

ABSTRACT
We previously showed that selective mineralocorticoid receptor (MR) blockade by eplerenone is cardioprotective in Dahl salt-sensitive (DS) rats. To clarify the consequences of glucocorticoid-mediated MR activation in these animals, we investigated the effects of exogenous corticosterone on blood pressure as well as cardiac remodeling and function after adrenalectomy. DS rats were subjected to adrenalectomy at 6 weeks of age and thereafter fed a high-salt diet and administered corticosterone (20 mg/kg per day) or vehicle. Systolic blood pressure was higher in the corticosterone group than in the vehicle group at 7 weeks and thereafter. By 11 weeks, corticosterone had reduced left ventricular (LV) mass and induced LV diastolic dysfunction. The ratio of collagen type I to type III mRNA levels in the left ventricle was increased in the corticosterone group compared with the vehicle group. Administration of a non-antihypertensive dose of the MR antagonist spironolactone (20 mg/kg per day) from 6 weeks inhibited the effects of corticosterone on both the collagen type I to type III mRNA ratio and diastolic function without affecting the decrease in LV mass. Spironolactone attenuated both the increase in NADPH oxidase activity in the left ventricle and coronary vascular inflammatory responses apparent in the corticosterone group. These results indicate that exogenous glucocorticoids induce hypertension, cardiac remodeling, and diastolic dysfunction in adrenalectomized DS rats fed a high-salt diet. The cardiac effects of exogenous glucocorticoids are likely attributable, at least in part, to myocardial oxidative stress and coronary vascular inflammation induced by glucocorticoid-activated MRs..
Key Words: glucocorticoids, mineralocorticoid receptor, diastolic function, oxidative stress, inflammation
INTRODUCTION
Abnormal activation of the renin-angiotensin-aldosterone system correlates directly with the incidence and extent of target organ damage. Indeed, a chronic increase in the plasma concentration of aldosterone contributes to the pathophysiology of heart failure. The harmful effects of aldosterone on the cardiovascular system include the induction of hypertrophy and fibrosis, a reduction in vascular compliance, ventricular diastolic dysfunction, and ventricular arrhythmia.1, 2)
Transgenic mice with a selective increase in the activity of mineralocorticoid receptors (MRs) in cardiac myocytes develop myocardial hypertrophy and interstitial fibrosis, resulting in progressive left ventricular (LV) dilation, heart failure, and death.3) All these effects were prevented by treatment with the selective MR antagonist eplerenone. Overactivity of the renin-angiotensin-aldosterone system is thus thought to be a risk factor for cardiovascular disease. Furthermore, we have previously shown that selective MR blockade with eplerenone is cardioprotective in the setting of hypertension associated with low renin and aldosterone levels.4)
The MR was originally thought to be activated only by the mineralocorticoid aldosterone and to act primarily by promoting sodium retention and potassium excretion in the kidney, thereby increasing blood pressure.5) It is now known to be expressed in nonepithelial cells, including cardiomyocytes, and to be activated by endogenous glucocorticoids, including cortisol in humans and corticosterone in rodents.6) In vivo, endogenous glucocorticoids bind both the MR and the glucocorticoid receptor (GR), whereas aldosterone specifically binds to the MR. Aldosterone and corticosterone possess similar high affinities for the MR, and glucocorticoids circulate at concentrations much higher than that of aldosterone under normal circumstances.6) Aldosterone action at MRs in epithelial tissues and vascular smooth muscle cells is ensured by the enzyme 11β-hydroxysteroid dehydrogenase type 2, which catalyzes the conversion of corticosterone to 11-dehydrocorticosterone, a metabolite with negligible activity at the MR. This enzyme is present at only low levels in cardiac tissue,4, 6) with the result that MRs in cardiomyocytes are normally occupied (but not activated) by high levels of intracellular glucocorticoids. Endogenous glucocorticoids normally do not mimic the effects of aldosterone at these unprotected MRs, however, but rather act as MR antagonists.6)
Redox regulation is thought to be an important determinant of the activity of transcription factors including nuclear hormone receptors. Cellular redox status thus modulates GR function in vivo,7) and oxidative stress and the generation of reactive oxygen species affect the expression of estrogen receptors.8) Given that nuclear nicotinamide adenine dinucleotide (NADH) activates transcriptional corepressors in vivo and attenuates transactivation,9) alterations in redox status during oxidative stress may be a driver for activation of the glucocorticoid-MR complex in the cardiovascular system.4)
We hypothesized that glucocorticoid-mediated MR activation may contribute to cardiac and coronary vascular injury associated with low-aldosterone hypertension. To test this hypothesis, we investigated the effects of exogenous corticosterone on blood pressure and cardiac remodeling and function, as well as the modulation of such effects by MR blockade, in adrenalectomized (ADX) Dahl salt-sensitive (DS) rats fed a high-salt diet.
