

Abstract
Aims
Acute lymphoblastic leukemia (ALL) is the most common of all paediatric cancers. Aside from predisposing to ALL, polymorphisms could also be associated with poor outcome. Indeed, genetic variations involved in drug metabolism could, at least partially, be responsible for heterogeneous responses to standardized leukemia treatments, hence requiring more personalized therapy. The aims of this study were to (a) to determine the prevalence of seven common genetic polymorphisms including those that affect the folate and/or thiopurine metabolic pathways, i.e. cyclin D1 (CCND1-G870A), γ-glutamyl hydrolase (GGH-C452T), methylenetetrahydrofolate reductase (MTHFR-C677T and MTHFR-A1298C), thymidylate synthase promoter (TYMS-TSER), thiopurine methyltransferase (TPMT*3A and TPMT*3C) and inosine triphosphate pyrophosphatase (ITPA-C94A), in Caucasian (n = 94, age < 20) and Vietnamese (n = 141, age < 16 years) childhood ALL and (b) to assess the impact of a multilocus genetic risk score (MGRS) on relapse-free survival (RFS) using a Cox proportional-hazards regression model.
Results
The prevalence of MTHFR-677TT genotype was significantly higher in Caucasians (P = 0.008), in contrast to the prevalence of TYMS-TSER*3R/3R and ITPA-94AA/AC genotypes which were significantly higher in Vietnamese (P < 0.001 and P = 0.02, respectively). Compared with children with a low MGRS (≤3), those with a high MGRS (≥4) were 2.06 (95% CI = 1.01, 4.22; P = 0.04) times more likely to relapse. Adding MGRS into a multivariate Cox regression model with race/ethnicity and four clinical variables improved the predictive accuracy of the model (AUC from 0.682 to 0.709 at 24 months).
Conclusion
Including MGRS into a clinical model improved the predictive accuracy of short and medium term prognosis, hence confirming the association between well determined pharmacogenotypes and outcome of paediatric ALL. Whether variants on other genes associated with folate metabolism can substantially improve the predictive value of current MGRS is not known but deserves further evaluation.
Keywords: Caucasian, leukemia, pharmacogenetics, single nucleotide polymorphisms, Vietnamese
What is already known about this subject
Current treatment protocols can cure approximately 80% of paediatric acute lymphoblastic leukemia (ALL) patients.
Several studies have shown the impact of racial and ethnic differences in the survival of children with ALL.
Some polymorphisms in genes involved in drug metabolism are associated with the clinical response in ALL children.
What this study adds
Large differences were found between Caucasian and Vietnamese ALL children in the frequency distribution of MTHFR-C677T, TYMS-TSER and ITPA-C94A polymorphisms.
A multilocus genetic risk score (MGRS) computed from seven polymorphisms (CCND1-G870A, GGH-C452T, MTHFR-C677T, MTHFR-A1298C, TYMS-TSER, TPMT, ITPA-C94A) identifies children with poor prognosis, irrespective of the racial/ethnic group.
Including MGRS into the clinical model improves the predictive accuracy of short and medium term prognosis.
Introduction
Nearly one-third of all paediatric cancers are acute lymphoblastic leukemia (ALL), making it the most common malignancy affecting children 1. Pharmacogenetics, which has become a major field of research in recent years 2–4, investigates genetic variations that affect pharmacokinetics and pharmacodynamics of drugs by changing the structure of proteins involved in drug absorption, distribution, metabolism, excretion and cellular transport, and their influence on drug response phenotypes 5. Although current treatment protocols can cure approximately 80% of paediatric ALL patients, a number of children will relapse 6, with a relapse rate shown to vary within a group of patients with similar risk even though their leukemia blasts carry the same characteristics. Children with ALL are most often homogeneously treated within collaborative studies according to the risk groups and the body surface area. Besides tumour and patient related factors, inter-individual variations in treatment responses and toxicity could also be attributable to genetic polymorphisms influencing drug disposition and treatment intensity.
While multiple drugs are used to treat children with ALL, mounting evidence indicates that gene polymorphisms may alter therapy responses, as they participate in folate and 6-mercaptopurine/azathioprine (6-MP/AZA) metabolism, steroid response, drug transport and detoxification 3. Polymorphisms in genes interacting with folate metabolism and methotrexate (MTX) activity such as the methylenetetrahydrofolate reductase (MTHFR) and the promoter enhancer region of TYMS (TYMS-TSER), or intervening in the 6-mercaptopurine (6-MP) metabolic pathway such as thiopurine methyltransferase (TPMT) and inosine triphosphate pyrophosphatase (ITPA) could explain at least part of the inconsistent clinical outcome in children with ALL receiving similar treatments 7–9. However, data regarding the Caucasian and Vietnamese allelic distribution of polymorphic variants involved in MTX metabolism in children with ALL are scarce. Polymorphisms within MTHFR, TYMS-TSER, TPMP and ITPA, as well as cyclin D1 (CCND1), γ-glutamyl hydrolase (GGH) genes were assessed in this study.
The CCND1 gene codes for cyclin D1, which is a key protein for the cellular division, as it regulates the transition from the G1 to the S phase of the cell cycle. The CCND1 gene harbours a G > A polymorphism (G870A) in exon 4. The CCND1-870AA genotype has been reported to increase the risk for childhood ALL in Chinese and Egyptian populations 10,11. It has been associated with a lower probability of event free survival 12 but also with a reduced MTX toxicity 13. A lower MTX efficacy in patients with CCND1-870AA genotype could explain these effects. The reported prevalence of 870AA homozygotes in Caucasian and in Asian children with ALL ranges from 22.3 to 25.7% and 32.4 to 38%, respectively 14–17.
GGH is a lysosomal peptidase that catalyzes the removal of γ-linked polyglutamates of folates and anti-folates such as MTX, thus allowing folates to be exported from the cells or converting long chain MTX polyglutamates (MTXPG) into short chain MTXPGs and ultimately to MTX 18,19. It has been previously shown that the T variant allele of GGH may result in decreased GGH activity, hence increasing intracellular MTXPGs. These observations suggest that GGH-C452T polymorphism monitoring could be used to predict side effects and resistance to MTX 20. Little is known however about MTX resistance in relapsed childhood ALL. Cells from relapsed precursor-B ALL have been shown to be threefold more resistant to MTX than those from newly diagnosed ALL 21. It can therefore be hypothesized that the GGH-C452T polymorphism could play a role in ALL relapse through its interaction with MTX metabolism. While the reported frequency of the GGH-452CT genotype in Caucasian and in Asian ALL patients varies from 6 to 15.5% and 10 to 19%, respectively, it ranges between 0 and 4% or 0.4 and 1.5% for the GGH-452TT homozygote variant in both ALL populations, respectively 16,20,22,23.
MTHFR catalyzes the reduction of 5,10-methylene-THF to 5,10-methyl-THF, being therefore a major determinant of the amount of folate available in the cell for DNA synthesis and methylation. The accumulation of 5,10-methyl-THF resulting from the MTHFR genetic polymorphism may play a role in MTX response 24. Specific MTHFR genotypes have allegedly been associated with MTX toxicity in leukemia patients 25,26 and with an increased risk of relapse in paediatric ALL patients 27. Recent meta-analyses and literature reviews were recently carried out regarding the relationship between MTHFR-C677T and -A1298C polymorphisms, and ALL patients outcome including MTX toxicity 28, relapse 29 and overall survival 30. Both polymorphisms were reported to be unsatisfactory predictors of MTX-related toxicity in paediatric ALL 28 whereas MTHFR-677CT/TT genotypes were possibly associated with higher relapse 29 and mortality rates 30. The prevalence of MTHFR-C677T and the A1298C variant alleles varies with ethnicity. The prevalence of 677TT genotype ranges between 9 to 11% and 2.5 to 8.5% in Caucasian and Asian ALL children, respectively 24,31,32, vs. 9% to 16.5% and 0% to 9%, respectively, for the prevalence of the 1298CC genotype 27,31,33.
