

Significance
The last decade has delivered astounding advances in DNA sequencing technology, which has led to the wide discovery of point mutations in genes associated with neuropsychiatric disorders. However, the mechanism by which the majority of mutations may contribute to the disease is unknown. Here we show that a reported autism mutation can dramatically inhibit a posttranslational modification, which leads to profound synaptic changes. These data underscore how a simple perturbation of a protein at the synapse can lead to dramatic changes in synaptic transmission and supports a model by which imbalances of excitatory/inhibitory currents may underlie the pathophysiology of autism.
Keywords: autism, neuroligin, synaptogenesis, development, synaptic adhesion molecule
Abstract
Autism spectrum disorders (ASDs) comprise a highly heritable, multifarious group of neurodevelopmental disorders, which are characterized by repetitive behaviors and impairments in social interactions. Point mutations have been identified in X-linked Neuroligin (NLGN) 3 and 4X genes in patients with ASDs and all of these reside in their extracellular domains except for a single point mutation in the cytoplasmic domain of NLGN4X in which an arginine is mutated to a cysteine (R704C). Here we show that endogenous NLGN4X is robustly phosphorylated by protein kinase C (PKC) at T707, and R704C completely eliminates T707 phosphorylation. Endogenous NLGN4X is intensely phosphorylated on T707 upon PKC stimulation in human neurons. Furthermore, a phospho-mimetic mutation at T707 has a profound effect on NLGN4X-mediated excitatory potentiation. Our results now establish an important interplay between a genetic mutation, a key posttranslational modification, and robust synaptic changes, which can provide insights into the synaptic dysfunction of ASDs.
In a 2014 report published by the Centers for Disease Control and Prevention (CDC), it was estimated that 1 in 68 children in the United States have an autism spectrum disorder (ASD) (1). These neuropsychiatric disorders have a strong genetic component consistent with high recurrence rates between siblings and a higher concordance frequency seen in monozygotic than dizygotic twins. Furthermore, deletions, insertions, and substitutions have been identified within the genome that increase the risk of these disorders (2, 3). These cytogenetic and genome sequencing studies have revealed that Neuroligins (NLGNs) are one of a subset of genes encoding synaptic proteins associated with ASDs (4, 5).
The NLGN gene family consists of five members (Nlgn1, 2, 3, 4X, and 4Y) within the human genome that encode transsynaptic cell adhesion molecules that are critical for synapse assembly, maintenance, and plasticity (5–7). Numerous studies have identified a variety of mutations in X-linked NLGN3 and 4X genes that range from copy number variants (8–11) to protein truncations and amino acid substitutions in patients with ASDs (4, 12, 13). Interestingly, all of the point mutations in NLGN3 and NLGN4X reside in their extracellular domains except for a single point mutation in the cytoplasmic domain (c-tail) of NLGN4X at arginine (R) 704, which is modified to a cysteine (C) (14). How this mutation or other NLGN disease mutations contribute to the pathophysiology is unknown.
Protein phosphorylation is a critical modulator of NLGN function (15, 16). Recently, we showed that Ca2+/CaM kinase II (CaMKII) phosphorylates the c-tail of NLGN1 in an isoform-specific and activity-dependent manner, which regulates its ability to enhance excitatory synapses (16). Next, we wondered if different kinases might regulate the function of other NLGN isoforms?
In the present study, we show in vitro and in vivo that NLGN4X is robustly phosphorylated by protein kinase C (PKC). We identified the dominant phosphorylation site as T707, a residue not conserved in NLGNs 1, 2, and 3. Intriguingly, PKC phosphorylation is eliminated with the autism-associated mutation R704C. Most dramatically, we found that the phospho-mimetic mutation at T707 profoundly enhances anatomical markers for synapses and potentiates NLGN4X-mediated excitatory synaptic transmission. This study establishes compelling evidence that NLGN4X can act at and regulate excitatory synapses. Furthermore, it demonstrates, strikingly, how a single point mutation in NLGNs can acutely adjust synaptic properties.
Results
PKC Phosphorylates NLGN4X at T707.
