

Abstract
Aim
Selisistat (SEN0014196), a first-in-class SirT1 inhibitor, is being developed as a disease-modifying therapy for Huntington's disease. This first-in-human study investigated the safety, pharmacokinetics and pharmacogenomics of single and multiple doses of selisistat in healthy male and female subjects.
Method
In this double-blind, randomized, placebo-controlled study, seven cohorts of eight subjects received a single dose of selisistat at dose levels of 5, 25, 75, 150, 300 and 600 mg and four cohorts of eight subjects were administered 100, 200 and 300 mg once daily for 7 days. Blood sampling and safety assessments were conducted throughout the study.
Results
Selisistat was rapidly absorbed and systemic exposure increased in proportion to dose in the 5–300 mg range. Steady-state plasma concentrations were achieved within 4 days of repeated dosing. The incidence of drug related adverse events showed no correlation with dose level or number of doses received and was comparable with the placebo group. No serious adverse events were reported and no subjects were withdrawn due to adverse events. There were no trends in clinical laboratory parameters or vital signs. No trends in heart rate or ECG parameters, including the QTc interval and T-wave morphology, were observed. There were no findings in physical or neurological examinations or postural control. Transcriptional alteration was observed in peripheral blood.
Conclusion
Selisistat was safe and well tolerated by healthy male and female subjects after single doses up to 600 mg and multiple doses up to 300 mg day−1.
Keywords: concentration−effect modelling, first-in-human, Huntington's disease, SirT1, transcriptional profile
What is Already Known about this Subject
Modulation of acetylation levels via non-selective histone deacetylase inhibitors has been shown to confer benefit in cellular and animal models of Huntington's disease.
The therapeutic potential of histone deacetylase inhibitors for chronic treatment has been limited by toxicity issues, requiring the development of compounds with improved selectivity and safety profiles.
What this Study Adds
This was a first-in-human study with selisistat, a first-in-class, selective SirT1 inhibitor, shown to exhibit disease-modifying potential in a series of preclinical Huntington's disease models.
Results demonstrate that selective SirT1 inhibition is safe and well tolerated in healthy male and female volunteers.
Introduction
Huntington's disease (HD) is a monogenetic, autosomal dominant disease that gives rise to progressive neural cell death resulting in chorea, behavioural disturbances and dementia. The disease runs a relentless degenerative course over a period of 10–25 years and the patient eventually succumbs to complications such as heart failure and aspiration pneumonia. The disease is caused by an increase in the length of a CAG (cytosine, adenine, guanine) triplet repeat, encoding for the amino acid glutamine, present in the N-terminal part of the ‘Huntington gene’ (‘huntingtin’, HTT). The HD gene is located on chromosome 4 and was identified in 1993 1. Clinical manifestations of HD can start at any age. Onset shows an inverse correlation to CAG repeat length, and although several studies have attempted to define a mean age of onset, no exact a priori definition of age of onset may be given. Many patients start developing symptoms at the age of 35–45 years. The presymptomatic phase gradually transforms into a pre-diagnostic phase, when patients start exhibiting subtle changes of personality, cognition and motor function. Clinical diagnosis occurs when these effects become more pronounced and specific. In this phase, individuals might become irritable or disinhibited and unreliable at work, multi-tasking becomes difficult and forgetfulness and anxiety mount. In the symptomatic phase, distinct chorea, dystonia, incoordination, motor impersistence and slowed saccadic eye movements develop. Cognitive dysfunction preferentially affects logistics, coordination, planning and decision making, while long term memory is often intact. Cognitive dysfunction tends to deteriorate gradually together with speech impairment. Depression with suicidal ideation is common and mania and psychotic symptoms may develop 2. Histopathological findings include marked cell loss and atrophy in the caudate nucleus and putamen. Striatal medium spiny GABAergic neurons are the most vulnerable and imaging studies have shown loss of striatal volume and increased size of the frontal horns of the lateral ventricles. Other affected areas include the substantia nigra, cortical layers 3, 5 and 6, the CA1 region of the hippocampus, the parietal angular gyrus, the cerebellum, the hypothalamus and the centro-medial-parafascicular complex of the thalamus 3. No disease-modifying treatment is available and current therapy is directed at symptoms associated with the disease 4.