Thymidylate synthase (TYMS) catalyzes the conversion of deoxyuridylate monophosphate (dUMP) to deoxythymidylate monophosphate (dTMP) which is required for DNA synthesis and repair 34. A 28-bp polymorphic tandem repeat sequence in the 5′ untranslated enhancer region of the human TSER gene has been shown to influence TYMS expression. Whereas homozygous TSER*3R correlates with increased TYMS expression in childhood ALL 35, this variant may also be associated with MTX resistance and with a higher risk of relapse 24,35. Accordingly, determining the TYMS-TSER genotype may help predict the patient response to cytotoxic treatment 36. The prevalence of homozygous TYMS-TSER*3R lies between 28 to 30% and 50 to 77% in Caucasian and Asian ALL children, respectively 24,27,37.
TPMT catalyzes the S-methylation of thiopurine drugs which are used within the routine treatment for patients with malignant (ALL) and inflammatory diseases (rheumatoid arthritis) 38–40. TPMT enzyme activity in human tissues is influenced by genetic polymorphisms 38–40. Variations in TPMT activity impact on treatment toxicity and efficacy. Patients with low or undetectable levels of TPMT activity have the highest risk of developing severe myelosuppression when treated with standard doses of thiopurines whereas patients with very high TPMT activity are more likely to have a reduced clinical response to these cytotoxic agents 41–44. Four common alleles have been identified in the TPMT gene (i.e. TPMT*2, TPMT*3A, TPMT*3B and TPMT*3C) which account for approximately 80% of Caucasians with low or intermediate TPMT activity. The TPMT*3A allele is mutated at both nucleotides G460A and A719G whereas TPMT*3B and TPMT*3C only have the G460A or A719G mutation, respectively 45–47. The prevalence of the TPMT*3A mutation ranges from 0.9 to 5.7% in Caucasians but is usually not found in Asians 48–50 whereas the prevalence of the TPMT*3C mutation varies between 0.6 and 3% in Asians vs. 0.3 and 1.6% in Caucasians 48,50–52.
Like TPMT, ITPA is an enzyme affecting 6-MP metabolism. ITPA catalyzes the hydrolysis of inosine triphosphate (ITP) to inosine monophosphate (IMP), protecting cells from the accumulation of harmful nucleotides such as ITP and deoxyinosine triphosphate 4,53. ITPA shows genetically determined polymorphic activity. The ITPA-C94A transversion reduces ITPA enzymatic activity to 25% and ∼0% in heterozygote and homozygote carriers, respectively 54. For patients without dose reduction of 6-MP according to TPMT genotype, those with ITPA-94AC/AA genotype were reported to have a higher probability of severe febrile neutropenia 54. Conversely, TPMT has been shown to have a higher impact than ITPA on 6-MP toxicity when the dose of 6-MP was not adjusted for TPMT genotype 55. In line with these data, it has been suggested that ITPA variants may have a stronger impact in Asians than in Caucasians as the prevalence of the TPMT*3A variant in Asians is lower than in Caucasians whereas the reverse is observed for the prevalence of ITPA variants 56,57. In contrast, Kim et al. showed that toxicity of 6-MP was related neither to TMPT nor to ITPA polymorphisms, but reported a lower event-free survival (EFS) rate in patients carrying the ITPA-94AC/AA genotypes 57. The effect of ITPA on relapse free survival (RFS) remains so far unclear. The prevalence of the ITPA-C94A transversion varies with ethnicity, ranging from 3.3% in Africans to 8.3% in Caucasians, 11–19% in Asians 54,56 and 3% in Chileans 58.
Accordingly, the first aim of this study was to compare the allele frequency distribution of the seven aforementioned loci in Caucasian and Vietnamese children with ALL. All these polymorphisms were common variants associated with drug toxicity and/or response to therapy. Except for CCND1, GGH and ITPA, they are all routinely assessed with validated methods in clinical practice. The second aim was to identify their effects on the clinical outcome of these children in both series, as measured by RFS.
Methods
Ethics statement
The comparative study of the long term outcome between Caucasian and Vietnamese children treated for acute ALL according to the FRALLE 2000 Protocol was approved by Professor J.M. Maloteaux, Chairman of the Faculty of Medicine and Health Sciences Research Ethics Committee of the Université catholique de Louvain (Belgium) and by Phan Nguyen Thanh Van, M.D., Ph.D., Chairman of the Health Sciences Research Ethics Committee of the Blood Transfusion and Hematology Hospital, Ho Chi Minh city, Vietnam. Following approval by both Ethics Committees, an informed consent was provided by each patient and samples were anonymized prior to analysis, as requested.
Caucasian and Vietnamese series of patients
The Caucasian series included 94 ALL patients diagnosed under the age of 21 (mean age 6.66 ± 4.57 years with a median of 5.0 years, range 1–20 years) between 2000 and 2011, and followed at the Cliniques Universitaires Saint-Luc (UCL), Brussels, Belgium. The Vietnamese series included 141 ALL patients diagnosed under the age of 16 (mean age 5.57 ± 3.52 years with a median of 5.0 years, range 1–15 years) between 2005 and 2011, and followed at the Blood Transfusion and Hematology Hospital, University of Medicine Pham Ngoc Thach (UPNT) at Ho Chi Minh city, Vietnam.
Treatment protocol
In both series, the patients were treated according to the FRALLE 2000 protocol (Figure 1, Supplementary files 1 and 2). The description of both clinical series and their respective clinical outcomes assessment has been thoroughly described previously 59. Comparison of groups originating in countries with differences in socio-economic and health care systems is highly challenging. Accordingly, efforts were made to limit this potential bias. Only Vietnamese children from families with sufficient financial resources were included in this study and the same treatments were applied accurately in both countries during induction and consolidation therapies. Patients who discontinued the treatment were excluded from the study.
Figure 1.
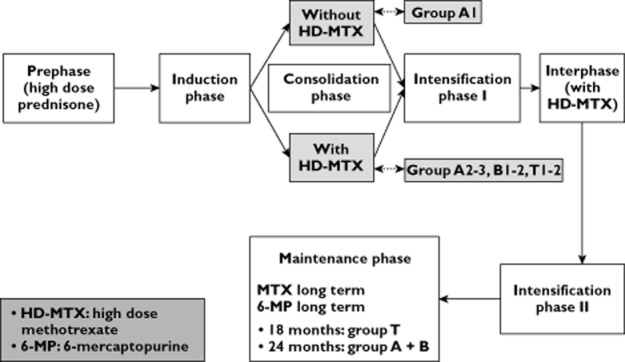
Diagram of FRALLE 2000 protocol. HD-MTX indicates high dose methotrexate; MTX, methotrexate; 6-MP, 6-mercaptopurine
Comparing respective therapeutic management between both series highlighted, however, some major differences as previously detailed 59. Firstly, both series differed in terms of timing of the first intrathecal (IT) therapy. In Belgium, the first lumbar puncture was combined with the first IT injection. In Vietnam, the first IT injection was delayed for a few days after diagnosis when the WBC count was high or the lumbar puncture traumatic. Secondly, the lack of pharmacokinetic monitoring of HD-MTX in Vietnam precluded any dosage adjustment during consolidation therapy. There was therefore no reduction in MTX dose but lederfoline was systematically administered to Vietnamese patients. During maintenance therapy, the Vietnamese children interrupted the treatment in case of toxicity but resumed it later and for a longer period whereas Caucasian children received a lower dose of 6-MP and MTX and discontinued the treatment according to the protocol. Irrespective of these socio-economic factors, health care system and therapeutic management differences, each Vietnamese patient received, however, the same FRALLE 2000 protocol and the total dose of each drug, but over a longer period of time which ultimately resulted in a significantly higher dose intensity (P < 0.001) 59.
DNA and RNA isolation
For each subject, two venous blood samples (7.5 ml) were collected in EDTA tubes. The extraction of genomic DNA was performed from peripheral blood lymphocytes using the EZ1 DNA Blood kit and BioRobot EZ1 (Qiagen, Leusden, The Netherlands), according to the manufacturer's protocol.
Total RNA was isolated from whole blood over Ficoll Hypaque and extracted with TRIzol® reagent (Invitrogen Life Sciences, Merelbeke, Belgium) according to the manufacturer's protocol.
Concentrations of extracted DNA and RNA were measured both using the NanoDrop® ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and were stored at −20°C and −80°C until use, respectively.