To test whether NLGNs are substrates for kinases other than CaMKII, we conducted an in vitro kinase assay with GST-fusion constructs with the c-tails of NLGNs 1, 2, 3, and 4X (Fig. 1A and Fig. S1) and found that NLGN4X was robustly phosphorylated by PKC as evaluated by radiography (Fig. 1B). Weak phosphorylation of NLGN1 and NLGN3 was observed compared with NLGN4X (Fig. 1B, see longer exposure), indicating NLGN4X to be the best NLGN substrate for PKC. To identify the individual PKC phosphorylation site(s), we subjected NLGN4X to the same in vitro kinase assay and evaluated phosphorylation using liquid LC coupled to tandem MS (LC-MS/MS) and identified the major phosphorylation site as T707 (Fig. 1C), a residue that is not conserved in NLGNs 1, 2, and 3 (Fig. 1A and Fig. S1 show complete alignment).
Fig. 1.

Autism-associated mutation eliminates PKC phosphorylation of NLGN4X. (A) Alignment of the transmembrane domains and partial c-tails of human NLGNs 1, 2, 3, 4X, and 4Y. The PKC phosphorylation site, T707, is boxed in orange; autism mutation, R704, boxed in blue; and pT707-Ab epitope boxed in yellow. The CaMKII phosphorylation site on NLGN1 is boxed in gray. (B and D) Purified PKC and [γ-P-32]ATP were incubated with GST-fusion proteins and analyzed by autoradiography. CBB protein staining was used to visualize total protein, and GST (negative) and GST-GluA1 c-tail (positive) functioned as phosphorylation controls. (C) ETD MS/MS spectrum of chymotrypsin digested phosphorylated NLGN4X peptide 704-RHETHRRPSPQ-714 found in GST-NLGN4X fusion proteins that were incubated with ATP and purified PKC. Samples were analyzed using the LC/MS/MS method. (E) Means ± SEM of phosphorylated NLGN4X by PKC normalized to WT (n = 4) for NLGN4X R704C (P = 0.0013, n = 4) and NLGN4X T707A (P = 0.0030, n = 4). (F) GST, GST-NLGNs 1, 2, 3, and 4X (WT, R704C, and T707A) were incubated with PKC, and phosphorylation was evaluated by immunoblotting with pT707-Ab. Total protein was evaluated with a GST-Ab. (G) GFP, FLAG-GluA1, or NLGN4X (WT, R704C, T707A, or T707D) were transfected in COS cells and treated with DMSO or PMA, a PKC activator. GluA1 pS831 served as control for PKC activation and the arrow denotes the NLGN4X specific band. (H) Means ± SEM of phosphorylated NLGN4X pT707 achieved by PMA activation normalized to WT (n = 3) and NLGN4X R704C (P = 0.0310, n = 3). (I) Means ± SEM of phosphorylated GluA1 S831 achieved by PMA activation (P = 0.0187, n = 3) normalized to no treatment (n = 3). Immunoblots (WB) were probed with indicated antibodies in F and G. *P < 0.05 **P < 0.01.
Autism-Associated Mutation Eliminates PKC Phosphorylation of NLGN4X.
Interestingly, T707 resides only a few amino acids away from the only known autism-associated mutation (R704C) in the c-tail of any NLGN protein (Fig. 1A). This substitution replaces a positive charge for a polar amino acid and occupies a critical residue in a potential PKC consensus sequence (RXXS/T, where X denotes any amino acid). Might the autism-related R704C mutation abolish PKC phosphorylation of NLGN4X at T707? To independently verify the MS results, we performed the same in vitro kinase assay with a nonphosphorylatable point mutation, T707A, and showed that T707 was the dominant NLGN4X PKC phosphorylation site, which resulted in an ∼75% reduction in NLGN4X phosphorylation. Likewise, NLGN4X R704C eliminated T707 phosphorylation (Fig. 1 D and E). Importantly, CaMKII phosphorylation of NLGN4X T718, the only other known phosphorylation site near T707, was not reduced by the R704C mutation, demonstrating that this mutation did not result in a generalized problem rendering the protein unphosphorylated (Fig. S2 A and B). Taken together, these results indicate that NLGN4X is a robust substrate for PKC, and the only autism-associated NLGN4X c-tail mutation abolishes T707 phosphorylation.