METHODS
Animals and experimental protocols
Male inbred DS rats were obtained from Japan SLC Inc. (Hamamatsu, Japan) and were handled in accordance with the guidelines of Nagoya University Graduate School of Medicine as well as with the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85–23, revised 1996). Weaning rats were fed laboratory chow containing 0.3% NaCl until 6 weeks of age, after which the diet was switched to chow containing 8% NaCl. DS rats fed an 8% NaCl diet after 6 weeks of age develop compensated LV hypertrophy attributable to hypertension at 11 weeks.10) DS rats on such a diet were randomly allocated to four groups: (1) the LV hypertrophy (LVH) group (n = 7); (2) the ADX+CTC group (n = 10), in which the animals underwent bilateral adrenalectomy and were administered corticosterone (CTC, 20 mg per kilogram of body weight per day; Sigma, St. Louis, MO); (3) the ADX+CTC+SPL group (n = 9), in which the animals underwent adrenalectomy and were administered both corticosterone (20 mg/kg per day) and spironolactone (SPL, 20 mg/kg per day; Sigma); and (4) the ADX+V group (n = 10), in which the animals underwent adrenalectomy and were administered vehicle. Adrenalectomy was performed with rats under pentobarbital anesthesia at 3 days after initiation of the high-salt diet. Corticosterone was injected subcutaneously twice a day and spironolactone was administered orally via a gastric tube, with drug treatment beginning on the day of adrenalectomy and ending when the animals reached 11 weeks of age. The doses of corticosterone and spironolactone were determined on the basis of results of previous studies.11, 12) DS rats fed a 0.3% NaCl diet after 6 weeks of age remain normotensive, and such animals served as age-matched controls (CONT group, n = 8). Both the diets and tap water were provided ad libitum throughout the experimental period. At 11 weeks of age, all of the rats were anesthetized by intraperitoneal injection of ketamine (50 mg/kg) and xylazine (10 mg/kg) and were subjected to hemodynamic and echocardiographic analyses. The heart was subsequently excised, and LV tissue was separated for analysis.
Echocardiographic and hemodynamic analyses
Systolic blood pressure (SBP) was measured weekly in conscious animals by tail-cuff plethysmography (BP-98A; Softron, Tokyo, Japan). At 11 weeks of age, rats were subjected to transthoracic echocardiography, as described previously.10) In brief, M-mode echocardiography was performed with a 12.5-MHz transducer (Aplio SSA-700A; Toshiba Medical Systems, Tochigi, Japan). LV end-diastolic (LVDd) and end-systolic (LVDs) dimensions as well as the thickness of LV posterior wall (LVPWT) were measured, and LV fractional shortening (LVFS) was calculated according to the formula: LVFS = [(LVDd – LVDs)/LVDd] × 100%. For assessment of LV diastolic function, we calculated the deceleration time (DcT) and the isovolumic relaxation time (IRT) from the pulsed Doppler echocardiographic data. After echocardiography, cardiac catheterization was performed as described previously.13) Tracings of LV pressure and the electrocardiogram were digitized to determine LV end-diastolic pressure (LVEDP). The time constant of isovolumic relaxation (tau) was calculated by the derivative method of Raff and Glantz as described previously.14)
Histology and immunohistochemistry
LV tissue was fixed in ice-cold 4% paraformaldehyde for 48 to 72 h, embedded in paraffin, and processed for histology as described.15) Transverse sections (thickness, 3 µm) were stained either with hematoxylin-eosin for routine histological examination or with Azan-Mallory solution for evaluation of the extent of fibrosis, as described previously.10) To evaluate macrophage infiltration into the myocardium, we performed immunostaining for the monocyte-macrophage marker CD68 with frozen sections (thickness, 5 µm) that had been fixed with acetone. Endogenous peroxidase activity was blocked by exposure of the sections to methanol containing 0.3% hydrogen peroxide. Sections were incubated at 4°C first overnight with mouse monoclonal antibodies to CD68 (1:100 dilution of clone ED1; Chemicon, Temecula, CA) and then for 30 min with Histofine Simple Stain Rat MAX PO (Nichirei Biosciences, Tokyo, Japan). Immune complexes were visualized with diaminobenzidine and hydrogen peroxide, and the sections were counterstained with hematoxylin. Image analysis was performed with NIH Scion Image software.
Assay of superoxide production
Nicotinamide adenine dinucleotide phosphate (NADPH)-dependent superoxide production by homogenates prepared from freshly frozen LV tissue was measured with an assay based on lucigenin-enhanced chemiluminescence as described previously.16) The chemiluminescence signal was sampled every minute for 10 min with a microplate reader (WALLAC 1420 ARVO MX/Light; Perkin-Elmer, Waltham, MA), and the respective background counts were subtracted from experimental values. Superoxide production in tissue sections was examined as described17) with the use of dihydroethidium (Sigma), which is rapidly oxidized by superoxide to yield fluorescent ethidium. Sections were examined with a fluorescence microscope equipped with a 585-nm long-pass filter. As a negative control, sections were incubated with superoxide dismutase (300 U/mL) before staining with dihydroethidium; such treatment prevented the generation of fluorescence signals (data not shown).
Quantitative RT-PCR analysis
Total RNA was extracted from LV tissue and treated with DNase with the use of a spin-vacuum isolation kit (Promega, Madison, WI). Complementary DNA (cDNA) was synthesized from 2 µg of the total RNA by reverse transcription (RT) with the use of random primers (Invitrogen, Carlsbad, CA) and MuLV reverse transcriptase (Applied Biosystems, Foster City, CA). Real-time polymerase chain reaction (PCR) analysis was performed with the use of a Prism 7700 Sequence Detector (Perkin-Elmer, Wellesley, MA), as previously described,18) and with primers and TaqMan probes specific for rat cDNAs encoding atrial natriuretic peptide (ANP),10) brain natriuretic peptide (BNP),10) collagen type I,19) collagen type III,19) insulin-like growth factor (IGF)–1,20) cyclooxygenase (COX)–2,21) osteopontin,4) monocyte chemoattractant protein (MCP)–1,4) and the p22phox,22) gp91phox,22) p47phox,23) p67phox,23) and Rac123) subunits of NADPH oxidase. Reagents for detection of human 18S rRNA (Applied Biosystems) were used to quantify rat 18S rRNA as an internal standard.