The monogenic nature of HD suggests that all pathological phenotypes arise from the mutation, which triggers a variety of both central and peripheral responses. Disease modification might therefore be achievable by interfering with the effects of the mutation. The development of transgenic in vivo models of HD has shown that the continuous production of mutant HTT is required for maintenance of histopathological and motor deficits, and suggests that therapeutic approaches aimed at inhibition, depletion or clearance of mutant HTT hold promise not only as means of slowing disease progression but potentially also delaying disease onset 5. SirT1 (silencing information regulator T1) is a member of the sirtuin deacetylase family of enzymes, removing acetyl groups from lysine residues in histones and other proteins including mutant HTT itself. SirT1 is one of the few deacetylases capable of deacetylating mutant HTT 6,7. Inhibition of SirT1 results in increased acetylation of specific lysine residues of the mutant protein, which increases the rate of autophagocytic clearance selectively of the mutant HTT protein 6.
A large body of experimental evidence suggests that transcriptional dysregulation is one of the key pathogenic mechanisms underlying HD. This includes the alteration of transcription factor activity as well as modulation of gene expression through histone hypoacetylation and decreased acetyltransferase activity 8–10. Dysfunctional protein acetylation is believed to be an important factor in HD and the restoration of normal protein acetylation levels is considered to be a possible efficacious therapeutic approach. Modulation of acetylation levels through the use of broad-spectrum histone deacetylase inhibitors has been demonstrated to confer benefit in both cellular and animal models of HD, demonstrating reversal of transcriptional dysregulation and at least partial reversal of motor dysfunction and alterations in body weight phenotypes. However, the therapeutic potential of histone deacetylase inhibitors for chronic treatment has been limited by toxicity issues, requiring the development of compounds with improved selectivity and safety profiles 10,11. The SirT1 inhibitor selisistat possesses an attractive pharmacological and biopharmaceutical profile 12,13 and appears suitable to test the hypothesis that modulation of the acetylation status of huntingtin may lead to preferential clearance of the mutant protein in human patients with HD.
Selisistat (racemic 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide, SEN0014196, EX-527, molecular weight 248.7 g mol−1) is a selective inhibitor of SirT1 under clinical development for HD. Selisistat inhibits recombinant human SirT1 protein with a 50% maximum inhibitory concentration of 98 nm, with >200 fold selectivity over silencing information regulator T2 (SirT2) and T3 (SirT3) 12. Selisistat is cytoprotective in PC-12 cells transfected with an inducible mutant HTT, and neuroprotective in primary rat neurons transfected with mutant human HTT. Selisistat has been shown to increase acetylation at specific lysine residues of mutant HTT in cell models, resulting in an increased rate of macroautophagic clearance of the mutant protein. Selisistat is neuroprotective in a Drosophila model of HD and phenocopies the effects of SirT1 haploinsufficiency. An in vivo efficacy study in the transgenic R6/2 mouse model showed statistically significant effects on life span, several psychomotor endpoints and histological endpoints, with a minimal effective dose of 5 mg kg−1 day−1 corresponding to an average steady-state plasma concentration of 370 nm 13.
The primary objective of the study reported herein was to assess the safety and tolerability of single and multiple ascending doses of selisistat in healthy male and female volunteers as this was the first time selisistat had been administered to human subjects. Secondary objectives were to characterize the single and multiple dose pharmacokinetics of the compound and to assess the transcriptional profile of selected gene targets associated with SirT1.
Methods
Subjects
Healthy male and female subjects as defined by standard criteria were included. Exclusion criteria included clinically significant abnormal medical history or physical examination. In the single dose part (part 1), the study population consisted of six groups of healthy male subjects, aged between 18 and 54 years and one group of healthy, surgically sterile (hysterectomy, bilateral tubal occlusion or bilateral oophorectomy) or post-menopausal female subjects between 44 and 64 years of age. In the multiple dose part (part 2), subjects were recruited into three groups of healthy male subjects, aged between 21 and 49 years and one group of female subjects of child-bearing potential between 23 and 65 years of age. All subjects were Caucasian except for four males (two Blacks, one Asian and one Afro-Caribbean-Caucasian) and one female (Black). Subjects had a body weight between 57 and 105 kg and a body mass index (BMI) between 20 and 31 kg m−2. The study was conducted in accordance with Good Clinical Practice guidelines and with the ethical principles outlined in the Declaration of Helsinki. The study was conducted at the Covance Clinical Research Unit, Springfield House, Leeds, UK and the protocol was approved by the Reading Independent Ethics Committee, Reading, Berkshire, UK.