To detect NLGN4X T707 phosphorylation in vivo, we produced a phosphorylation state-specific Ab, pT707-Ab, against residues 703–712 in NLGN4X (Fig. 1A). The pT707 Ab only detected NLGN4X that was phosphorylated on T707 as detected by a PKC in vitro kinase assay that was resolved by SDS/PAGE and subsequent immunoblotting (Fig. 1F). Critically, the pT707-Ab did not show any immunoreactivity with the nonphosphorylated NLGN4X, the nonphosphorylatable mutants (R704C or T707A), or any of the other NLGN isoforms that were tested.
The in vitro experiments were performed with fusion proteins of NLGN c-tail isoforms. To test whether full-length NLGN4X can be phosphorylated in mammalian cells and modulated by PKC activity, we transfected WT NLGN4X or various NLGN4X mutants (R704C, T707A, or T707D) in COS-7 cells and immunoblotted cell lysates with the pT707-Ab. Under basal conditions, NLGN4X was not phosphorylated at T707, but transient activation of PKC by phorbol 12-myristate 13-acetate (PMA) triggered a robust increase in pT707 phosphorylation (Fig. 1G). NLGN4X R704C resulted in an ∼95% reduction in pT707 phosphorylation, whereas NLGN4X T707A or the phospho-mimetic mutation, T707D, was not detected by pT707-Ab (Fig. 1 G and H). Phosphorylation of serine (S) 831 on GluA1 was used as a positive control for PMA activation of PKC (17) (Fig. 1 G and I). Additionally, the pT707-Ab had astonishing specificity for immunoprecipitating (IP) only NLGN4X phosphorylated on T707 as validated by Western blotting with two independent NLGN4X Abs and a pan NLGN-Ab (Fig. S3). Collectively, these data demonstrate that NLGN4X T707 is phosphorylated in heterologous cells, and the pT707-Ab is a dynamic and isoform-specific reagent that faithfully distinguishes the phosphorylated form of NLGN4X on T707.
Phosphorylation of NLGN4X at T707 Induces Synaptogenesis.
Previously, two autism-associated mutations in NLGN3 (R451C) and NLGN4X (R87W) were shown to disrupt surface expression (18, 19). Moreover, phosphorylation of the NLGN1 c-tail regulates its forward trafficking (16). Does T707 regulate NLGN4X surface expression? To test this possibility, cultured rat hippocampal neurons were transfected with NLGN4X (WT, R704C, T707A, or T707D) and visualized with immunofluorescence confocal microscopy. To prevent potential heterodimerization, we performed these experiments on a reduced endogenous NLGN background using an exogenous chained microRNA against NLGNs 1, 2, and 3 (NLmiRs) as previously described (20). Human NLGN4X and NLGN4Y are absent in rats (21). Consequently, rat neurons were used in all imaging and physiology experiments to ensure an NLGN4-null background to avoid any complications that might occur between dimerization of WT and mutant NLGN4 receptors. Phosphorylation at T707 did not affect the trafficking of NLGN4X to the cell surface (Fig. 2 A and B). However, we noticed that expression of a phospho-mimetic mutation of NLGN4X T707 (T707D) resulted in a robust increase in dendritic protrusions compared with NLGN4X WT and the nonphosphorylatable mutants, R704C and T707A (Fig. 2 A and C). To examine whether the increase in spines resulted in new excitatory synapses, we coexpressed NLmiRs with NLGN4X (WT, R704C, T707A, or T707D) in rat hippocampal cultures and assayed for anatomical measures of functional synapses, namely increases in presynaptic VGLUT1 and postsynaptic PSD-95. Using immunofluorescence confocal microscopy, we found that the NLGN4X phospho-mimetic (T707D) mutation resulted in an increase in both VGLUT1 and PSD-95 staining, an enhancement that was absent in nonphosphorylated NLGN4X protein (Fig. 2 D–F). These results indicate that T707 phosphorylation promotes NLGN4X-induced synaptogenesis independent of regulating its surface expression.
Fig. 2.