Plasma hormone analysis
Blood was collected from the right carotid artery of rats after hemodynamic measurements. Plasma renin activity was measured by radioimmunoassay (RIA) with the use of renin RIA beads (Abbott Japan, Tokyo, Japan). Plasma aldosterone concentration was determined by RIA with the use of a DPC aldosterone kit (Mitsubishi Chemical Medience, Tokyo, Japan).
Statistical analysis
Data are presented as means ± SEM. Differences among groups of rats at 11 weeks of age were assessed by one-way factorial analysis of variance (ANOVA) followed by Fisher’s multiple-comparison test. The time course of SBP was compared among groups by two-way repeated-measures ANOVA. A P value of <0.05 was considered statistically significant.
RESULTS
Plasma renin activity and aldosterone concentration
Plasma renin activity was markedly reduced in rats of the LVH, ADX+V, ADX+CTC, and ADX+CTC+SPL groups at 11 weeks of age (Figure 1A). Plasma aldosterone concentration was also greatly reduced in LVH rats, whereas it was below the detection limit in ADX+V, ADX+CTC, and ADX+CTC+SPL rats (Figure 1B), indicative of the complete removal of the adrenal glands in these animals.
Fig. 1.
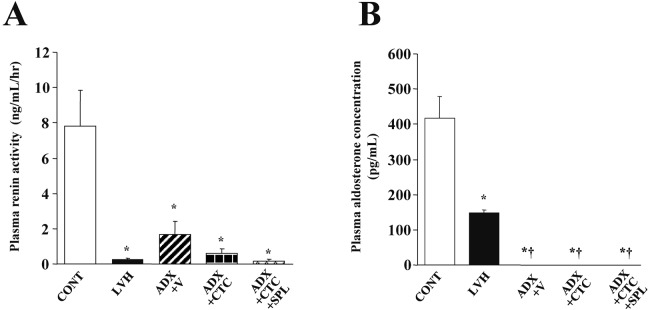
Plasma renin activity and aldosterone concentration in DS rats of the five experimental groups at 11 weeks of age. Renin activity (A) and aldosterone concentration (B) are presented as means ± SEM for animals in each group (n = 8, 7, 10, 10, and 9 for CONT, LVH, ADX+V, ADX+CTC, and ADX+CTC+SPL groups, respectively). *P < 0.05 versus CONT group; †P < 0.05 versus LVH group.
Hemodynamics and LV geometry and function
At 11 weeks of age, there was no significant difference in body weight between LVH and CONT rats (Table 1). However, body weight of ADX+CTC rats was significantly reduced compared with that of ADX+V rats. Treatment of ADX+CTC rats with SPL resulted in a further decrease in body weight. LVH rats progressively developed severe hypertension (Figure 2, Table 1). In contrast, ADX+V rats maintained a normal SBP, just like CONT rats. However, ADX+CTC rats showed a substantial increase in SBP at 8 weeks of age and then maintained a constant high blood pressure until 11 weeks. This increase in SBP apparent in ADX+CTC rats was not affected by treatment with SPL. Heart rate was similar among the animals of the five experimental groups (Table 1). LV weight was significantly increased in LVH rats compared with CONT rats, and this effect was abolished in ADX+V rats. ADX+CTC rats showed a significant decrease in LV weight compared with ADX+V rats, and treatment of ADX+CTC rats with SPL did not significantly affect this parameter.
Table 1.
Physiological parameters for DS rats in the five experimental groups at 11 weeks of age.
| Parameter | CONT | LVH | ADX+V | ADX+CTC | ADX+CTC+SPL |
|---|---|---|---|---|---|
| BW (g) | 321.0 ± 5.0 | 310.6 ± 6.5 | 289.1 ± 6.4*† | 234.3 ± 2.9*†‡ | 221.3 ± 2.3*†‡§ |
| HR (bpm) | 406.8 ± 5.8 | 421.2 ± 8.7 | 406.3 ± 8.1 | 415.0 ± 6.7 | 422.4 ± 5.7 |
| SBP (mmHg) | 127.4 ± 1.4 | 191.0 ± 2.7* | 138.3 ± 8.1† | 161.9 ± 2.3*†‡ | 161.1 ± 3.5*†‡ |
| LVW (mg) | 706.6 ± 17.9 | 955.4 ± 14.4* | 714.0 ± 37.0† | 642.1 ± 53.6†‡ | 587.7 ± 12.9*†‡ |
Abbreviations not defined in text: BW, body weight; HR, heart rate; LVW, left ventricular weight. Data are means ± SEM for animals in each group. *P < 0.05 versus CONT group; †P < 0.05 versus LVH group; ‡P < 0.05 versus ADX+V group; §P < 0.05 versus ADX+CTC group.
Fig. 2.

Time course of SBP in DS rats of the five experimental groups. Data are means ± SEM for animals in each group. *P < 0.05 versus CONT group; †P < 0.05 versus LVH group; ‡P < 0.05 versus ADX+V group.
Echocardiography revealed that both LVPWT and LVFS were significantly increased in LVH rats compared with CONT rats (Table 2). The increase in LVPWT seen in LVH rats was attenuated in ADX+V rats. There was no significant difference in LVDd among these three groups. CTC significantly reduced both LVPWT and LVDd, whereas it did not affect LVFS, in ADX rats. DcT, IRT, and tau, all of which are indices of LV relaxation, as well as the ratio of LVEDP to LVDd, an index of LV diastolic stiffness, were all increased in LVH rats compared with CONT rats, and these effects were attenuated in ADX+V rats. ADX+CTC rats showed significant increases in DcT, IRT, tau, and the LVEDP to LVDd ratio compared with ADX+V rats, and treatment of ADX+CTC rats with SPL attenuated all of these changes in parameters of LV diastolic function.