Pyrosequencing
Identification of selected polymorphisms in CCNDA-G870A, GGH-C452T, MTHFR-A1298C and ITPA-C94A was performed by polymerase chain reaction (PCR) followed by pyrosequencing (PyroMark Q96 ID Sequencer from Qiagen). Forward and reverse primers for each of the four SNPs, as well as primers for pyrosequencing were designed using the Pyrosequencing™ Assay Design or the Bionumerics software and synthesized by Eurogentec (Liège, Belgium). Each pyro-sequencing run included a negative control. The cumulated genotype call rate was 100%. The primers used in PCR and pyrosequencing (QIAGENE GmbH, Hilden, Germany), and size of corresponding amplicons are given in Table 1. The PCR reaction mixture was as follows: 5 μl buffer (10 mm Tris hydrochloride, and 50 mm potassium hydrochloride, pH 8.3), 3 mm MgCl2, 1U Taq Gold polymerase, 200 μm of each deoxynucleotide triphosphate (dNTP), 10 pmol of each primer and 5 μl (∼150–200 ng) of genomic DNA in a final volume of 50 μl. The cycling conditions were 95°C for 5 min, followed by 40 cycles at 95°C for 40 s, 60°C for 40 s, and 72°C for 80 s, with a final extension step at 72°C for 7 min.
Table 1.
Target genes and related single nucleotide polymorphisms: primers and probes used in the various pharmacogenetic assays
| Target gene and related polymorphism | Genbank (NCBI) accession | Location | RS number | Forward and reverse primers (5′ – 3′) used in polymerase chain reaction | Target size (bp) | Pyrosequencing primer, Taqman probes (5′–3′) |
|---|---|---|---|---|---|---|
| CCND1 (G870A) | NG_007375.1 | 11q13 | rs603965 | F: Biot- CAC ACG CTT CCT CTC CAG AGT | 108 | S: GGACATCACCCTCACTTA |
| R: CAT TTC CGT GGC ACT AGG TG | ||||||
| GGH (C452T) | NG_028126.1 | 8p12.3 | rs11545078 | F: GAG CTT TCA CTG CTG ATT AGT GG | 142 | S: ATTAGTGGAGAGTGCTTAT |
| S: Bio-TAC TTA CTA ATC CTG CCC AGC AA | ||||||
| MTHFR (A1298C) | NG_013351.1 | 1p36.3 | rs1801133 | F: GGGAGGAGCTGACCAGTG | 422 | S: AGGAGCTGACCAGTGAA-3 |
| R: Biot-CGCAGCAGCTCCTCCT | ||||||
| MTHFR (C677T) | rs1801131 | F: AAG CAC TTG AAG GAG AAG GTG TC | 64 | VIC-ATGAAATCGGCTCCC-MGB | ||
| R: CCT CAA AGA AAA GCT GCG TGA | FAM-ATGAAATCGACTCCCGC-MGB | |||||
| TSER (2R, 3R, 4R) | NG_028255.1. | 18p11.32 | Not applicable | F: CTG GCG CAC GCT CTC T | 315 | Not applicable |
| R: FAM-TTG GAT CTG CCC CAG GT | ||||||
| TPMT (G460A and/or A719G ) | NM_000367.2 | 6p22.3 | rs1800460 | F: GGAAGACATATGCTTGTGAGACA | 818 | Not applicable |
| rs1142345 | R: AAAAACATGTCAGTGTGATTTTATTTT | |||||
| ITPA (C94A) | NC_018931.2 | 20p13 | rs1127354 | F: Biot- CTGAGTTGGTAAGCTTTAGGAGA | 483 | GCCACCAAAGTGCAT |
| R: CCAATATTAACTGAACCCTAGAGG |
Biot, biotine; Fam, mutated probe; MGB, minor groove binding; NCBI, National Center for Biotechnology Information; VIC, wild type probe.
Taqman real-time PCR assay using Minor Groove Binding (MGB) probes
The MTHFR-C677T polymorphism was studied with two Taqman probes added together in one PCR tube, each consisting of an oligonucleotide with a fluorescent reporter dye, a non-fluorescent quencher and an MGB group (Applied Biosystems, Foster City, CA, USA). VIC and FAM reporter dyes were used for the WT and mutated probes, respectively. A distinct emission wavelength maximum for each probe allowed the detection of allele-specific cleavage. PCR primers and MGB probes are detailed in Table 1. The reaction was carried out on the ABI PRISM 7900 machine with SDS 2.4.0 software (Applied Biosystems) in a final volume of 25 μl with 900 nm of forward and reverse primers, 200 nm of probes labelled with VIC and FAM, 12.5 μl of TaqMan® 2XUniversal Master Mix (Applied Biosystems) and 2.5 μl (∼76–100 ng) of DNA sample as detailed previously 60.
Polymerase chain reaction amplification
TYMS-TSER polymorphisms were analyzed by PCR using: buffer 1x, 1.5 mm MgCl2, 2U Taq Gold polymerase, 200 μm dNTP, 10 pmol of each primer, 2.5 μl DMSO (dimethylsulfoxide) and H2O to reach a final volume of 50 μl. The cycling conditions were 95°C for 5 min, followed by 35 cycles at 95°C for 40 s, 60°C for 40 s and 72°C for 1 min 20 s with a final extension step at 72°C for 7 min. The forward and reverse primers used for classic PCR of the TYMS-TSER gene are described in Table 2. DNA fragments for TSER*2R and *3R genotypes were run in a 2.5% agarose gel with ethidium bromide and visualized on a UV transilluminator. Sequencing analysis was performed in the assay validation phase as well as to confirm any unusual genotype (TSER*4R). The DNA sequence of the purified product was determined on an automated ABI 3130 Genetic Analyzer using the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems).
Table 2.
Genotype frequencies of folate and thiopurine pathways associated polymorphisms in Caucasian and Vietnamese children with ALL
| Polymorphic loci | Genotype | Protective vs. risk | Caucasian (n = 94) | Vietnamese (n = 141) | Raw | Adjusted | Reported distribution |
|---|---|---|---|---|---|---|---|
| n (%) | n* (%) | P value £ | P value ¥ | ||||
| CCND1 G870A | 0.24 | 1.00 | 14–17 | ||||
| G/G | P | 28 (29.8) | 22 (18.6) | ||||
| G/A | P | 45 (47.9) | 60 (50.9) | ||||
| A/A | R | 21 (22.3) | 36 (30.5) | ||||
| GGH C452T | 0.14 | 0.97 | 16,20,22,23 | ||||
| C/C | P | 74 (78.7) | 103 (87.3) | ||||
| C/T | R | 20 (21.3) | 15 (12.7) | ||||
| MTHFR C677T | 0.001 | 0.008 | 24,31,32 | ||||
| C/C | P | 42 (44.7) | 80 (67.8) | ||||
| C/T | R | 43 (45.7) | 36 (30.5) | ||||
| T/T | R | 9 (9.6) | 2 (1.7) | ||||
| MTHFR A1298C | 0.153 | 1.00 | 27,31,33 | ||||
| A/A | P | 54 (57.4) | 55 (46.6) | ||||
| A/C | R | 31 (33) | 54 (45.8) | ||||
| C/C | R | 9 (9.6) | 9 (7.6) | ||||
| TYMS (TSER) | <0.001 | <0.001 | 24,31,37 | ||||
| *2R/*2R | P | 20 (21.3) | 3 (2.5) | ||||
| *2R/*3R | P | 41 (43.6) | 35 (29.7) | ||||
| *3R/*3R | R | 33 (35.1) | 79 (67.0) | ||||
| *3R/*4R | R | 0 | 1 (0.8) | ||||
| TPMT(*3A and*3C) | 0.31 | 1.00 | 49,51 | ||||
| No mutation | P | 90 (95.7) | 116 (91.3) | ||||
| Mutation | R | 4 (4.3) | 11 (8.7) | ||||
| TPMT*3A | 4 (4.3) | 0 | |||||
| TPMT*3C | 0 | 11(8.7) | |||||
| ITPA A94C | 0.003 | 0.02 | 54,56,59 | ||||
| A/A | P | 1 (1.1) | 4 (3.4) | ||||
| A/C | P | 13 (14.0) | 36 (30.5) | ||||
| C/C | R | 79 (84.9) | 78 (66.1) |
Sum of n not equals to 141 because of missing data. £Chi-squared test to compare reference with associated genotype between Caucasian and Vietnamese. ¥Bonferroni adjusted P values for multiple (n = 7) testing.