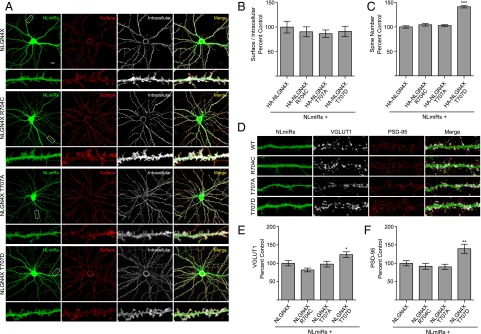
NLGN4X phosphorylation at T707 induces synaptogenesis. (A) Coexpression of NLmiRs and NLGN4X (WT, R704C, T707A, or T707D) in rat hippocampal neurons. Surface and intracellular receptors were labeled with an anti-hemagglutinin (HA) antibody, which recognized a tag inserted downstream of the signal peptide. (Scale bar, 20 μm.) (B) Means ± SEM normalized to NLGN4X (n = 27), NLGN4X R704C (P > 0.05, n = 29), NLGN4X T707A (P > 0.05, n = 30), and NLGN4X T707D (P > 0.05, n = 29). (C) Means of spine number ± SEM normalized to NLGN4X (n = 30), NLGN4X R704C (P > 0.05, n = 30), NLGN4X T707A (P > 0.05, n = 30), and NLGN4X T707D (P = 0.001, n = 30). (D) Same transfections as in A except NLGN4X constructs did not contain an HA tag. Endogenous VGLUT1 and PSD-95 were stained and visualized as described in Experimental Procedures. (E) Means ± SEM of VGLUT1 normalized to NLGN4X (n = 30) for NLGN4X R704C (P > 0.05, n = 30), NLGN4X T707A (P > 0.05, n = 30), and NLGN4X T707D (P = 0.0466, n = 30). (F) Means ± SEM of PSD-95 normalized to NLGN4X (n = 30) for NLGN4X R704C (P > 0.05, n = 27), NLGN4X T707A (P > 0.05, n = 28), and NLGN4X T707D (P = 0.0049, n = 30). *P < 0.05 **P < 0.01, ***P < 0.001.
To test whether these new synapses were functional, we biolistically coexpressed NLmiRs with NLGN4X (WT, R704C, T707A, or T707D) in rat organotypic hippocampal slice cultures and used a dual whole cell recording configuration to simultaneously record evoked excitatory postsynaptic currents (EPSCs) in both transfected and neighboring control untransfected cells. Expression of NLGN4X T707D resulted in a profound increase in both AMPA receptor (AMPAR)- and NMDA receptor (NMDAR)-mediated currents compared with a control cell or NLGN4X (WT, R704C, or T707A; Fig. 3 A–D). Previously, we, as well as others, reported that NLGN4X expression on a WT background decreased postsynaptic excitatory currents (19, 20). However, on a reduced NLGN background, which alone reduces AMPAR- and NMDAR-mediated currents by about 50% (Fig. S4), NLGN4X expression induced a modest, but significant, increase in AMPAR transmission, an enhancement that was absent in R704C and T707A (Fig. 3 A–D). Together, these results show that phosphorylation at T707 dynamically regulates NLGN4X’s ability to robustly induce the genesis of functional excitatory synapses.
Fig. 3.

NLGN4X T707D dramatically enhances excitatory postsynaptic currents. (A) AMPAR-mediated EPSC scatter plots. Expression of either NLGN4X or NLGN4X T707D results in a potentiation of AMPAR-mediated currents compared with control, untransfected cells (P = 0.0023, n = 18; P = 0.0010, n = 11). The enhancement was absent in NLGN4X R704C (P > 0.05, n = 13) and NLGN4X T707A (P > 0.05, n = 11) expressing cells. Experiments were performed in rat organotypic slice cultures on a reduced NLGN background (NLmiRs). Open circles are individual pairs; filled (in red) are mean ± SEM. Black sample traces are control; green are transfected. (Scale bars, 15 pA and 10 ms.) (B) Summary graph of data in A. Expression of NLGN4X T707D resulted in a greater enhancement of AMPAR-mediated currents compared with NLGN4X (P = 0.0143), NLGN4X R704C (P = 0.0025), or NLGN4X T707A (P = 0.0066). (C) NMDAR-mediated EPSC scatter plots. Expression of NLGN4X (n = 12) or phosphodeficient mutants, NLGN4X R704C (n = 11), or NLGN4X T707A (n = 13), did not enhance NMDAR-mediated currents compared with control untransfected cells (P > 0.05), whereas expression of NLGN4X T707D significantly potentiated NMDA-mediated currents (P = 0.0117, n = 9). Open circles are individual pairs, filled (in red) are mean ± SEM. Black sample traces are control; green are transfected. (Scale bars, 30 pA and 20 ms.) (D) Summary graph of data in C. Expression of NLGN4X T707D resulted in an enhancement of NMDAR-mediated currents compared with NLGN4X (P = 0.0409), NLGN4X R704C (P = 0.0250), and NLGN4X T707A (P = 0.0138). *P < 0.05 **P < 0.01, ***P < 0.001.
