidal gold (EM.STP10; BBI Solutions) in 50 mM phosphate buffer (pH 7.0) containing 0.5% BSA and 0.05% Tween 20. After 3 h at 25 °C, the grids were washed twice using phosphate buffer and then negatively stained with 1% phosphotungstic acid-NaOH (pH 7.0). The grids were observed under an electron microscope 1400EX.
For LM observations, the reaction mixtures were applied to silanized glass slides (S3003; DAKO Cytomation Co.), dried overnight in a 37 °C dry box, and stained with 1% Congo red dye (23). These samples were examined for apple-green colored birefringence under polarized light using a LM.
LC-MS/MS Analysis.
Aliquots of reaction mixtures were centrifuged, and the pellets were stored in a −80 °C freezer immediately after the supernatants were removed. The pellets were redissolved in 99% formic acid and diluted with 2% (vol/vol) formic acid/2% (vol/vol) acetonitrile, and analyzed by LC-MS/MS as described previously (23, 52). The sample solutions were applied to a Paradigm MS4 (Microm BioResources, Inc.) HPLC system fitted with an HTC-PAL automatic sampler (CHROMSYS LLC). A reverse-phase capillary column (Develosil ODS-HG3, 0.075 mm i.d. × 50 mm; Nomura Chemical Co. Ltd.) was used at a flow rate of 200 nL/min with a 4–72% (vol/vol) linear gradient of acetonitrile in 0.1% formic acid. Eluted apoA-II partial peptides were directly detected with an ion trap mass spectrometer, LXQ (Thermo Electron Corporation). The data were analyzed with SEQUEST (Thermo Electron Corporation).
Suppression of AApoAII Amyloidosis in Mice by the C-Terminal Peptide of apoA-II.
Osmotic pumps (ALZET micro-osmotic pump, no. 1004; DURECT Corp.) were filled with 1.0 mM a48/65(N62K) or a48/65 peptide solutions in 5% (vol/vol) DMSO or vehicle according to the manufacturer’s instructions. Two-month-old Apoa2c-Tg+/− mice were anesthetized with pentobarbital sodium (Somnopentyl; Kyoritsu Seiyaku Corp.), and the osmotic pumps were implanted into their abdominal cavities. One day after the implantation, the mice were injected i.v. with 5 µg of sonicated AApoAII fibrils. After 27 d, the mice were killed by cardiac puncture under deep anesthesia using diethyl ether. Several organs were collected and used to detect amyloid deposits.
Chemical Reagents.
Unless otherwise specified, chemical reagents in this section were obtained from Wako Pure Chemical Industries, Ltd.
Statistical Analysis.
We used the R software package (The R Development Core Team, Vienna University of Economics and Business). The Mann–Whitney U test was used to analyze the AI for amyloid deposits. The Tukey–Kramer method for multiple comparisons was performed to test for any significant differences in various pairwise comparisons in blood biochemical parameters. The Student’s t test was used to analyze the inhibitory abilities of the C-terminal peptides on polymerization of APOA2A peptides. The one-way ANOVA was used to test for any significant differences in the stability of amyloid fibrils in the presence of additional C-terminal peptides. P values <0.05 were regarded as statistically significant.
Supplementary Material
Acknowledgments
We thank Prof. Kiyoshi Matsumoto, Ms. Ayako Nishio, Mr. Kiyokazu Kametani, and Ms. Kayo Suzuki (Research Support Center for Human and Environmental Sciences, Shinshu University) for animal care and technical assistance with TEM. We greatly appreciate the helpful suggestions and comments from Prof. Yuji Goto (Osaka University) and Prof. Vittorio Bellotti (University of Pavia). This research was supported in part by Grants-in-Aid for Priority Areas (22020015) and Scientific Research (B) (20300144, 23390093, and 26293084) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and by grants for the Amyloidosis Research Committee from the Ministry of Health, Labour and Welfare, Japan.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1416363112/-/DCSupplemental.