Table 2.
Morphological and functional parameters for DS rats in the five experimental groups at 11 weeks of age.
| Parameter | CONT | LVH | ADX+V | ADX+CTC | ADX+CTC+SPL |
|---|---|---|---|---|---|
| LVPWT (mm) | 1.39±0.03 | 2.21±0.04* | 1.60±0.04*† | 1.45±0.04†‡ | 1.43±0.07†‡ |
| LVDd (mm) | 8.02±0.19 | 7.69±0.28 | 7.79±0.10 | 6.97±0.11*†‡ | 7.09±0.05*†‡ |
| LVFS (%) | 39.0±1.3 | 41.0±2.1* | 43.2±1.0* | 44.6±0.7*† | 44.4±0.6*† |
| DcT (ms) | 36.1±0.8 | 55.8±1.0* | 44.6±1.4*† | 47.3±0.8*‡ | 46.3±1.1*§ |
| IRT (ms) | 16.8±0.4 | 27.0±1.8* | 24.3±1.1*† | 28.4±0.7*‡ | 24.4±1.0†§ |
| Tau (ms) | 24.9±2.8 | 33.5±2.1* | 23.5±3.1*† | 34.7±4.4*‡ | 21.8±2.2*†§ |
| LVEDP/LVDd (mmHg/mm) | 0.47±0.03 | 0.91±0.07* | 0.70±0.02*† | 0.91±0.05*‡ | 0.65±0.07*†§ |
Data are means ± SEM for animals in each group. *P < 0.05 versus CONT group; †P < 0.05 versus LVH group; ‡P < 0.05 versus ADX+V group; §P < 0.05 versus ADX+CTC group.
Cardiomyocyte growth and cardiac fibrosis
Microscopic analysis revealed that the cross-sectional area of cardiac myocytes was increased in LVH rats compared with CONT rats and that this effect was abolished in ADX+V rats (Figure 3A, B). CTC significantly reduced LV cardiomyocyte size in ADX rats and this effect was not significantly influenced by treatment with SPL. Hemodynamic overload resulted in marked upregulation of ANP and BNP gene expression in the left ventricle of LVH rats and these effects were prevented by adrenalectomy (Figure 3C, D). The abundance of IGF-1 mRNA did not differ between CONT and LVH rats but was reduced by adrenalectomy (Figure 3E). CTC downregulated the expression of both fetal-type cardiac genes as well as that of the IGF-1 gene in ADX rats and these effects were not modified by treatment with SPL.
Fig. 3.

Cardiomyocyte size and expression of ANP, BNP, and IGF-1 genes in the left ventricle of DS rats in the five experimental groups at 11 weeks of age. (A) Hematoxylin-eosin staining of transverse sections of the LV myocardium. Scale bars, 50 µm. (B) Cross-sectional area of cardiac myocytes determined from sections similar to those in (A). (C–E) Quantitative RT-PCR analysis of ANP, BNP, and IGF-1 mRNAs, respectively. The amount of each mRNA was normalized by that of 18S rRNA and then expressed relative to the corresponding mean value for the CONT group. Data in (B) through (D) are means ± SEM for animals in each group. *P < 0.05 versus CONT group; †P < 0.05 versus LVH group; ‡P < 0.05 versus ADX+V group.
Azan-Mallory staining revealed that fibrosis in perivascular and interstitial regions of the LV myocardium was increased in LVH rats compared with CONT rats, and that this effect was abolished in ADX+V rats (Figure 4A–C). Perivascular fibrosis, but not interstitial fibrosis, was significantly increased by CTC in ADX rats, and this increase in perivascular fibrosis was inhibited by treatment with SPL. The ratio of the amount of collagen type I mRNA to that of collagen type III mRNA, which correlates with myocardial stiffness, was increased in LVH rats compared with CONT rats and this effect was abolished in ADX+V rats (Figure 4D). CTC significantly increased the collagen type I to type III mRNA ratio in a manner sensitive to SPL in ADX rats.
Fig. 4.

Cardiac fibrosis and expression of collagen genes in the left ventricle of DS rats in the five experimental groups at 11 weeks of age. (A) Collagen deposition as revealed by Azan-Mallory staining in perivascular (upper panels) or interstitial (lower panels) regions of the LV myocardium. Scale bars, 200 µm. (B, C) Relative extents of perivascular and interstitial fibrosis, respectively, in the LV myocardium as determined from sections similar to those in (A). (D) Ratio of the amount of collagen type I mRNA to that of collagen type III mRNA. Data in (B) through (D) are means ± SEM for animals in each group. *P < 0.05 versus CONT group; †P < 0.05 versus LVH group; ‡P < 0.05 versus ADX+V group; §P < 0.05 versus ADX+CTC group.
Myocardial oxidative stress
Superoxide production in myocardial tissue sections revealed by staining with dihydroethidium as well as the activity of NADPH oxidase in LV homogenates were both significantly increased in LVH rats compared with CONT rats, and these effects were abolished in ADX+V rats (Figure 5A, B). CTC increased myocardial superoxide production and NADPH oxidase activity in a manner sensitive to SPL in ADX rats. The expression of genes for the p22phox, gp91phox, p67phox and Rac1 but not for p47phox subunits of NADPH oxidase was also upregulated in the left ventricle of LVH rats compared with CONT rats and these effects were prevented by ADX (Figure 5C–G). CTC did not affect the expression levels of p22phox, gp91phox, or Rac1 genes, but it significantly upregulated the expression of p47phox and p67phox genes in ADX rats.