Endogenous NLGN4X Is Phosphorylated by PKC in Human Neurons.
Because our NLGN4X pT707-Ab was so specific, we were able to investigate endogenous NLGN4X T707 phosphorylation in cultured human embryonic neurons. These cultures consisted primarily of MAP2 positive neurons (Fig. 4A), and transient activation of PKC with PMA resulted in approximately an eightfold increase in NLGN4X pT707 phosphorylation (Fig. 4 B and C). Consistent with previous reports, NLGN4X was expressed in human embryonic neurons, as detected with two independent NLGN4X Abs and a pan NLGN Ab (22). Taken together, these results show that endogenous NLGN4X is phosphorylated in human neurons by PKC.
Fig. 4.

PKC phosphorylates endogenous NLGN4X in human neurons. (A) Immunofluorescence image of human embryonic neurons stained with the neuronal marker MAP2. (B) Regulation of NLGN4X phosphorylation at T707 in human embryonic neurons ± PMA treatment. Arrow denotes the NLGN4X pT707-specific band. Immunoblots (WB) were probed with indicated antibodies. (C) Means ± SEM of phosphorylated NLGN4X T707 achieved by PMA activation normalized to no treatment (n = 4) and + PMA treatment (P = 0.0148, n = 4).
Discussion
We set out to examine whether other NLGN isoforms, like NLGN1, were regulated by phosphorylation. We identified a PKC phosphorylation site on human NLGN4X at T707, which establishes a novel interplay between two critical constituents of the synapse. It was to our surprise, R704, a residue mutated in a familial case of autism (14), resided within the PKC consensus sequence in the c-tail of NLGN4X. However, after identification, it was not unexpected that the autism mutation (R704C) abolished the kinase’s ability to chemically modify NLGN4X at T707. Using a diverse combination of techniques, we show that phosphorylated NLGN4X can dynamically enhance excitatory synapses.
Before this report, the importance of protein phosphorylation on NLGN1 was established (15, 16). CaMKII phosphorylation of T739 regulates NLGN1’s ability to potentiate excitatory synaptic transmission by regulating its surface expression. This current study demonstrates that PKC phosphorylation of T707 dramatically enhances NLGN4X’s ability to potentiate excitatory currents independent of modulating its surface expression. Therefore, phosphorylation is a key regulatory modification modulating both NLGN1 and NLGN4X’s function but by different kinases, at different sites, and likely by different molecular mechanisms. Collectively, these studies raise two exciting questions: Might other kinases phosphorylate NLGN2 and NLGN3 and regulate their synaptic functions similarly to NLGN1 and NLGN4X? Second, what is the molecular mechanism by which T707 phosphorylation induces the genesis of functional synapses independent of surface expression? These answers remain to be elucidated but are currently an area of future investigation.
Interestingly, human NLGN4X is not well conserved in rodents, unlike NLGNs 1, 2, and 3. NLGN4 is absent in Rattus norvegicus and highly divergent and on an autosome in Mus musculus (21). These facts have made the investigation of human autism mutations in NLGN4X challenging and led some researchers to study the R704C point mutation in NLGN3. Although R704 is conserved in all human NLGNs, the residue analogous to NLGN4X T707 is not conserved. A knock-in mutation in NLGN3 resulted in minor synaptic phenotypes (23). Therefore, we believe it is imperative to study the R704C mutation in NLGN4X and more generally for all disease mutations to be studied in their respective isoforms independent of residue(s) conservation.
Canonically, NLGN1 is known as a critical component of the excitatory synapse based on its cellular localization (24, 25), regulation by CaMKII (16), and its ability to robustly potentiate AMPAR and NMDAR postsynaptic currents (20, 26). Conversely, mouse NLGN4 is known to localize to and modulate inhibitory transmission (27). However, the human and mouse isoforms are not well conserved. It raises the possibility that these proteins might perform distinct functions in different species. Here, we show that like NLGN1, human NLGN4X can enhance excitatory synapses, suggesting that NLGN4X may associate with excitatory synapses in humans. At a minimum, this underscores the reservations of studying human NLGN4X and rodent NLGN4 interchangeably. However, it is important to note our study does not preclude the possibility that human NLGN4X may act or be expressed at inhibitory synapses in humans.