References
- 1.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol. 1998;8(1):101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 2.Westermark P, et al. Amyloid fibril protein nomenclature: 2002. Amyloid. 2002;9(3):197–200. doi: 10.3109/13506120209114823. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen HD, Hall CK. Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc Natl Acad Sci USA. 2004;101(46):16180–16185. doi: 10.1073/pnas.0407273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindgren M, Sörgjerd K, Hammarström P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys J. 2005;88(6):4200–4212. doi: 10.1529/biophysj.104.049700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olzscha H, et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144(1):67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- 6.Tjernberg L, Hosia W, Bark N, Thyberg J, Johansson J. Charge attraction and β propensity are necessary for amyloid fibril formation from tetrapeptides. J Biol Chem. 2002;277(45):43243–43246. doi: 10.1074/jbc.M205570200. [DOI] [PubMed] [Google Scholar]
- 7.Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J Mol Med (Berl) 2003;81(11):678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 8.Selkoe DJ. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat Cell Biol. 2004;6(11):1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- 9.Xing Y, Higuchi K. Amyloid fibril proteins. Mech Ageing Dev. 2002;123(12):1625–1636. doi: 10.1016/s0047-6374(02)00098-2. [DOI] [PubMed] [Google Scholar]
- 10.Sekijima Y, et al. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121(1):73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 11.Blanco-Vaca F, Escolà-Gil JC, Martín-Campos JM, Julve J. Role of apoA-II in lipid metabolism and atherosclerosis: Advances in the study of an enigmatic protein. J Lipid Res. 2001;42(11):1727–1739. [PubMed] [Google Scholar]
- 12.Higuchi K, et al. Purification and characterization of a senile amyloid-related antigenic substance (apoSASSAM) from mouse serum: apoSASSAM is an apoA-II apolipoprotein of mouse high density lipoproteins. J Biol Chem. 1986;261(27):12834–12840. [PubMed] [Google Scholar]
- 13.Xing Y, et al. Induction of protein conformational change in mouse senile amyloidosis. J Biol Chem. 2002;277(36):33164–33169. doi: 10.1074/jbc.M111570200. [DOI] [PubMed] [Google Scholar]
- 14.Zhang H, et al. Transmissibility of mouse AApoAII amyloid fibrils: Inactivation by physical and chemical methods. FASEB J. 2006;20:1012–1014. doi: 10.1096/fj.05-4890fje. [DOI] [PubMed] [Google Scholar]
- 15.Ge F, et al. Amyloidosis in transgenic mice expressing murine amyloidogenic apolipoprotein A-II (Apoa2c) Lab Invest. 2007;87(7):633–643. doi: 10.1038/labinvest.3700559. [DOI] [PubMed] [Google Scholar]
- 16.Kitagawa K, et al. Polymorphisms of mouse apolipoprotein A-II: Seven alleles found among 41 inbred strains of mice. Amyloid. 2003;10(4):207–214. doi: 10.3109/13506120309041737. [DOI] [PubMed] [Google Scholar]
- 17.Higuchi K, Yonezu T, Tsunasawa S, Sakiyama F, Takeda T. The single proline-glutamine substitution at position 5 enhances the potency of amyloid fibril formation of murine apo A-II. FEBS Lett. 1986;207(1):23–27. doi: 10.1016/0014-5793(86)80006-0. [DOI] [PubMed] [Google Scholar]
- 18.Fu X, et al. Induction of AApoAII amyloidosis by various heterogeneous amyloid fibrils. FEBS Lett. 2004;563(1-3):179–184. doi: 10.1016/S0014-5793(04)00295-9. [DOI] [PubMed] [Google Scholar]
- 19.Higuchi K, et al. Polymorphism of apolipoprotein A-II (apoA-II) among inbred strains of mice: Relationship between the molecular type of apoA-II and mouse senile amyloidosis. Biochem J. 1991;279(Pt 2):427–433. doi: 10.1042/bj2790427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Higuchi K, Naiki H, Kitagawa K, Hosokawa M, Takeda T. Mouse senile amyloidosis: ASSAM amyloidosis in mice presents universally as a systemic age-associated amyloidosis. Virchows Arch B Cell Pathol Incl Mol Pathol. 1991;60(4):231–238. [PubMed] [Google Scholar]