Study design
This was a double-blind, placebo-controlled, ascending single and multiple oral dose, sequential group study conducted in two parts. Part 1 comprised an ascending single dose, sequential group study, incorporating a single group, two-period crossover arm to investigate the effect of food. Part 2 comprised an ascending multiple dose, sequential group study. For both parts of the study, a sequential group, ascending dose design was chosen for safety reasons, as this was the first time selisistat was administered to humans. A two-period crossover design was chosen for the food effect arm as this allowed a within-subject assessment of the influence of food on the pharmacokinetics of selisistat. The study was double-blind and placebo-controlled in order to avoid bias in the collection and evaluation of data during its conduct. Oral doses were chosen as this is the intended clinical route of administration. No specific sample size calculation was conducted as the planned number of subjects who completed the study was considered sufficient to achieve the study objectives.
The no-observable-adverse-effect dose level (NOAEL) in the rat and dog were 20 mg kg−1 day−1 and 30 mg kg−1 day−1, respectively, corresponding to human equivalent doses (HEDs) of 3.2 mg kg−1 and 16.7 mg kg−1, respectively. For part 1 of the study, the starting dose was 5 mg, representing a dose of 0.1 mg kg−1 in a 50 kg subject, giving an approximately 32-fold safety margin compared with the HED for the NOAEL in the rat, the more sensitive species. For the subsequent dose levels in part 1, dose increments were no higher than five-fold for predicted non-pharmacologically active dose levels and no higher than two-fold for dose levels predicted to be pharmacologically active based on preclinical data. Dose escalation was to be stopped at a systemic exposure level corresponding to the NOAEL in the rat (220 μm h). Actual dose level increments following the 5 mg starting level were five-, three-, two-, two- and two-fold, corresponding to doses of 5, 25, 75, 150, 300 and 600 mg. Following examination of safety, tolerability and pharmacokinetic data from the previous groups, the dose increment for subsequent groups was to be adjusted to ensure that the pharmacokinetic stopping criterion was not exceeded. There was a minimum of 10 days between dose escalations. Female subjects received a dose that was shown to be safe and well tolerated in the male subjects. Within each group, six subjects received selisistat and two subjects received placebo. Dose escalation was continued until the pharmacokinetic stopping criterion was reached and the maximum dose administered to male subjects was 600 mg selisistat. One group comprised eight male subjects who participated in two treatment periods separated by 10 days. These subjects received 150 mg selisistat after an overnight fast and a high fat breakfast in treatment periods 1 and 2, respectively. The high fat breakfast was prepared according to the FDA specifications for this type of meal. Each subject received selisistat or placebo in both dietary states according to the study randomization schedule. A single dose of 300 mg selisistat was selected for the post-menopausal or surgically sterile female subjects based on the safety data from male subjects. Within this group six female subjects received 300 mg selisistat and two female subjects received placebo. In part 1, each subject participated in a single treatment period, with the exception of subjects in the crossover food effect group. Subjects resided at the Clinical Research Unit from day –1 (the day before dosing) to day 3 (48 h post-dose). All subjects returned for a post-study visit 5 to 7 days after their final dose.
In part 2 of the study, 32 subjects were studied in four groups, where each group consisted of eight subjects. Three groups included male subjects and one group included female subjects of child-bearing potential. Doses were administered to the male subjects in an escalating manner. Following receipt of safety, tolerability and pharmacokinetic data from the earlier male groups, the dose increment for subsequent groups was to be adjusted. Female subjects received a dose that was safe and well tolerated by the male subjects. In male subjects, once daily dosing occurred on days 1 to 7 inclusive at doses of 100 mg and 300 mg, respectively. The systemic exposure to selisistat on day 7 for the 300 mg dose group was close to the pharmacokinetic stopping criterion (220 μm h), and no further dose escalation was therefore possible. Consequently, a dosing regimen of 100 mg twice daily was selected for both males and females, where subjects were given doses in 12 h intervals on days 1 to 6 with a single dose on the morning of day 7. In each dose group, six subjects received selisistat and two subjects received placebo following an overnight fast. Subjects remained fasted until 4.5 h post-dose on day 1 and day 7 but were given a standard breakfast approximately 1 h post-dose on days 2 to 6. Each subject participated in one treatment period only. Subjects resided at the Clinical Research Unit from the evening of day –2 until the morning of day 9 (48 h after the final dose on day 7 under the once daily regime and 36 h after the final dose under the twice daily regime). All subjects returned for a post-study visit 7 to 10 days after their final dose.