TPMT cDNA amplification
Total RNA was isolated from venous blood using Trizol reagent (Roche) and retro-transcribed by Superscript™ II RNase H-reverse transcriptase (Invitrogen) according to the manufacturers' instructions. TPMT cDNA was amplified by PCR using Ampli Taq DNA polymerase (Applied Biosystems) and a pair of primers that anneal to sequences within exon 4 and exon 10 of the published sequence of the TPMT cDNA (NCBI: accession number BC009596) (Table 2), as previously reported 61. cDNA sequencing was performed on both strands with the Big Dye® terminator cycle sequencing kit (Applied Biosystems), using an automated ABI3130 capillary sequencer. Sequences were compared with the wild-type sequence using the SeqScap version 2.0 software, which identifies variant and sequence matches from an allele library. In addition, sequences identify variant and sequence matches from an allele library.
Statistical analysis
The Caucasian and Vietnamese series were compared for seven polymorphisms using the chi-squared test. RFS was computed for each patient as the time from diagnosis of disease until first relapse or death. Considering that Vietnamese families confronted with a relapsing child can rarely afford further drug therapy because of the financial burden of leukemia care, this study was not pursued after the first relapse. Within the Caucasian series, all patients were diagnosed for ALL after the beginning of the study in Belgium (2001). In contrast, some patients within the Vietnamese series were diagnosed for ALL before the beginning of the study in Vietnam (2009). Time between the diagnosis of ALL and inclusion into the series was introduced in the modelling as a left-truncated time in all time-to-event analyses in order to avoid overestimation of post-diagnostic survival probabilities and likely underestimation of hazard ratios in the Cox regression models 62. A multilocus genetic risk score (MGRS) was computed for each patient by summing the number of risk genotypes among the seven tested polymorphisms. Survival free of relapse curves were compared between low (≤3) and high (≥4) MGRS using the log-rank test. The effect of the MGRS was adjusted for clinical variables including race, age at diagnosis, white blood cell (WBC) initial count, abnormal cytogenetics, corticosensitivity at day 8 (D8) using a multivariate Cox proportional hazards regression model. The predictive accuracy of the models was compared with and without the MGRS for time points ranging from 1 to 60 months. Accuracy was assessed by computing area under the time-dependent ROC curves after leave-one-out cross-validation 63. All statistical analyses were adjusted for multiple testing using Bonferroni correction, with the exception of regression analyses as recommended 62. Statistical analysis was performed using R 2.15.0 (http://www.r-project.org) 64.
Results
The distribution of each genotype studied in 94 Caucasian and 141 Vietnamese children with ALL is listed in Table 2.
The prevalence of CCND1-A870G, GGH-C452T, MTHFR-A1298C and TPMT polymorphisms between Caucasian and Vietnamese series was not statistically different. Although TMPT*3A and TMPT*3C were detected in the Caucasian and Vietnamese series, respectively, their frequency was very low. Accordingly, they were pooled together within the TPMT group to allow the statistical analysis.
The prevalence of MTHFR-677TT genotype was significantly higher in the Caucasian (9.6%) than in the Vietnamese (1.7%) series (P = 0.008). Conversely, the prevalence of TYMS-TSER*3R/3R and ITPA-94AC/AA genotypes were significantly higher (P < 0.001 and P = 0.02, respectively) in Vietnamese (67.0% and 33.9%, respectively) than in Caucasian (35.1% and 15.1%, respectively) children with ALL. The hazard ratio of each genetic variant was computed within each clinical series and for the pooled series by using Cox proportional hazards (Cox PH) models (Table 3). For the pooled series, the hazard ratio of each genetic variant was adjusted for race/ethnicity. As shown in Table 3, the individual impact of each variant was not statistically significant, except for TPMT mutation in the Caucasian series (HR = 6.07; 95% CI = 1.36, 27.0; raw P value = 0.02) before Bonferroni correction for multiple testing.
Table 3.
Univariate analysis of relapse free survival (RFS) according to pharmacogenotypes
| Polymorphisms | Caucasian series (n = 17/94)* | Vietnamese series (n = 33/141)* | Pooled series (n = 50/235)* | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Hazard ratios (95% CI) | Raw P value | Adjusted P value¥ | Hazard ratios (95% CI) | Raw P value | Adjusted P-value¥ | Hazard ratios (95% CI) | Raw P value | Adjusted P value¥ | |
| CCND1 G870A | |||||||||
| G/G – G/A | 1 | 1 | 1 | ||||||
| A/A | 0.70 (0.20, 2.42) | 0.57 | 1.00 | 2.05 (0.77, 5.45) | 0.15 | 1.00 | 1.20 (0.57, 2.53) | 0.63 | 1.00 |
| GGH C452T | |||||||||
| C/C | 1 | 1 | 1 | ||||||
| C/T | 1.79 (0.63, 5.09) | 0.28 | 1.00 | 0.73 (0.17, 3.19) | 0.67 | 1.00 | 1.32 (0.57, 3.06) | 0.51 | 1.00 |
| MTHFR C677T | |||||||||
| C/C | 1 | 1 | 1 | ||||||
| C/T – T/T | 1.09 (0.41, 2.84) | 0.87 | 1.00 | 1.05 (0.39, 2.8) | 0.93 | 1.00 | 1.08 (0.54, 2.15) | 0.83 | 1.00 |
| MTHFR A1298C | |||||||||
| A/A | 1 | 1 | 1 | ||||||
| A/C – C/C | 1.18 (0.45, 3.06) | 0.73 | 1.00 | 1.14 (0.45–2.91) | 0.78 | 1.00 | 1.14 (0.59, 2.23) | 0.69 | 1.00 |
| TSER | |||||||||
| 2R/2R – 2R/3R | 1 | 1 | 1 | ||||||
| 3R/3R – 3R/4R | 1.08 (0.40, 2.91) | 0.89 | 1.00 | 2.37 (0.68–8.27) | 0.18 | 1.00 | 1.48 (0.72, 3.05) | 0.28 | 1.00 |
| TPMT (*3A and *3C) | |||||||||
| No mutation | 1 | 1 | 1 | ||||||
| Mutation | 6.07 (1.36, 27.0) | 0.02 | 0.37 | 0.66 (0.09–5.04) | 0.69 | 1.00 | 1.51 (0.45, 5.05) | 0.50 | 1.00 |
| ITPA | |||||||||
| A/A A/C | 1 | 1 | 1 | ||||||
| CC | 2.91 (0.40, 22.0) | 0.30 | 1.00 | 1.39 (0.5–3.91) | 0.53 | 1.00 | 1.69 (0.69, 4.14) | 0.25 | 1.00 |
Number of relapse/number of children. ¥Bonferroni adjusted P values for multiple (n = 21) testing.
The MGRS was computed for each patient as the number of risk genotypes. Because of the limited number of patients, the original MGRS ranging between 0 and 6 was dichotomized in two categories (≤3 and ≥4). Compared with children with a low MGRS (≤3), a high MGRS (≥4) significantly increased the risk of relapse (univariate HR = 2.06; 95% CI = 1.01, 4.21; log rank P value = 0.042) (Figure 2). The adjusted hazard ratio of MGRS between children with a low (≤3) and high (≥4) MGRS computed from the multivariate model integrating race/ethnicity and four clinical variables was equivalent (HR = 2.09, 95% CI = 1.06, 4.13, P value = 0.03) to the univariate hazard ratio (Table 4). Adding MGRS to the model only slightly impacted on the highly significant hazard ratio of the single race/ethnicity variable. Compared with Caucasian children, the risk of relapse in Vietnamese children was increased by 4.39 (95% CI = 2.28, 8.46, P value <0.001) times, irrespective of MGRS.
Figure 2.