- 21.HogenEsch H, et al. Gastrointestinal AAPOAII and systemic AA-amyloidosis in aged C57BL/Ka mice: Amyloid-type dependent effect of long-term immunosuppressive treatment. Virchows Arch B Cell Pathol Incl Mol Pathol. 1993;64(1):37–43. doi: 10.1007/BF02915094. [DOI] [PubMed] [Google Scholar]
- 22.Gruys E, Tooten PCJ, Kuijpers MHM. Lung, ileum and heart are predilection sites for AApoAII amyloid deposition in CD-1 Swiss mice used for toxicity studies: Pulmonary amyloid indicates AApoAII. Lab Anim. 1996;30(1):28–34. doi: 10.1258/002367796780745018. [DOI] [PubMed] [Google Scholar]
- 23.Sawashita J, et al. Amyloid fibrils formed by selective N-, C-terminal sequences of mouse apolipoprotein A-II. Biochim Biophys Acta. 2009;1794(10):1517–1529. doi: 10.1016/j.bbapap.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 24.Hasegawa K, Yamaguchi I, Omata S, Gejyo F, Naiki H. Interaction between A β(1-42) and A β(1-40) in Alzheimer’s β-amyloid fibril formation in vitro. Biochemistry. 1999;38(47):15514–15521. doi: 10.1021/bi991161m. [DOI] [PubMed] [Google Scholar]
- 25.Kozhukh GV, et al. Investigation of a peptide responsible for amyloid fibril formation of β2-microglobulin by achromobacter protease I. J Biol Chem. 2002;277(2):1310–1315. doi: 10.1074/jbc.M108753200. [DOI] [PubMed] [Google Scholar]
- 26.Modler AJ, et al. Polymerization of proteins into amyloid protofibrils shares common critical oligomeric states but differs in the mechanisms of their formation. Amyloid. 2004;11(4):215–231. doi: 10.1080/13506120400014831. [DOI] [PubMed] [Google Scholar]
- 27.Kammerer RA, et al. Exploring amyloid formation by a de novo design. Proc Natl Acad Sci USA. 2004;101(13):4435–4440. doi: 10.1073/pnas.0306786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen HD, Hall CK. Kinetics of fibril formation by polyalanine peptides. J Biol Chem. 2005;280(10):9074–9082. doi: 10.1074/jbc.M407338200. [DOI] [PubMed] [Google Scholar]
- 29.Higuchi K, et al. Fibrilization in mouse senile amyloidosis is fibril conformation-dependent. Lab Invest. 1998;78(12):1535–1542. [PubMed] [Google Scholar]
- 30.Higuchi K, et al. Systemic senile amyloid in senescence-accelerated mice: A unique fibril protein demonstrated in tissues from various organs by the unlabeled immunoperoxidase method. Lab Invest. 1983;48(2):231–240. [PubMed] [Google Scholar]
- 31.Korenaga T, et al. Tissue distribution, biochemical properties, and transmission of mouse type A AApoAII amyloid fibrils. Am J Pathol. 2004;164(5):1597–1606. doi: 10.1016/S0002-9440(10)63718-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, et al. ApoA-I deficiency in mice is associated with redistribution of apoA-II and aggravated AApoAII amyloidosis. J Lipid Res. 2011;52(8):1461–1470. doi: 10.1194/jlr.M013235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higuchi K, et al. Accelerated senile amyloidosis induced by amyloidogenic Apoa-II gene shortens the life span of mice but does not accelerate the rate of senescence. J Gerontol A Biol Sci Med Sci. 1996;51(4):B295–B302. doi: 10.1093/gerona/51a.4.b295. [DOI] [PubMed] [Google Scholar]
- 34.Higuchi K, et al. Development of congenic strains of mice carrying amyloidogenic apolipoprotein A-II (Apoa2c): Apoa2c reduces the plasma level and the size of high density lipoprotein. FEBS Lett. 1993;317(3):207–210. doi: 10.1016/0014-5793(93)81277-7. [DOI] [PubMed] [Google Scholar]