In both parts of the study, treatments were administered orally with a glass (240 ml) of water while the subjects were in a standing position according to the treatment randomization. Following dosing, subjects were not allowed to lie supine for 2 h, except for study procedures or if clinically indicated. Dosing of all subjects took place at a designated time between 07.00 h and 10.30 h with an approximately 10 min interval between subjects. The second dose of the twice daily regimen was given 12 h after the first daily dose.
Pharmacokinetic methods
Blood samples for determination of selisistat plasma concentrations were collected into 4 ml lithium heparin Vacutainer™ tubes (Becton Dickinson UK, Ltd, Oxford, UK) and, after mixing, were placed on ice and centrifuged within 1 h of collection at 1500 g for 10 min at approximately 4°C. Harvested plasma was stored within 2 h of collection at approximately −70°C until analysis. In part 1, blood samples were collected prior to dosing and at 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 48, 72 and 96 h post-dose. In part 2, blood samples were collected at pre-dose, 0.5, 1, 2, 3, 4, 6, 8, 12 h on day 1, pre-dose each morning of days 2–6 and at pre-dose, 0.5, 1, 2, 3, 4, 6, 8, 12, 24 and 48 h after the last dose. Urine samples were collected into polyethylene containers at pre-dose, from 0–6, 6–12 and 12–24 h post-dose. During each collection period, the containers were stored in a refrigerator at 2°C to 8°C. Aliquots of urine were removed and stored within 4 h of the end of the collection at approximately −70°C until analyzed.
Plasma and urine samples were analyzed using a fully validated LC-MS/MS procedure, with a lower limit of quantitation of 0.1 ng ml−1 in plasma and 10 ng ml−1 in urine. The accuracy values, based upon the calibration standards across the range, were between 97.0% and 103% over the QC range.
The pharmacokinetic analysis was conducted using WinNonlin Enterprise Version 4.0.1 (Pharsight Corporation, Mountain View, California, USA). Pharmacokinetic parameters were determined from the plasma and urine concentrations of selisistat using non-compartmental procedures. In addition, dose- and body weight-normalized (norm) values for AUC(0,τ), AUC(0,tlast), AUC(0,∞), AUC(0,24 h) and Cmax were determined by dividing the original pharmacokinetic parameter by dose per kg body weight. Body weight normalized [norm] values for CL/F, Vz/F and CLR were calculated by dividing the original pharmacokinetic parameter by body weight. Actual blood sampling times post-dosing were used in the computation of the pharmacokinetic parameters, while Cmax and tmax were obtained directly from the plasma concentration–time profiles. The terminal plasma half-life, 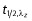 was calculated by least squares linear regression of the terminal portion of the log transformed plasma concentration–time curve. The start of the terminal elimination phase for each subject was defined by visual inspection. AUC(0,τ), AUC(0,24 h) and AUC(0,tlast) were calculated using the linear trapezoidal method with increasing concentrations and the logarithmic trapezoidal method with decreasing concentrations. The urinary excretion (Ae) for each urinary collection interval was calculated as the product of urine concentration and urine volume. Ae 0–24 h was calculated by summing the Ae values for each collection interval over the 0–24 h period. The fraction of the dose excreted (fe) was calculated for each urinary collection interval (i) and over 0–24 h.
was calculated by least squares linear regression of the terminal portion of the log transformed plasma concentration–time curve. The start of the terminal elimination phase for each subject was defined by visual inspection. AUC(0,τ), AUC(0,24 h) and AUC(0,tlast) were calculated using the linear trapezoidal method with increasing concentrations and the logarithmic trapezoidal method with decreasing concentrations. The urinary excretion (Ae) for each urinary collection interval was calculated as the product of urine concentration and urine volume. Ae 0–24 h was calculated by summing the Ae values for each collection interval over the 0–24 h period. The fraction of the dose excreted (fe) was calculated for each urinary collection interval (i) and over 0–24 h.