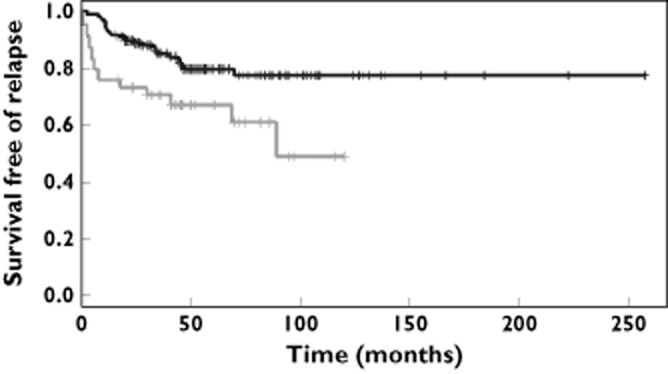
Relapse free survival according to multilocus genetic risk score (MGRS). Compared with those with a low MGRS (≤3), ALL children with a high MGRS (≥4) presented a significantly increased risk of relapse (HR = 2.06; 95% CI = 1.01, 4.21; log rank P value = 0.042).  , multilocus genetic risk score <=3;
, multilocus genetic risk score <=3;  , multilocus genetic risk score >=4
, multilocus genetic risk score >=4
Table 4.
Multivariate Cox proportional-hazards regression model in the pooled series with and without multilocus genetic risk score (MGRS)
| Cox PH model without MGRS | Cox PH model with MGRS | |||
|---|---|---|---|---|
| Hazard ratios (95% CI) | P value | Hazard ratios (95% CI) | P value | |
| Race | ||||
| Belgian | 1 | 1 | ||
| Vietnamese | 4.19 (2.20, 7.97) | <0.001 | 4.39 (2.28, 8.46) | <0.001 |
| Age at diagnosis | ||||
| < 10 years | 1 | 1 | ||
| ≥ 10 years | 1.26 (0.60, 2.65) | 0.54 | 1.37 (0.64, 2.92) | 0.42 |
| Initial WBC count | ||||
| < 50.000/mm3 | 1 | 1 | ||
| ≥ 50.000/mm3 | 1.82 (0.91, 3.64) | 0.09 | 1.57 (0.77, 3.17) | 0.21 |
| Abnormal cytogenetics | ||||
| Good prognosis | 1 | 1 | ||
| Poor prognosis | 3.66 (1.31, 10.24) | 0.01 | 3.51 (1.23, 9.99) | 0.02 |
| Corticoresistance on D8 | ||||
| Sensitive | 1 | 1 | ||
| Resistant | 1.41 (0.66, 3.00) | 0.36 | 1.38 (0.64, 2.98) | 0.42 |
| Multilocus genetic risk score | ||||
| ≤ 3 | 1 | |||
| ≥ 4 | 2.09 (1.06, 4.13) | 0.03 | ||
Predictive accuracy of the model was higher with than without MGRS on the whole range of tested time points. The predictive accuracy of both Cox regression models slightly decreased with longer time points (Figure 3).
Figure 3.
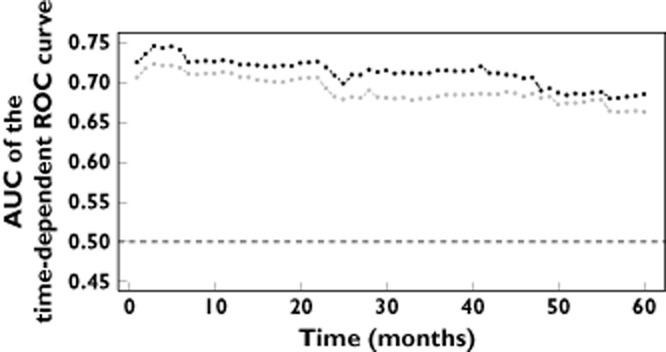
AUC of the time-dependent ROC curves obtained on the cross-validated predictions of the model with (black) and without (grey) multilocus genetic risk score.  , Cox reression equation including the indicator of a multilocus genetic risk score >=4;
, Cox reression equation including the indicator of a multilocus genetic risk score >=4;  , Cox reression equation without the indicator of a multilocus genetic risk score >=4
, Cox reression equation without the indicator of a multilocus genetic risk score >=4
Discussion
This study showed differences between Caucasian and Vietnamese children with ALL in the frequency distribution of MTHFR-C677T, TYMS-TSER and ITPA-C94A polymorphisms. Indeed, Vietnamese children had a much higher proportion of TYMS-TSER*3R/3R and of ITPA-94AC/AA genotypes, whereas the prevalence of the MTHFR-677TT genotype was very low. No significant difference was found between both series in the frequency distribution of CCND1-G870A, GGH-C452T, MTHFR-A1298C and TPMT polymorphisms. However TPMT*3A (G460A and A719G) was only found in Caucasian children while TPMT*3C (A719G) was only found in Vietnamese children. TPMT*3B (G460A) was found neither in Caucasian nor in Vietnamese children.
Vietnamese children had a two-fold higher prevalence of TSER*3R/3R than Caucasian children. These results are in line with data from Dutch 31 and Indonesian children with and without leukemia 24. It is of note that a very rare TSER*3R/4R genotype was identified in a single case from the Vietnamese clinical series (one out of 141 children) and was associated with a poor short term outcome after chemotherapy. The prevalence of MTHFR-677TT genotype in Caucasian and Vietnamese ALL children was 9.6% and 1.7%, respectively. While not found in Caucasian children, TPMT*3C is known to be more prevalent among Vietnamese (∼10%) and Thai (11%) than among other Asian patients (i.e. Chinese, Japanese, Indian and Malaysian) where prevalence ranges between 0.8% and 3% 50,51. While GGH-452TT genotype was not found in this study, this may well be biased by the moderate sample size. It is indeed a rare variant being only infrequently reported in a Japanese population (one out of 269) 20, in Chinese leukemia children (one in 203) 65, but not found in Mexican paediatric patients with ALL (n = 70) 23. The prevalence of ITPA-94AC/AA genotypes in Caucasian (15.1%) and Vietnamese (33.9%) children with ALL was in line with previous studies 37,54,58.
The univariate analyses show that the individual impact of each variant was not statistically significant probably due to a limited association with RFS and a small sample size. This univariate analysis model showed, however, the association between heterozygous TPMT*3A and a six-fold increase in RFS of Caucasian paediatric ALL. Nevertheless, it should be pointed out that this finding was only based on four cases heterozygous for TPMT*3A and 17 ALL relapses, all from the Caucasian series. All P values were adjusted for multiple testing with Bonferroni correction because of the number of hypothesis tests (n = 21) which were simultaneously conducted. The Bonferroni-adjusted P value obtained with the TPMT*3A genotype in the Caucasian series was not statistically significant and should therefore be confirmed in larger clinical series. Whereas the association between TPMT polymorphisms and 6-MP cytotoxicity is widely accepted 40,41,50, reports on a potential association between TPMT mutations and RFS are extremely scarce. In a hospital-based cohort study of 172 childhood ALL patients 44, a 6-thioguanine nucleotide (6-TGN) concentration lower than the median concentration value (i.e. 284 pmol per 8 × 108 erythrocytes) was associated with a 63% RFS at 5 years whereas RFS was 84% at 5 years in patients with the median concentration value (RFS difference 21%; 95% CI = 3, 39%; P = 0.0018). As the 6-MP metabolic pathway is influenced both by TPMT and ITPA genotypes, the effect of ITPA genotyping on RFS was also assessed in this study. Regarding ITPA variants, it is worth emphasizing that evidence on their impact in terms of toxicity and survival are scarce and often controversial 53,57, which explains why the impact of ITPA on RFS remains so far unknown. ITPA-94CA/AA genotypes appeared both in the Caucasian and Vietnamese series as a protective genotype in this study, whereas the wild-type ITPA-94CC genotype was associated with a significantly poorer survival. ITPA-94CC was therefore considered as a risk factor in univariate analysis of RFS, as well as in the computation of the MGRS (Tables 2 and 3). While the pharmacologic explanation of the effect of ITPA-C94A on RFS is so far unclear, it could be related to the unfavourable effect on the pool of 6-TGN. 6-TGN is indeed incorporated in nucleic acids or with phosphate in 6-thioinosine triphosphate, a process that determines the clinical efficacy but can be reversed by ITPA 53.