Notably, all of the NLGN-associated autism point mutations reported thus far reside in their extracellular or transmembrane domains, except for NLGN4X R704C. The lack of c-tail autism mutations may be a result of chance or may highlight the critical importance of T707 phosphorylation in regulating NLGN4X’s synaptic properties. It is tempting to speculate whether this phosphorylation occurs at a critical developmental period to shape synaptic properties or whether it is a global switch that happens continually at excitatory synapses. The profound effect the phospho-mimetic mutation (T707D) has on excitatory synapses may suggest the prior.
Mouse models of autism have increasingly revealed underlying imbalances of excitatory/inhibitory transmission, which are believed to play a central role in the etiology of ASDs (18, 28, 29). Our results are consistent with this hypothesis, which support a pathophysiological model by which NLGN4X R704C decreases excitatory transmission by abolishing PKC-mediated NLGN4X potentiation of excitatory transmission. Furthermore, we believe given the overlap of synaptic proteins found to be candidate genes in ASDs that by understanding the biology of specific mutations such as R704C has implications for clinical, as well as future therapeutic treatment for ASDs.
Experimental Procedures
Plasmids and Antibodies.
Human pCAG-NLGN4X (WT, R704C, T707A, or T707D)-IRES-mCherry, pCAG-eGFP, pCAG-NLmiRs-GFP, and pRK5-FLAG-GluA1 plasmids were used for biochemical, electrophysiological, and imaging (synaptic markers) experiments. Human pCAG-HA-NLGN4X (WT, R704C, T707A, or T707D) were used in surface expression imaging experiments. pGEX-GST-NLGN c-tail constructs were made as previously described (16). Point mutations were introduced using QuikChange Site-Directed mutagenesis. The primers used to construct NLGN4X R704C were as follows: forward (FOR) 5′-ACAAAAAGGACAAGAGGTGCCATGAGACTCACAGG-3′ and reverse (REV) 5′-CCTGTGAGTCTCATGGCACCTCTTGTCCTTTTTGT-3′, NLGN4X T707A were FOR 5′-AGGCGCCATGAGGCTCACAGGCGCC-3′ and REV 5′-GGCGCCTGTGAGCCTCATGGCGCCT-3′, and NLGN4X T707D were FOR 5′- GAGGCGCCATGAGGATCACAGGCGCCCC-3′ and REV 5′-GGGGCGCCTGTGACCTCATGGCGCCTC-3′. The primers used to insert the HA-tag in pCAG-NLGN4X were FOR 5′-TATCCATACGACGTTCCGGACTACGCTCCAGTTGTCAACACAAATTATGGC-3′ and REV 5′-AGCGTAGTCCGGAACGTCGTATGGATAATACTGTGCTTGGCTGTCAATGAG-3′. To generate the rabbit NLGN4X pT707-Ab (against residues 703–712 in NLGN4X), animals were immunized with synthetic phosphopeptide Ac-CKRRHE(pT)HRRP-amide (New England Peptide). All immunoblotting with the NLGN4X T707-Ab began with blocking in 5% PhosphoBLOCKER (CELL BIOLABS) in TBS-T at room temperature, followed by 1% PhosphoBLOCKER in the primary and secondary Ab incubations. Antibodies used in the study were anti-NLGN4X (Sigma), anti-NLGN4X (abcam), anti-pan NLGN 4F9 (Synaptic Systems), anti-GST (Bethyl Laboratories), anti-FLAG (Sigma), anti-HA rat (Roche), anti-HA rabbit (abcam), anti-GluA1 pS831 (Covance), anti-PSD-95 (Neuromab), anti-VGLUT1 (Millipore), anti-MAP2 (Cell Signaling), and anti-actin (ABM). All antibodies were used at 1 μg/μL.
GST-Fusion Protein Production, In Vitro Phosphorylation, and MS.
Reagents were prepared and assays were performed and analyzed as previous described (16).
Immunoblotting.