Fig. 5.

Superoxide production as well as NADPH oxidase activity and gene expression in the left ventricle of rats in the five experimental groups at 11 weeks of age. (A) Superoxide production as revealed by dihydroethidium staining in perivascular (upper panels) or interstitial (lower panels) regions of the LV myocardium. Scale bars, 100 µm. (B) NADPH-dependent superoxide production in LV homogenates. Results are expressed as relative light units (RLU) per milligram of protein. (C–G) Quantitative RT-PCR analysis of p22phox, gp91phox, p47phox, p67phox, and Rac1 mRNAs, respectively. The amount of each mRNA was normalized by that of 18S rRNA and then expressed relative to the corresponding mean value for the CONT group. Data in (B) through (G) are means ± SEM for animals in each group. *P < 0.05 versus CONT group; †P < 0.05 versus LVH group; ‡P < 0.05 versus ADX+V group; §P < 0.05 versus ADX+CTC group.
Myocardial inflammation
Immunostaining of the LV myocardium for the monocyte-macrophage marker CD68 revealed that macrophage infiltration in the myocardium was increased in LVH rats compared with CONT rats and that this effect was attenuated in ADX+V rats (Figure 6A, B). CTC increased macrophage infiltration in ADX rats in a manner sensitive to treatment with SPL. The expression of MCP-1, osteopontin, and COX-2 genes in the left ventricle was increased in LVH rats compared with CONT rats and these effects were abolished in ADX+V rats (Figure 6C–E). CTC upregulated the expression of these inflammatory marker genes in ADX rats in a manner sensitive to SPL.
Fig. 6.

Macrophage infiltration as well as expression of MCP-1, osteopontin, and COX-2 genes in the left ventricle of rats in the five experimental groups at 11 weeks of age. (A) Immunohistochemical staining for the monocyte-macrophage marker CD68. Scale bars, 50 µm. (B–D) Quantitative RT-PCR analysis of MCP-1, osteopontin, and COX-2 mRNAs, respectively. The amount of each mRNA was normalized by that of 18S rRNA and then expressed relative to the corresponding mean value for the CONT group. Data in (B) through (D) are means ± SEM for animals in each group. *P < 0.05 versus CONT group; †P < 0.05 versus LVH group; ‡P < 0.05 versus ADX+V group; §P < 0.05 versus ADX+CTC group.
DISCUSSION
We have shown that exogenous corticosterone induces hypertension, cardiac remodeling, and diastolic dysfunction in adrenalectomized DS rats fed a high-salt diet, and that spironolactone attenuates these effects without lowering blood pressure. Our data thus suggest that exogenous glucocorticoids activate cardiac MRs and thereby contribute to cardiac remodeling and diastolic dysfunction in this model of no-aldosterone hypertension. Increased oxidative stress may underlie activation of the glucocorticoid-MR complex in the heart of these animals. The beneficial effects of spironolactone in this model are likely attributable to attenuation of myocardial oxidative stress and coronary vascular inflammation induced by glucocorticoid-activated MRs.
Cardiac hypertrophy induced by hypertension is associated with increases in cardiomyocyte cell volume, changes in cardiac gene transcription and translation, and enhanced myofibrillar assembly.24) In the present study, corticosterone-treated ADX rats showed cardiac atrophy despite the presence of sustained LV pressure overload, and this effect was not inhibited by spironolactone treatment. These data suggest that MRs may not be responsible for the corticosterone-induced inhibition of LV myocardial growth.20) Downregulation of IGF-1 in muscle has been implicated in the skeletal muscle atrophy induced by glucocorticoids.25) The corticosterone-induced cardiac atrophy observed in the present study may thus have resulted from inhibition of the IGF-1 system in a manner independent of MRs.