Regarding the impact of the folate pathway, many but not all studies have reported the association of genetic variations in the selected genes with the effect of MTX and relapse risk. In a cohort study 27 and case-control study 8 of 201 and 137 paediatric patients with ALL, respectively, MTHFR-C677T and MTHFR-A1298C variants were indeed associated with an increased risk of relapse. In a prospective 9 and case-control 66 study of 247 and 80 childhood ALL patients, respectively, homozygous TSER*3R/3R variants showed a greater risk of haematological relapse related to drug resistance. However, the assessment of 20 polymorphisms located in 11 genes (i.e. GSTM1, GSTT1, GSTP1, NQO1, MTHFR, MTHFD1, SLC19A1, ABCB1, TSER, CRC5 and IL15) in a cohort study of 463 paediatric ALL patients failed to demonstrate any association between MTHFR and TSER variants with event-free survival (EFS). Conversely, ABCB1 3435TT and CCR5 246GA were found to predict lower EFS probability than their 3435CC (HR = 2.496; P = 0.012) and 246GG (HR = 1.82; P = 0.033) genotype counterparts 67. In a Cox regression model of EFS adjusted for clinical variables such as ethnicity, gender, age, lineage, initial WBC and cytogenetic subtypes, a significant difference (P = 0.0015) was found only with cytogenetic subtypes. This is in agreement with our own findings, i.e., a strong association between abnormal cytogenetics (associated with a poor prognosis group), Vietnamese ethnicity and the risk of relapse 59.
In the current study, the univariate analysis of RFS showed a significant impact of MGRS and multivariate analysis indicated being Vietnamese, having a high MGRS and having abnormal cytogenetics significantly increased the risk of relapse. MGRS improved the predictive accuracy of the clinical model integrating ethnicity and four relevant clinical variables. The MGRS benefit was particularly marked during the first 4 years but tended to decrease with longer time points. However, there was a restricted number of polymorphisms and patients assessed in both clinical case series. Furthermore, the association between selected polymorphisms and adverse drug reaction to MTX and 6-MP was not assessable.
Although patients in both series were treated according to the same protocol, there were major differences in the therapeutic management 59. Limitations concerned the day of first intrathecal therapy and the way MTX toxicity was managed (resulting in a higher dose intensity in Vietnamese children), which could explain in part the difference of RFS between the series. However, it is of note that significant differences in the prevalence of the MTHFR, TYMS-TSER and ITPA polymorphism were also detected, suggesting that MGRS may improve the predictive accuracy of the clinical model. To strengthen the current observation, the impact of pharmacogenetic determinants affecting the folate and/or thiopurine pathway genes on RFS in different ethnic groups deserves further study in a larger and more homogeneous clinical series of patients. Additional SNPs, such as those previously reported (i.e. ABCB1, CCR5) 67, could also be integrated as potential predictive factors when further assessing and evaluating the strength of association between well determined pharmacogenotypes and outcome of paediatric ALL.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
This study was supported by a grant from the Belgian government: CUD-CUI-UPNT04 (http://www.cud.be) and by a grant from the Fondation Salus Sanguinis. The authors would like to acknowledge Dr Chi Dung PHU, director of Blood Transfusion Hospital at Ho Chi Minh city for contributing to data collection in Vietnam. We are also grateful to the Cliniques universitaires Saint-Luc, Brussels, Belgium, and the Pediatric Clinical Service II of Blood Transfusion and Hematology Hospital, Ho Chi Minh city, Vietnam, for collecting data.
Supporting Information
Annex 1
Protocol FRALLE 2000: details of successive treatments (dose and schedule)
Annex 2
FRALLE 2000 protocol: definition of groups and subgroups
References
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Cheok MH, Evans WE. Acute lymphoblastic leukaemia: a model for the pharmacogenomics of cancer therapy. Nat Rev Cancer. 2006;6:117–129. doi: 10.1038/nrc1800. [DOI] [PubMed] [Google Scholar]
- Kager L, Evans WE. Pharmacogenomics of acute lymphoblastic leukemia. Curr Opin Hematol. 2006;13:260–265. doi: 10.1097/01.moh.0000231424.46148.f9. [DOI] [PubMed] [Google Scholar]
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol. 2012;68:1233–1242. doi: 10.1007/s00228-012-1251-4. [DOI] [PubMed] [Google Scholar]
- Evans WE, Relling MV. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429:464–468. doi: 10.1038/nature02626. [DOI] [PubMed] [Google Scholar]
- Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- Stanulla M, Schaeffeler E, Flohr T, Cario G, Schrauder A, Zimmermann M, Welte K, Ludwig WD, Bartram CR, Zanger UM, Eichelbaum M, Schrappe M, Schwab M. Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA. 2005;293:1485–1489. doi: 10.1001/jama.293.12.1485. [DOI] [PubMed] [Google Scholar]
- Aplenc R, Thompson J, Han P, La M, Zhao H, Lange B, Rebbeck T. Methylenetetrahydrofolate reductase polymorphisms and therapy response in pediatric acute lymphoblastic leukemia. Cancer Res. 2005;65:2482–2487. doi: 10.1158/0008-5472.CAN-04-2606. [DOI] [PubMed] [Google Scholar]
- Rocha JC, Cheng C, Liu W, Kishi S, Das S, Cook EH, Sandlund JT, Rubnitz J, Ribeiro R, Campana D, Pui CH, Evans WE, Relling MV. Pharmacogenetics of outcome in children with acute lymphoblastic leukemia. Blood. 2005;105:4752–4758. doi: 10.1182/blood-2004-11-4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Wang S, Zhou Y, Xu Z, Zou Y, Zhu X, Han M, Pang T, Han ZC. Cyclin D1 gene polymorphism and susceptibility to childhood acute lymphoblastic leukemia in a Chinese population. Int J Hematol. 2005;82:206–209. doi: 10.1532/IJH97.A10418. [DOI] [PubMed] [Google Scholar]
- Bedewy AM, Mostafa MH, Saad AA, El-Maghraby SM, Bedewy MM, Hilal AM, Kandil LS. Association of cyclin D1 A870G polymorphism with two malignancies: acute lymphoblastic leukemia and breast cancer. J BUON. 2013;18:227–238. [PubMed] [Google Scholar]
- Costea I, Moghrabi A, Krajinovic M. The influence of cyclin D1 (CCND1) 870A>G polymorphism and CCND1-thymidylate synthase (TS) gene-gene interaction on the outcome of childhood acute lymphoblastic leukaemia. Pharmacogenetics. 2003;13:577–580. doi: 10.1097/00008571-200309000-00006. [DOI] [PubMed] [Google Scholar]
- Costea I, Moghrabi A, Laverdiere C, Graziani A, Krajinovic M. Folate cycle gene variants and chemotherapy toxicity in pediatric patients with acute lymphoblastic leukemia. Haematologica. 2006;91:1113–1116. [PubMed] [Google Scholar]
- Onay UV, Aaltonen K, Briollais L, Knight JA, Pabalan N, Kilpivaara O, Andrulis IL, Blomqvist C, Nevanlinna H, Ozcelik H. Combined effect of CCND1 and COMT polymorphisms and increased breast cancer risk. BMC Cancer. 2008;8:6. doi: 10.1186/1471-2407-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabalan N, Bapat B, Sung L, Jarjanazi H, Francisco-Pabalan O, Ozcelik H. Cyclin D1 Pro241Pro (CCND1-G870A) polymorphism is associated with increased cancer risk in human populations: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2008;17:2773–2781. doi: 10.1158/1055-9965.EPI-08-0169. [DOI] [PubMed] [Google Scholar]