- 35.Suto J, Takahashi Y, Sekikawa K. Quantitative trait locus analysis of plasma cholesterol and triglyceride levels in C57BL/6J x RR F2 mice. Biochem Genet. 2004;42(9-10):347–363. doi: 10.1023/b:bigi.0000039809.40228.d3. [DOI] [PubMed] [Google Scholar]
- 36.Yonezu T, et al. A molecular-pathologic approach to murine senile amyloidosis: Serum precursor-apolipoprotein A-II variant (Pro5----Gln) presents only in the senile amyloidosis-prone SAM-P/1 and SAM-P/2 mice. Lab Invest. 1987;57(1):65–70. [PubMed] [Google Scholar]
- 37.Valleix S, et al. Hereditary systemic amyloidosis due to Asp76Asn variant β2-microglobulin. N Engl J Med. 2012;366(24):2276–2283. doi: 10.1056/NEJMoa1201356. [DOI] [PubMed] [Google Scholar]
- 38.Biasini E, et al. The hydrophobic core region governs mutant prion protein aggregation and intracellular retention. Biochem J. 2010;430(3):477–486. doi: 10.1042/BJ20100615. [DOI] [PubMed] [Google Scholar]
- 39.Tzotzos S, Doig AJ. Amyloidogenic sequences in native protein structures. Protein Sci. 2010;19(2):327–348. doi: 10.1002/pro.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Torbeev VY, Hilvert D. Both the cis-trans equilibrium and isomerization dynamics of a single proline amide modulate β2-microglobulin amyloid assembly. Proc Natl Acad Sci USA. 2013;110(50):20051–20056. doi: 10.1073/pnas.1310414110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexandrescu AT. Amide proton solvent protection in amylin fibrils probed by quenched hydrogen exchange NMR. PLoS ONE. 2013;8(2):e56467. doi: 10.1371/journal.pone.0056467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pawar AP, et al. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J Mol Biol. 2005;350(2):379–392. doi: 10.1016/j.jmb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 43.Tartaglia GG, et al. Prediction of aggregation-prone regions in structured proteins. J Mol Biol. 2008;380(2):425–436. doi: 10.1016/j.jmb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 44.Tartaglia GG, Vendruscolo M. The Zyggregator method for predicting protein aggregation propensities. Chem Soc Rev. 2008;37(7):1395–1401. doi: 10.1039/b706784b. [DOI] [PubMed] [Google Scholar]
- 45.Hamada D, et al. Competition between folding, native-state dimerisation and amyloid aggregation in β-lactoglobulin. J Mol Biol. 2009;386(3):878–890. doi: 10.1016/j.jmb.2008.12.038. [DOI] [PubMed] [Google Scholar]
- 46.Fu L, et al. Extrahepatic expression of apolipoprotein A-II in mouse tissues: Possible contribution to mouse senile amyloidosis. J Histochem Cytochem. 2001;49(6):739–748. doi: 10.1177/002215540104900607. [DOI] [PubMed] [Google Scholar]
- 47.Pras M, Zucker-Franklin D, Rimon A, Franklin EC. Physical, chemical, and ultrastructural studies of water-soluble human amyloid fibrils: Comparative analyses of nine amyloid preparations. J Exp Med. 1969;130(4):777–796. doi: 10.1084/jem.130.4.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puchtler H, Sweat F, Levine M. On the binding of Congo red by amyloid. J Histochem Cytochem. 1962;10:355–364. [Google Scholar]
- 49.Ravid M, Gafni J, Sohar E, Missmahl H-P. Incidence and origin of non-systemic microdeposits of amyloid. J Clin Pathol. 1967;20(1):15–20. doi: 10.1136/jcp.20.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal Biochem. 1989;177(2):244–249. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 51.Yan J, et al. Cross-seeding and cross-competition in mouse apolipoprotein A-II amyloid fibrils and protein A amyloid fibrils. Am J Pathol. 2007;171(1):172–180. doi: 10.2353/ajpath.2007.060576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kametani F. Secretion of long Abeta-related peptides processed at ε-cleavage site is dependent on the α-secretase pre-cutting. FEBS Lett. 2004;570(1-3):73–76. doi: 10.1016/j.febslet.2004.06.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.