HEK293T cells were maintained and transfected, and proteins were isolated as previously described (16). Treatment with 200 ng/μL PMA (Tocris) or DMSO (Sigma) began 30–60 min before protein isolation. For IPs, cell lysates were incubated with 6 μg of NLGN4X pT707-Ab and protein A-Sepharose beads (GE Healthcare) at 4 °C overnight. The following day, the IPs were washed in TBS buffer containing 150 mM NaCl, 50 mM Tris⋅HCl, 1 mM EDTA, protease (Roche), and phosphatase (Sigma) inhibitors. The antibody conjugated beads were resuspended in SDS/PAGE sample buffer and subjected to Western blotting.
Neuronal Cultures and Immunocytochemistry.
Hippocampal neurons were prepared from E18 Sprague–Dawley rats and used for all immunocytochemistry experiments. The use and care of animals used in this study followed the guidelines of the National Institutes of Health Animal Research Advisory Committee. Neurons were grown on precoated poly-d-lysine (Sigma) glass coverslips and cotransfected (Liptofectamine 2000) at days in vitro (DIV) 10 with NLmiRs and HA-NLGN4X constructs for surface expression experiments and without the HA tag for staining with synaptic markers. Cells were fixed at DIV14. Spine number was counted using the GFP signal from the NLmiRs construct from three to four regions of 30 μm/cell. Methods, reagents, and analysis for imaging were done as previously described (16).
Electrophysiology.
Hippocampal organotypic slice cultures were prepared from 6- to 8-d-old rats as described previously (30). All experiments were performed in accordance with established protocols approved by the University of California San Francisco Institutional Animal Care and Use Committee.
Sparse biolistic transfections of organotypic slice cultures were performed 4 d after culturing as previously described (31). Briefly, 100 μg total of mixed plasmid DNA was coated on 1-μm-diameter gold particles in 0.5 mM spermidine, precipitated with 0.1 mM CaCl2, and washed four times in pure ethanol. The gold particles were coated onto PVC tubing, dried using ultra-pure N2 gas, and stored at 4 °C in desiccant. DNA-coated gold particles were delivered with a Helios Gene Gun (BioRad). When biolistically expressing two plasmids, gold particles were coated with equal amounts of each plasmid, and plasmids always expressed different fluorescent markers. Observed frequency of coexpression was nearly 100%. Slices were maintained at 34 °C with media changes every other day.
Recordings were performed at DIV 7–8 after 3–4 d of expression. Dual whole-cell recordings in area CA1 were done by simultaneously recording responses from a fluorescent transfected neuron and neighboring untransfected control neuron. Synaptic responses were evoked by stimulating with a monopolar glass electrode filled with artificial CSF in stratum radiatum of CA1. Typically each pair of neurons is from a separate slice, whereas on rare occasions, two pairs may come from one slice. For all paired recordings, the number of experiments (n) reported in the figure legends refer to the number of pairs. Pyramidal neurons were identified by morphology and location. To ensure stable recording, membrane holding current, input resistance, and pipette series resistance were monitored throughout recording. All recordings were made at 20–25 °C using glass patch electrodes filled with an internal solution consisting of 135 mM CsMeSO4, 8 mM NaCl, 10 mM Hepes, 0.3 mM EGTA, 4 mM Mg-ATP, 0.3 mM Na-GTP, 5 mM QX-314, and 0.1 mM spermine and an external solution containing 119 mM NaCl, 2.5 mM KCl, 4 mM MgSO4, 4 mM CaCl2, 1 mM NaH2PO4, 26.2 mM NaHCO3, and 11 mM glucose bubbled continuously with 95% O2 and 5% CO2. Recordings were made in the presence of picrotoxin (100 μM) to block inhibitory currents and a small (50 nM) amount of NBQX to reduce epileptiform activity at −70 mV (AMPA) and +40 mV (NMDA). AMPAR-mediated currents were measured at the peak of the current, whereas NMDAR-mediated currents were measured 150 ms after the stimulation.
Human Neuron Cultures.
Human fetal neural cells were cultured as previously reported (32). Total NLGN4X protein was quantified using the anti-NLGN4X Ab (Sigma).
Statistical Analysis.
Statistical significance of immunoblots was tested using a t test, immunocytochemistry with a one-way ANOVA, paired whole-cell recordings with a Wilcoxon signed-rank test, and comparisons between sets of paired data with a Mann–Whitney U test. All experiments were done at least three independent times.