- Yang YL, Lin DT, Chang SK, Lin SR, Lin SW, Chiou RJ, Yen CT, Lin KH, Jou ST, Lu MY, Chang HH, Chang WH, Lin KS, Hu CY. Pharmacogenomic variations in treatment protocols for childhood acute lymphoblastic leukemia. Pediatr Blood Cancer. 2010;54:206–211. doi: 10.1002/pbc.22292. [DOI] [PubMed] [Google Scholar]
- Pakakasama S, Phaitchichinda P, Kajanaschumpol S, Udomsubpayakul U, Apibal S, Hongeng S. Cyclin DI genetic polymorphism and clinical presentation of childhood acute lymphoblastic leukemia. Australasian J Cancer. 2009;8:191–194. [Google Scholar]
- Panetta JC, Wall A, Pui CH, Relling MV, Evans WE. Methotrexate intracellular disposition in acute lymphoblastic leukemia: a mathematical model of gamma-glutamyl hydrolase activity. Clin Cancer Res. 2002;8:2423–2429. [PubMed] [Google Scholar]
- Rhee MS, Lindau-Shepard B, Chave KJ, Galivan J, Ryan TJ. Characterization of human cellular gamma-glutamyl hydrolase. Mol Pharmacol. 1998;53:1040–1046. [PubMed] [Google Scholar]
- Hayashi H, Fujimaki C, Inoue K, Suzuki T, Itoh K. Genetic polymorphism of C452T (T127I) in human gamma-glutamyl hydrolase in a Japanese population. Biol Pharm Bull. 2007;30:839–841. doi: 10.1248/bpb.30.839. [DOI] [PubMed] [Google Scholar]
- Rots MG, Pieters R, Peters GJ, Noordhuis P, Van Zantwijk CH, Henze G, Janka-Schaub GE, Veerman AJ, Jansen G. Methotrexate resistance in relapsed childhood acute lymphoblastic leukaemia. Br J Haematol. 2000;109:629–634. doi: 10.1046/j.1365-2141.2000.02071.x. [DOI] [PubMed] [Google Scholar]
- Tiseo M, Giovannetti E, Tibaldi C, Camerini A, Di Costanzo F, Barbieri F, Burgers JA, Vincent A, Peters GJ, Smit EF, Ardizzoni A. Pharmacogenetic study of patients with advanced non-small cell lung cancer (NSCLC) treated with second-line pemetrexed or pemetrexed-carboplatin. Lung Cancer. 2012;78:92–99. doi: 10.1016/j.lungcan.2012.07.009. [DOI] [PubMed] [Google Scholar]
- Organista-Nava J, Gomez-Gomez Y, Saavedra-Herrera MV, Rivera-Ramirez AB, Teran-Porcayo MA, Alarcon-Romero Ldel C, Illades-Aguiar B, Leyva-Vazquez MA. Polymorphisms of the gamma-glutamyl hydrolase gene and risk of relapse to acute lymphoblastic leukemia in Mexico. Leuk Res. 2010;34:728–732. doi: 10.1016/j.leukres.2009.11.027. [DOI] [PubMed] [Google Scholar]
- Giovannetti E, Ugrasena DG, Supriyadi E, Vroling L, Azzarello A, de Lange D, Peters GJ, Veerman AJ, Cloos J. Methylenetetrahydrofolate reductase (MTHFR) C677T and thymidylate synthase promoter (TSER) polymorphisms in Indonesian children with and without leukemia. Leuk Res. 2008;32:19–24. doi: 10.1016/j.leukres.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Robien K, Schubert MM, Chay T, Bigler J, Storb R, Yasui Y, Potter JD, Ulrich CM. Methylenetetrahydrofolate reductase and thymidylate synthase genotypes modify oral mucositis severity following hematopoietic stem cell transplantation. Bone Marrow Transplant. 2006;37:799–800. doi: 10.1038/sj.bmt.1705330. [DOI] [PubMed] [Google Scholar]
- Yang L, Hu X, Xu L. Impact of methylenetetrahydrofolate reductase (MTHFR) polymorphisms on methotrexate-induced toxicities in acute lymphoblastic leukemia: a meta-analysis. Tumour Biol. 2012;33:1445–1454. doi: 10.1007/s13277-012-0395-2. [DOI] [PubMed] [Google Scholar]
- Krajinovic M, Lemieux-Blanchard E, Chiasson S, Primeau M, Costea I, Moghrabi A. Role of polymorphisms in MTHFR and MTHFD1 genes in the outcome of childhood acute lymphoblastic leukemia. Pharmacogenomics J. 2004;4:66–72. doi: 10.1038/sj.tpj.6500224. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez E, Martin-Guerrero I, Ballesteros J, Garcia-Orad A. A systematic review and meta-analysis of MTHFR polymorphisms in methotrexate toxicity prediction in pediatric acute lymphoblastic leukemia. Pharmacogenomics J. 2013;13:498–506. doi: 10.1038/tpj.2012.44. [DOI] [PubMed] [Google Scholar]
- He HR, Chen SY, You HS, Hu SS, Sun JY, Dong YL, Lu J. Association between methylenetetrahydrofolate reductase polymorphisms and the relapse of acute lymphoblastic leukemia: a meta-analysis. Pharmacogenomics J. 2014 doi: 10.1038/tpj.2014.10. doi: 10.1038/tpj.2014.10. [DOI] [PubMed] [Google Scholar]
- Ojha RP, Gurney JG. Methylenetetrahydrofolate reductase C677T and overall survival in pediatric acute lymphoblastic leukemia: a systematic review. Leuk Lymphoma. 2014;55:67–73. doi: 10.3109/10428194.2013.792336. [DOI] [PubMed] [Google Scholar]
- de Jonge R, Hooijberg JH, van Zelst BD, Jansen G, van Zantwijk CH, Kaspers GJ, Peters GJ, Ravindranath Y, Pieters R, Lindemans J. Effect of polymorphisms in folate-related genes on in vitro methotrexate sensitivity in pediatric acute lymphoblastic leukemia. Blood. 2005;106:717–720. doi: 10.1182/blood-2004-12-4941. [DOI] [PubMed] [Google Scholar]
- Paluku They-They THK, Mazabraud A, Nadifi S. Frequency of C677T polymorphism of methylene tetrahyrofolate reductase (MTHFR) gene among Berber and Arabic Moroccan populations. Antropo. 2009;20:11–17. [Google Scholar]
- Sirachainan N, Wongruangsri S, Kajanachumpol S, Pakakasama S, Visudtibhan A, Nuchprayoon I, Lusawat A, Phudhicharoenrat S, Shuangshoti S, Hongeng S. Folate pathway genetic polymorphisms and susceptibility of central nervous system tumors in Thai children. Cancer Detect Prev. 2008;32:72–78. doi: 10.1016/j.cdp.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Radparvar S, Houghton PJ, Houghton JA. Characteristics of thymidylate synthase purified from a human colon adenocarcinoma. Arch Biochem Biophys. 1988;260:342–350. doi: 10.1016/0003-9861(88)90458-4. [DOI] [PubMed] [Google Scholar]
- Krajinovic M, Costea I, Chiasson S. Polymorphism of the thymidylate synthase gene and outcome of acute lymphoblastic leukaemia. Lancet. 2002;359:1033–1034. doi: 10.1016/S0140-6736(02)08065-0. [DOI] [PubMed] [Google Scholar]
- Welsh SJ, Titley J, Brunton L, Valenti M, Monaghan P, Jackman AL, Aherne GW. Comparison of thymidylate synthase (TS) protein up-regulation after exposure to TS inhibitors in normal and tumor cell lines and tissues. Clin Cancer Res. 2000;6:2538–2546. [PubMed] [Google Scholar]
- Marsh S, Ameyaw MM, Githang'a J, Indalo A, Ofori-Adjei D, McLeod HL. Novel thymidylate synthase enhancer region alleles in African populations. Hum Mutat. 2000;16:528. doi: 10.1002/1098-1004(200012)16:6<528::AID-HUMU11>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32:651–662. [PMC free article] [PubMed] [Google Scholar]
- McLeod HL, Krynetski EY, Wilimas JA, Evans WE. Higher activity of polymorphic thiopurine S-methyltransferase in erythrocytes from neonates compared to adults. Pharmacogenetics. 1995;5:281–286. doi: 10.1097/00008571-199510000-00003. [DOI] [PubMed] [Google Scholar]
- Krynetski EY, Tai HL, Yates CR, Fessing MY, Loennechen T, Schuetz JD, Relling MV, Evans WE. Genetic polymorphism of thiopurine S-methyltransferase: clinical importance and molecular mechanisms. Pharmacogenetics. 1996;6:279–290. doi: 10.1097/00008571-199608000-00001. [DOI] [PubMed] [Google Scholar]
- Lennard L, Lilleyman JS, Van Loon J, Weinshilboum RM. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336:225–229. doi: 10.1016/0140-6736(90)91745-v. [DOI] [PubMed] [Google Scholar]