Cardiac fibrosis is a pathological feature associated with hypertension and is responsible for LV diastolic dysfunction, likely as a result of increased LV diastolic stiffness.4) In addition, MR activation in the myocardium and coronary vasculature results in cardiac remodeling and fibrosis, myocardial oxidative stress, and coronary vascular inflammation.4, 5) Excess reactive oxygen species (ROS) may contribute to impairment of LV diastolic function through inhibition of Ca2+ handling proteins.26, 27) Increased oxidative stress might also result in an increase in the ratio of collagen type I to type III mRNA levels, which has been associated with LV diastolic stiffness.22) In the present study, spironolactone ameliorated LV perivascular fibrosis, the shift in collagen isoforms, and LV diastolic dysfunction as well as attenuated LV oxidative stress and inflammation, without lowering blood pressure, in corticosterone-treated ADX rats. These data suggest that glucocorticoid-mediated MR activation may be important in corticosterone-induced LV remodeling and diastolic dysfunction.20)
In previous studies, the cardiac effect of glucocorticoids during experimental myocardial infarction has been controversial depending on their types or doses.28, 29) Our results are consistent with a recent study showing that glucocorticoids aggravate cardiac damage by activation of MRs during ischemia-reperfusion in the isolated perfused rat heart.30) Although glucocorticoids have been thought to act as antagonists of the MR in nonepithelial cells such as cardiomyocytes and brain cells, glucocorticoid-MR complexes are activated as a result of the generation of ROS.6) Given that ROS regulate the sumoylation-desumoylation equilibrium in vitro, posttranslational modification of MRs by the small ubiquitin-related modifier might be involved in ROS-induced MR activation.31) In the present study, salt loading or LV pressure overload associated with corticosterone-induced hypertension may have been the stimulus for ROS production in the heart. ROS-induced MR activation results in activation of NADPH oxidase4) and induces ROS production and tissue injury, thereby forming a vicious circle. MR inhibition has been shown to attenuate cardiac oxidative stress and inflammation in rodent models of chronic pressure overload.4, 32, 33) Furthermore, spironolactone treatment partially suppressed cardiac inflammatory-fibrogenic and oxidative stress responses in vivo after angiotensin II infusion in normotensive Sprague-Dawley rats.34) ADX+CTC rats are a model of no-aldosterone hypertension, an extreme condition distinct from low-aldosterone hypertension (e.g. LVH rats) that is usually detected in humans. In the present study, spironolactone may thus have protected the heart from corticosterone-induced damage and diastolic dysfunction through the inhibition of aldosterone-independent, ROS-induced activation of corticosterone-MR complexes. The increase in NADPH oxidase activity was accompanied by upregulation of the p47phox and p67phox genes but not p22phox, gp91phox, or Rac1 genes in the left ventricle of ADX+CTC rats. These data suggest that the abundance of these cytosolic components (p47phox and p67phox) may have contributed to the activation of NADPH oxidase.35, 36)
Macrophage infiltration into the perivascular space of intramural coronary vessels as well as the interstitial space was accompanied by upregulation of the expression of genes for the proinflammatory proteins MCP-1, osteopontin, and COX-2 in the heart of ADX+CTC rats. The observation that the proinflammatory effects of corticosterone were attenuated by treatment with spironolactone suggests that coronary vascular inflammation induced by corticosterone-activated MRs may have contributed to the cardiac injury and perivascular fibrosis apparent in the present study. Our results are in good agreement with previous results showing that activation of MRs by exogenous glucocorticoids can lead to the development of cardiovascular inflammatory responses in adrenalectomized rats.37) MR- and salt-induced cardiac pathology is characterized by an early vascular inflammatory response involving MCP-1, adhesion molecules, and macrophages before the onset of collagen deposition.38) Administration of MR antagonists such as eplerenone or spironolactone reduces macrophage accumulation in various disease models, including those of myocardial infarction and angiotensin II– or aldosterone-induced vascular inflammation and damage.38-40) In the present study, the spironolactone-induced inhibition of coronary vascular inflammation may thus have contributed to the attenuation of cardiac remodeling and led to the amelioration of LV diastolic dysfunction in corticosterone-treated ADX rats.
Treatment of ADX rats with corticosterone induced a substantial increase in SBP that was maintained until 11 weeks of age. This increase in SBP was not affected by treatment with spironolactone, however, indicating that the adrenal glands are necessary for the development of salt-induced hypertension in this model and suggesting that MRs may not be responsible for corticosterone-induced hypertension in ADX rats. The present results are consistent with those of previous studies showing that adrenocorticotropic hormone– or corticosterone–induced hypertension in rats was not prevented by MR blockade with spironolactone.20, 41) Impairment of leukocyte-endothelium interactions in spontaneously hypertensive rats was found to be dependent on glucocorticoids.42) Oxidative stress and deficiency of nitric oxide are emerging as key factors in the pathogenesis of glucocorticoid-induced hypertension.43) MR-independent vascular inflammation and oxidative stress may thus have played a role in the pathogenesis of glucocorticoid-induced hypertension in ADX rats in the present study.
In conclusion, exogenous corticosterone induced hypertension, cardiac remodeling, and diastolic dysfunction in adrenalectomized DS rats fed a high-salt diet. The cardiac effects of exogenous corticosterone were likely attributable, at least in part, to myocardial oxidative stress and coronary vascular inflammation induced by glucocorticoid-activated MRs. Further studies are required to identify the precise mechanisms of ROS-induced activation of glucocorticoid-MR complexes in the cardiovascular system.
ACKNOWLEDGEMENTS
We thank Koji Tsuboi, Yuichiro Yamada, Chisa Inoue, and Masafumi Sakai for technical assistance.
Source of Funding
This work was supported by unrestricted research grants from Banyu Pharmaceutical Co. Ltd. (Tokyo, Japan) and Mitsubishi Tanabe Pharma Corporation (Osaka, Japan), and by Management Expenses Grants from the government to Nagoya University.