Supplementary Material
Acknowledgments
We thank Dr. Antonio Sanz-Clemente and John D. Badger, as well as other members of K.W.R. laboratory, for technical assistance and for discussions on the project and manuscript. We thank Dr. Avindra Nath for providing human embryonic neuron cultures, and the National Institute of Neurological Disorders and Stroke sequencing facility and light imaging facility for their expertise.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1500501112/-/DCSupplemental.
References
- 1.Anonymous; Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators Centers for Disease Control and Prevention (CDC) Prevalence of autism spectrum disorder among children aged 8 years-autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63(2):1–21. [PubMed] [Google Scholar]
- 2.Abrahams BS, Geschwind DH. Advances in autism genetics: On the threshold of a new neurobiology. Nat Rev Genet. 2008;9(5):341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012;4(3):a009886. doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jamain S, et al. Paris Autism Research International Sibpair Study Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34(1):27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455(7215):903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dean C, Dresbach T. Neuroligins and neurexins: Linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006;29(1):21–29. doi: 10.1016/j.tins.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Craig AM, Kang Y. Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol. 2007;17(1):43–52. doi: 10.1016/j.conb.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas NS, et al. Xp deletions associated with autism in three females. Hum Genet. 1999;104(1):43–48. doi: 10.1007/s004390050908. [DOI] [PubMed] [Google Scholar]
- 9.Marshall CR, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82(2):477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy D, et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70(5):886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 11.Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70(5):863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laumonnier F, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74(3):552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawson-Yuen A, Saldivar JS, Sommer S, Picker J. Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur J Hum Genet. 2008;16(5):614–618. doi: 10.1038/sj.ejhg.5202006. [DOI] [PubMed] [Google Scholar]
- 14.Yan J, et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol Psychiatry. 2005;10(4):329–332. doi: 10.1038/sj.mp.4001629. [DOI] [PubMed] [Google Scholar]
- 15.Giannone G, et al. Neurexin-1β binding to neuroligin-1 triggers the preferential recruitment of PSD-95 versus gephyrin through tyrosine phosphorylation of neuroligin-1. Cell Reports. 2013;3(6):1996–2007. doi: 10.1016/j.celrep.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 16.Bemben MA, et al. CaMKII phosphorylation of neuroligin-1 regulates excitatory synapses. Nat Neurosci. 2014;17(1):56–64. doi: 10.1038/nn.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16(6):1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 18.Tabuchi K, et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318(5847):71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang C, et al. A neuroligin-4 missense mutation associated with autism impairs neuroligin-4 folding and endoplasmic reticulum export. J Neurosci. 2009;29(35):10843–10854. doi: 10.1523/JNEUROSCI.1248-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shipman SL, et al. Functional dependence of neuroligin on a new non-PDZ intracellular domain. Nat Neurosci. 2011;14(6):718–726. doi: 10.1038/nn.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolliger MF, et al. Unusually rapid evolution of Neuroligin-4 in mice. Proc Natl Acad Sci USA. 2008;105(17):6421–6426. doi: 10.1073/pnas.0801383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marei HE, et al. Gene expression profile of adult human olfactory bulb and embryonic neural stem cell suggests distinct signaling pathways and epigenetic control. PLoS ONE. 2012;7(4):e33542. doi: 10.1371/journal.pone.0033542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Etherton MR, Tabuchi K, Sharma M, Ko J, Südhof TC. An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor-mediated synaptic transmission in hippocampus. EMBO J. 2011;30(14):2908–2919. doi: 10.1038/emboj.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song JY, Ichtchenko K, Südhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci USA. 1999;96(3):1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chih B, Engelman H, Scheiffele P. Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005;307(5713):1324–1328. doi: 10.1126/science.1107470. [DOI] [PubMed] [Google Scholar]
- 26.Chubykin AA, et al. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54(6):919–931. doi: 10.1016/j.neuron.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoon M, et al. Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc Natl Acad Sci USA. 2011;108(7):3053–3058. doi: 10.1073/pnas.1006946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Földy C, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron. 2013;78(3):498–509. doi: 10.1016/j.neuron.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rothwell PE, et al. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158(1):198–212. doi: 10.1016/j.cell.2014.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37(2):173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 31.Schnell E, et al. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA. 2002;99(21):13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang T, et al. Granzyme B mediates neurotoxicity through a G-protein-coupled receptor. FASEB J. 2006;20(8):1209–1211. doi: 10.1096/fj.05-5022fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.