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child. 1993;69:577–579. doi: 10.1136/adc.69.5.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr. 1991;119:985–989. doi: 10.1016/s0022-3476(05)83063-x. [DOI] [PubMed] [Google Scholar]
- Lilleyman JS, Lennard L. Mercaptopurine metabolism and risk of relapse in childhood lymphoblastic leukaemia. Lancet. 1994;343:1188–1190. doi: 10.1016/s0140-6736(94)92400-7. [DOI] [PubMed] [Google Scholar]
- Szumlanski C, Otterness D, Her C, Lee D, Brandriff B, Kelsell D, Spurr N, Lennard L, Wieben E, Weinshilboum R. Thiopurine methyltransferase pharmacogenetics: human gene cloning and characterization of a common polymorphism. DNA Cell Biol. 1996;15:17–30. doi: 10.1089/dna.1996.15.17. [DOI] [PubMed] [Google Scholar]
- Yates CR, Krynetski EY, Loennechen T, Fessing MY, Tai HL, Pui CH, Relling MV, Evans WE. Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med. 1997;126:608–614. doi: 10.7326/0003-4819-126-8-199704150-00003. [DOI] [PubMed] [Google Scholar]
- Otterness D, Szumlanski C, Lennard L, Klemetsdal B, Aarbakke J, Park-Hah JO, Iven H, Schmiegelow K, Branum E, O'Brien J, Weinshilboum R. Human thiopurine methyltransferase pharmacogenetics: gene sequence polymorphisms. Clin Pharmacol Ther. 1997;62:60–73. doi: 10.1016/S0009-9236(97)90152-1. [DOI] [PubMed] [Google Scholar]
- Ameyaw MM, Collie-Duguid ES, Powrie RH, Ofori-Adjei D, McLeod HL. Thiopurine methyltransferase alleles in British and Ghanaian populations. Hum Mol Genet. 1999;8:367–370. doi: 10.1093/hmg/8.2.367. [DOI] [PubMed] [Google Scholar]
- Tumer TB, Ulusoy G, Adali O, Sahin G, Gozdasoglu S, Arinc E. The low frequency of defective TPMT alleles in Turkish population: a study on pediatric patients with acute lymphoblastic leukemia. Am J Hematol. 2007;82:906–910. doi: 10.1002/ajh.20947. [DOI] [PubMed] [Google Scholar]
- Zhou S. Clinical pharmacogenomics of thiopurine S-methyltransferase. Curr Clin Pharmacol. 2006;1:119–128. doi: 10.2174/157488406784111627. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Goto E, Konno Y, Hiratsuka M, Mizugaki M. Three novel single nucleotide polymorphisms of the human thiopurine S-methyltransferase gene in Japanese individuals. Drug Metab Pharmacokinet. 2006;21:332–336. doi: 10.2133/dmpk.21.332. [DOI] [PubMed] [Google Scholar]
- Spire-Vayron de la Moureyre C, Debuysere H, Mastain B, Vinner E, Marez D, Lo Guidice JM, Chevalier D, Brique S, Motte K, Colombel JF, Turck D, Noel C, Flipo RM, Pol A, Lhermitte M, Lafitte JJ, Libersa C, Broly F. Genotypic and phenotypic analysis of the polymorphic thiopurine S-methyltransferase gene (TPMT) in a European population. Br J Pharmacol. 1998;125:879–887. doi: 10.1038/sj.bjp.0702152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam de Beaumais T, Fakhoury M, Medard Y, Azougagh S, Zhang D, Yakouben K, Jacqz-Aigrain E. Determinants of mercaptopurine toxicity in paediatric acute lymphoblastic leukemia maintenance therapy. Br J Clin Pharmacol. 2011;71:575–584. doi: 10.1111/j.1365-2125.2010.03867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocco G, Cheok MH, Crews KR, Dervieux T, French D, Pei D, Yang W, Cheng C, Pui CH, Relling MV, Evans WE. Genetic polymorphism of inosine triphosphate pyrophosphatase is a determinant of mercaptopurine metabolism and toxicity during treatment for acute lymphoblastic leukemia. Clin Pharmacol Ther. 2009;85:164–172. doi: 10.1038/clpt.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocco G, Crews KR, Evans WE. Genetic polymorphism of inosine-triphosphate-pyrophosphatase influences mercaptopurine metabolism and toxicity during treatment of acute lymphoblastic leukemia individualized for thiopurine-S-methyl-transferase status. Expert Opin Drug Saf. 2010;9:23–37. doi: 10.1517/14740330903426151. [DOI] [PubMed] [Google Scholar]
- Marsh S, Van Booven DJ. The increasing complexity of mercaptopurine pharmacogenomics. Clin Pharmacol Ther. 2009;85:139–141. doi: 10.1038/clpt.2008.219. [DOI] [PubMed] [Google Scholar]
- Kim H, Kang HJ, Kim HJ, Jang MK, Kim NH, Oh Y, Han BD, Choi JY, Kim CW, Lee JW, Park KD, Shin HY, Ahn HS. Pharmacogenetic analysis of pediatric patients with acute lymphoblastic leukemia: a possible association between survival rate and ITPA polymorphism. PLoS ONE. 2012;7:e45558. doi: 10.1371/journal.pone.0045558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfan MJ, Salas C, Canales C, Silva F, Villarroel M, Kopp K, Torres JP, Santolaya ME, Morales J. Prevalence of TPMT and ITPA gene polymorphisms and effect on mercaptopurine dosage in Chilean children with acute lymphoblastic leukemia. BMC Cancer. 2014;14 doi: 10.1186/1471-2407-14-299. 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu Hoang PT, Ambroise J, Dang Chi VL, Dekairelle AF, Dupont S, Huynh N, Nguyen TB, Robert A, Gala JL, Vermylen C. Comparison of long-term outcome between White and Vietnamese children treated for acute lymphoblastic leukemia according to the FRALLE 2000 Protocol. J Pediatr Hematol Oncol. 2013 doi: 10.1097/MPH.0000000000000062. doi: 10.1097/MPH.0000000000000062. [DOI] [PubMed] [Google Scholar]
- Louis M, Dekairelle AF, Gala JL. Rapid combined genotyping of factor V, prothrombin and methylenetetrahydrofolate reductase single nucleotide polymorphisms using minor groove binding DNA oligonucleotides (MGB probes) and real-time polymerase chain reaction. Clin Chem Lab Med. 2004;42:1364–1369. doi: 10.1515/CCLM.2004.255. [DOI] [PubMed] [Google Scholar]
- Dewit O, Moreels T, Baert F, Peeters H, Reenaers C, de Vos M, Van Hootegem P, Muls V, Veereman G, Mana F, Van Outryve M, Holvoet J, Naegels S, Piessevaux H, Horsmans Y, Gala JL. Limitations of extensive TPMT genotyping in the management of azathioprine-induced myelosuppression in IBD patients. Clin Biochem. 2011;44:1062–1066. doi: 10.1016/j.clinbiochem.2011.06.079. [DOI] [PubMed] [Google Scholar]
- Vittinghoff EGDV, Shiboski SC, McCulloch CE. Regression Methods in Biostatistics: Linear, Logistic, Survival, and Repeated Measures Models. New York: Springer; 2012. [Google Scholar]
- Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56:337–344. doi: 10.1111/j.0006-341x.2000.00337.x. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. 2005. R: a language and environment for statistical computing. Available at http://www.R-project.org (last accessed 27 August 2014)
- Chen X, Wen F, Yue L, Li C. Genetic polymorphism of gamma-glutamyl hydrolase in Chinese acute leukemia children and identification of a novel double nonsynonymous mutation. Pediatr Hematol Oncol. 2012;29:303–312. doi: 10.3109/08880018.2012.657767. [DOI] [PubMed] [Google Scholar]
- Lauten M, Asgedom G, Welte K, Schrappe M, Stanulla M. Thymidylate synthase gene polymorphism and its association with relapse in childhood B-cell precursor acute lymphoblastic leukemia. Haematologica. 2003;88:353–354. [PubMed] [Google Scholar]
- Lu Y, Kham YKS, Hany A, Tan AM, Quah TC, Juh Yeoh AE. Host pharmacogenetic factors significantly contribute to refinement of prognosis in children with Acute Lymphoblastic Leukemia (ALL): result from the Malaysia-Singapore (Ma-Spore) ALL 2003 Study. 2011. ASH.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Annex 1
Protocol FRALLE 2000: details of successive treatments (dose and schedule)
Annex 2
FRALLE 2000 protocol: definition of groups and subgroups