Conflict of Interest
None
REFERENCES
- 1).Muto T, Ueda N, Opthof T, Ohkusa T, Nagata K, Suzuki S, Tsuji Y, Horiba M, Lee JK, Honjo H, Kamiya K, Kodama I, Yasui K. Aldosterone modulates I(f) current through gene expression in cultured neonatal rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2007; 293: H2710–2718. [DOI] [PubMed]
- 2).Rocha R, Rudolph AE, Frierdich GE, Nachowiak DA, Kekec BK, Blomme EA, McMahon EG, Delyani JA. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol. 2002; 283: H1802–1810. [DOI] [PubMed]
- 3).Qin W, Rudolph AE, Bond BR, Rocha R, Blomme EA, Goellner JJ, Funder JW, McMahon EG. Transgenic model of aldosterone-driven cardiac hypertrophy and heart failure. Circ Res. 2003; 93: 69–76. [DOI] [PubMed]
- 4).Nagata K, Obata K, Xu J, Ichihara S, Noda A, Kimata H, Kato T, Izawa H, Murohara T, Yokota M. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and failure in low-aldosterone hypertensive rats. Hypertension. 2006; 47: 656–664. [DOI] [PubMed]
- 5).Nagata K. Mineralocorticoid antagonism and cardiac hypertrophy. Curr Hypertens Rep. 2008; 10: 216–221. [DOI] [PubMed]
- 6).Funder JW. RALES, EPHESUS and redox. J Steroid Biochem Mol Biol. 2005; 93: 121–125. [DOI] [PubMed]
- 7).Makino Y, Yoshikawa N, Okamoto K, Hirota K, Yodoi J, Makino I, Tanaka H. Direct association with thioredoxin allows redox regulation of glucocorticoid receptor function. J Biol Chem. 1999; 274: 3182–3188. [DOI] [PubMed]
- 8).Tamir S, Izrael S, Vaya J. The effect of oxidative stress on ERalpha and ERbeta expression. J Steroid Biochem Mol Biol. 2002; 81: 327–332. [DOI] [PubMed]
- 9).Zhang Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science2002; 295: 1895–1897. [DOI] [PubMed]
- 10).Nagata K, Somura F, Obata K, Odashima M, Izawa H, Ichihara S, Nagasaka T, Iwase M, Yamada Y, Nakashima N, Yokota M. AT1 receptor blockade reduces cardiac calcineurin activity in hypertensive rats. Hypertension. 2002; 40: 168–174. [DOI] [PubMed]
- 11).Mangos GJ, Turner SW, Fraser TB, Whitworth JA. The role of corticosterone in corticotrophin (ACTH)-induced hypertension in the rat. J Hypertens. 2000; 18: 1849–1855. [DOI] [PubMed]
- 12).Robert V, Heymes C, Silvestre JS, Sabri A, Swynghedauw B, Delcayre C. Angiotensin AT1 receptor subtype as a cardiac target of aldosterone: role in aldosterone-salt-induced fibrosis. Hypertension. 1999; 33: 981–986. [DOI] [PubMed]
- 13).Kato MF, Shibata R, Obata K, Miyachi M, Yazawa H, Tsuboi K, Yamada T, Nishizawa T, Noda A, Cheng XW, Murate T, Koike Y, Murohara T, Yokota M, Nagata K. Pioglitazone attenuates cardiac hypertrophy in rats with salt-sensitive hypertension: role of activation of AMP-activated protein kinase and inhibition of Akt. J Hypertens. 2008; 26: 1669–1676. [DOI] [PubMed]
- 14).Nagata K, Iwase M, Sobue T, Yokota M. Differential effects of dobutamine and a phosphodiesterase inhibitor on early diastolic filling in patients with congestive heart failure. J Am Coll Cardiol. 1995; 25: 295–304. [DOI] [PubMed]
- 15).Miyachi M, Yazawa H, Furukawa M, Tsuboi K, Ohtake M, Nishizawa T, Hashimoto K, Yokoi T, Kojima T, Murate T, Yokota M, Murohara T, Koike Y, Nagata K. Exercise training alters left ventricular geometry and attenuates heart failure in dahl salt-sensitive hypertensive rats. Hypertension. 2009; 53: 701–707. [DOI] [PubMed]
- 16).Ichihara S, Noda A, Nagata K, Obata K, Xu J, Ichihara G, Oikawa S, Kawanishi S, Yamada Y, Yokota M. Pravastatin increases survival and suppresses an increase in myocardial matrix metalloproteinase activity in a rat model of heart failure. Cardiovasc Res. 2006 69: 726–735. [DOI] [PubMed]
- 17).Elmarakby AA, Loomis ED, Pollock JS, Pollock DM. NADPH oxidase inhibition attenuates oxidative stress but not hypertension produced by chronic ET-1. Hypertension. 2005; 45: 283–287. [DOI] [PubMed]
- 18).Somura F, Izawa H, Iwase M, Takeichi Y, Ishiki R, Nishizawa T, Noda A, Nagata K, Yamada Y, Yokota M. Reduced myocardial sarcoplasmic reticulum Ca(2+)-ATPase mRNA expression and biphasic force-frequency relations in patients with hypertrophic cardiomyopathy. Circulation. 2001; 104: 658–663. [DOI] [PubMed]
- 19).Sakata Y, Yamamoto K, Mano T, Nishikawa N, Yoshida J, Hori M, Miwa T, Masuyama T. Activation of matrix metalloproteinases precedes left ventricular remodeling in hypertensive heart failure rats: its inhibition as a primary effect of Angiotensin-converting enzyme inhibitor. Circulation. 2004; 109: 2143–2149. [DOI] [PubMed]
- 20).Hattori T, Murase T, Iwase E, Takahashi K, Ohtake M, Tsuboi K, Miyachi M, Murohara T, Nagata K. Glucocorticoid-induced hypertension and cardiac injury: effects of mineralocorticoid and glucocorticoid receptor antagonism. Nagoya J Med Sci. 2013; 75: 81–92. [PMC free article] [PubMed]
- 21).Murase T, Hattori T, Ohtake M, Nakashima C, Takatsu M, Murohara T, Nagata K. Effects of estrogen on cardiovascular injury in ovariectomized female DahlS.Z-Leprfa/Leprfa rats as a new animal model of metabolic syndrome. Hypertension. 2012; 59: 694–704. [DOI] [PubMed]