Pharmacogenomic methods
Whole blood samples (2.5 ml) for transcriptional investigations were collected in PAXgene® RNA blood tubes. For part 1, the transcriptional profile was investigated by using an Affymetrix microarray (HG-U133 Plus 2.0, Affymetrix Inc. Santa Clara, CA, USA) and samples from the 75 mg and 150 mg or placebo dose groups on day –1 and day 1 at 3 and 6 post-dose. These samples with the addition of samples coming from part 2 (100 mg once daily, 100 mg twice daily and 300 mg once daily on day –1 and day 1 at 3 and 6 h post-dose and on day 7 at pre-dose, 1, 3 and 6 h post-dose) were analyzed by a RT-qPCR method. RNA sample preparation and analysis were performed as previously described 14.
Safety
Safety assessments including adverse event (AE) monitoring, physical examinations, 12-lead ECGs, vital signs (blood pressure, pulse rate and oral body temperature) and laboratory safety tests were conducted at intervals throughout the study. All observed or reported AEs were recorded for all subjects throughout the study. AEs were classified as mild, moderate or severe and the relationship to study drug was determined.
Appropriate descriptive statistics for the safety data were determined using SAS Version 8.2 (SAS Institute Inc., Cary, North Carolina, USA). Adverse events were coded according to the Medical Dictionary for Regulatory Activities (MedDRA; Version 12.0). Concomitant medications were coded using the World Health Organization (WHO; Version March 2009) drug dictionary.
Cardiovascular function
ECGs were obtained digitally using a continuous 12-lead ECG recorder (ELA SpiderView Plus). The ECGs were stored on a flash card and were not available for review until the card was received by the central ECG laboratory (iCardiac Technologies, Rochester, NY, USA) and analyzed. ECGs to be used in the cardiac safety analysis were extracted from predetermined nominal time points: part 1: −1 h, −0.75, −0.5 h pre-dose and 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 h post-dose and in part 2 at pre-dose and 0.5, 1, 2, 3, 4, 6, 8 and 12 h post-dose on day 1 and day 7 and at the same time points on day –1. On the baseline day of part 2 (day –1), experimental conditions were the same as post-dosing and subjects were resting supine at the same nominal time points as the post-dose time points on day 1 and 7. At each of these time points, subjects were resting supine for at least 10 min before and 5 min after the nominal time. ECG extractions at each time point were to be performed before the pharmacokinetic blood sampling occurred.
The method for ECG extraction and interval measurements has been described previously 15. ECGs were extracted from the continuous 24 h Holter recordings using the TQTPlus® algorithm. During protocol-specified ECG extraction windows (the nominal time points as detailed above), 10 s digital 12-lead ECG tracings were extracted from continuous Holter recordings. The ECGs were extracted based on actual times of dosing, using criteria that included heart rate stability and optimal signal-to-noise ratio. At each time point, 10 ECG replicates were extracted and QT and RR intervals were measured using the COMPAS software, as previously described 15. Categorical T-wave morphology analysis (as described below) as well as the measurements for PR, QRS and presence of U-waves was performed fully manually utilizing three out of the 10 replicates at each time point. The ECG core laboratory staff were blinded to treatment, time and study day/subject identifiers. ECGs for each subject were read by a single reader and were over-read by a cardiologist for both interval measurements and overall interpretation.
QTc concentration–effect modelling
The relationship between ΔΔQTcF and selisistat concentrations was investigated by linear mixed-effects modelling. Data from all male subjects dosed with single doses of 5 to 600 mg selisistat in part 1 (six groups) and with multiple doses of 100 mg once or twice daily and 300 mg daily in part 2 (three groups) were included in the analysis. Subjects dosed with placebo were analyzed as a pooled group with 12 subjects in part 1 and six subjects in part 2. For the placebo adjustment, the mean ΔQTcF (baseline adjusted QTcF) in the placebo group calculated at a specific time point was subtracted from ΔQTcF for each subject on treatment at the same time point to generate ΔΔQTcF. Time-matched concentrations were included in the model as a covariate and subjects as a random effect for both intercept and slope, when applicable. The model was used for predicting population average ΔΔQTcF and its corresponding upper 95% one-sided confidence interval bound at the geometric mean maximum plasma concentrations. A plot of the observed median-decile drug concentrations and associated mean ΔΔQTcF (90% CI) together with the mean model-predicted ΔΔQTcF was used to evaluate the adequacy of the model fit to the assumption of linearity and the impact on quantifying the concentration-response relationship.