- 22).Yamada T, Nagata K, Cheng XW, Obata K, Saka M, Miyachi M, Naruse K, Nishizawa T, Noda A, Izawa H, Kuzuya M, Okumura K, Murohara T, Yokota M. Long-term administration of nifedipine attenuates cardiac remodeling and diastolic heart failure in hypertensive rats. Eur J Pharmacol. 2009; 615: 163–170. [DOI] [PubMed]
- 23).Murase T, Hattori T, Ohtake M, Abe M, Amakusa Y, Takatsu M, Murohara T, Nagata K. Cardiac remodeling and diastolic dysfunction in DahlS.Z-Leprfa/Leprfa rats: a new animal model of metabolic syndrome. Hypertens Res. 2012; 35: 186–193. [DOI] [PubMed]
- 24).Sugden PH, Clerk A. Cellular mechanisms of cardiac hypertrophy. J Mol Med. 1998; 76: 725–746. [DOI] [PubMed]
- 25).Schakman O, Gilson H, de Coninck V, Lause P, Verniers J, Havaux X, Ketelslegers JM, Thissen JP. Insulin-like growth factor-I gene transfer by electroporation prevents skeletal muscle atrophy in glucocorticoid-treated rats. Endocrinology. 2005; 146: 1789–1797. [DOI] [PubMed]
- 26).Zhang X, Klein AL, Alberle NS, Norby FL, Ren BH, Duan J, Ren J. Cardiac-specific overexpression of catalase rescues ventricular myocytes from ethanol-induced cardiac contractile defect. J Mol Cell Cardiol. 2003; 35: 645–652. [DOI] [PubMed]
- 27).Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004; 10: 1200–1207. [DOI] [PubMed]
- 28).Pearl JM, Nelson DP, Schwartz SM, Wagner CJ, Bauer SM, Setser EA, Duffy JY. Glucocorticoids reduce ischemia-reperfusion-induced myocardial apoptosis in immature hearts. Ann Thorac Surg. 2002; 74: 830–836. [DOI] [PubMed]
- 29).Scheuer DA, Mifflin SW. Chronic corticosterone treatment increases myocardial infarct size in rats with ischemia-reperfusion injury. Am J Physiol. 1997; 272: R2017–2024. [DOI] [PubMed]
- 30).Mihailidou AS, Loan Le TY, Mardini M, Funder JW. Glucocorticoids activate cardiac mineralocorticoid receptors during experimental myocardial infarction. Hypertension. 2009; 54: 1306–1312. [DOI] [PubMed]
- 31).Bossis G, Melchior F. Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol Cell. 2006; 21: 349–357. [DOI] [PubMed]
- 32).Kuster GM, Kotlyar E, Rude MK, Siwik DA, Liao R, Colucci WS, Sam F. Mineralocorticoid receptor inhibition ameliorates the transition to myocardial failure and decreases oxidative stress and inflammation in mice with chronic pressure overload. Circulation. 2005; 111: 420–427. [DOI] [PubMed]
- 33).Kobayashi N, Yoshida K, Nakano S, Ohno T, Honda T, Tsubokou Y, Matsuoka H. Cardioprotective mechanisms of eplerenone on cardiac performance and remodeling in failing rat hearts. Hypertension. 2006; 47: 671–679. [DOI] [PubMed]
- 34).Zhao W, Ahokas RA, Weber KT, Sun Y. ANG II-induced cardiac molecular and cellular events: role of aldosterone. Am J Physiol Heart Circ Physiol. 2006; 291: H336–343. [DOI] [PubMed]
- 35).Wilson P, Morgan J, Funder JW, Fuller PJ, Young MJ. Mediators of mineralocorticoid receptor-induced profibrotic inflammatory responses in the heart. Clin Sci (Lond). 2009; 116: 731–739. [DOI] [PubMed]
- 36).Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, Miyoshi J, Takai Y, Fujita T. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008; 14: 1370–1376. [DOI] [PubMed]
- 37).Young MJ, Morgan J, Brolin K, Fuller PJ, Funder JW. Activation of mineralocorticoid receptors by exogenous glucocorticoids and the development of cardiovascular inflammatory responses in adrenalectomized rats. Endocrinology. 2010; 151: 2622–2628. [DOI] [PubMed]
- 38).Young MJ, Moussa L, Dilley R, Funder JW. Early inflammatory responses in experimental cardiac hypertrophy and fibrosis: effects of 11 beta-hydroxysteroid dehydrogenase inactivation. Endocrinology. 2003; 144: 1121–1125. [DOI] [PubMed]
- 39).Fraccarollo D, Galuppo P, Schraut S, Kneitz S, van Rooijen N, Ertl G, Bauersachs J. Immediate mineralocorticoid receptor blockade improves myocardial infarct healing by modulation of the inflammatory response. Hypertension. 2008; 51: 905–914. [DOI] [PubMed]
- 40).Neves MF, Amiri F, Virdis A, Diep QN, Schiffrin EL. Role of aldosterone in angiotensin II-induced cardiac and aortic inflammation, fibrosis, and hypertrophy. Can J Physiol Pharmacol. 2005; 83: 999-1006. [DOI] [PubMed]
- 41).Li M, Wen C, Fraser T, Whitworth JA. Adrenocorticotrophin-induced hypertension: effects of mineralocorticoid and glucocorticoid receptor antagonism. J Hypertens. 1999; 17: 419–426. [DOI] [PubMed]
- 42).Suzuki H, Schmid-Schonbein GW, Suematsu M, DeLano FA, Forrest MJ, Miyasaka M, Zweifach BW. Impaired leukocyte-endothelial cell interaction in spontaneously hypertensive rats. Hypertension. 1994; 24: 719–727. [DOI] [PubMed]
- 43).Whitworth JA, Schyvens CG, Zhang Y, Andrews MC, Mangos GJ, Kelly JJ. The nitric oxide system in glucocorticoid-induced hypertension. J Hypertens. 2002; 20: 1035–1043. [DOI] [PubMed]