Neurological examinations
The neurological examination included the abbreviated Mini Mental State Examination, observations for muscular fibrillations and tremor, pupillary light reaction, eye movement assessment, including nystagmus during eye movement (other cranial nerves if indicated), motor assessments (muscle strength, tone, reflex), cerebellar assessments (finger nose test, finger tapping, rapid alternating movements, heel shin test and gait, sensory assessment (crude touch and sense of vibration). In addition, the modified Clinical Test for the Sensory Integration in Balance (mCTSIB), an extended version of the Romberg's test, was performed using a Basic Master Balance™ sway platform (Biosense Medical, Chelmsford, UK) on day –1 and at 3 and 48 h post-dose during the single dose part and on day -1 and at 3 h post-dose on day 1 and on day 7 as well as 48 h after the last dose. Triplicate 10 s assessments were made in four different conditions (eyes open, firm surface; eyes closed, firm surface; eyes open, foam surface; eyes closed, foam surface).
Statistical analysis
No formal statistical assessment of sample size was conducted, as this was the first time selisistat had been administered to man. The number of subjects who participated in this study is common in early clinical pharmacology studies and was considered sufficient to achieve the objectives of the study. Treatment randomization was achieved using a computer-generated pseudo-random permutation procedure. For each group, two subjects were randomly assigned to receive placebo. Subjects in group D (food effect group at 150 mg) were randomized to the same treatment in treatment periods 1 and 2. All statistical analysis of the ECGs was performed using the statistical software R for Windows (version 2.8.1). Two types of baselines were used: the average of three pre-dose time points for part 1 and the day –1 time-matched time points for part 2. Subjects dosed with placebo were analyzed as a pooled group with 12 subjects in part 1 and six subjects in part 2, respectively. The placebo-corrected change from baseline was computed using the two-sample t-test with unequal variances. Microarray CEL files were pre-processed with R program (The R Project for Statistical Computing http://www.r-project.org/) making use of the RMA Bioconductor package [16 ]. Differential expression analysis on microarray- and RT-qPCR data was performed in MATLAB with repeated measure anova considering ‘treatment’ and ‘time’ as independent variables and ‘subject’ as random effect, after log transformation. Profiles of differentially expressed genes were clustered in order to select only profiles of interest.
Results
Subjects
Eighty-eight subjects entered the study, with 56 subjects in part 1 and 32 subjects in part 2. All subjects completed the study in accordance with the protocol, amendments and the treatment randomization (see Table 1 for baseline demographics and subject disposition). All 88 subjects were included in the safety population and were used for the safety analyses. The pharmacokinetic population included all 66 subjects assigned to selisistat treatment (42 subjects in part 1, and 24 subjects in part 2). All subjects in part 1 were Caucasian except for a male of mixed Afro-Caribbean-Caucasian race and an Asian male. In part 2, all subjects were Caucasian except for two male and one female Blacks. In part 1, the females in group H were older than the males as this group comprised post-menopausal or surgically sterile female subjects. The mean weight and BMI was generally similar across all male treatment groups in part 1. Female subjects in group H had a lower mean body weight than male treatment groups while the BMI was similar between male and female subjects. In part 2, male subjects in group J had a lower mean body weight and BMI than groups I and K, and was similar to that of the female subjects in group L. Female subjects tended to be older than male subjects even though women of child bearing potential were not excluded from this group.
Table 1.
Subject demographics, disposition and dose levels, mean ± SD
| Group | Treatment | Gender | Number of subjects | Age (years) | Body weight (kg) | BMI (kg m−2) |
|---|---|---|---|---|---|---|
| A, B, C, E, F | Placebo | Male | 10 | 33 ± 9.5 | 79.8 ± 12 | 25.7 ± 3.4 |
| D | Placebo | Male | 2 | 24, 48 | 74.1, 81.1 | 25.6, 26.8 |
| H | Placebo | Female | 2 | 61, 64 | 56.9, 73.0 | 23.4, 28.5 |
| A | 5 mg selisistat | Male | 6 | 34 ± 13 | 75.7 ± 7.0 | 25.8 ± 2.5 |
| B | 25 mg selisistat | Male | 6 | 39 ± 10 | 77.7 ± 5.6 | 26.8 ± 2.6 |
| C | 75 mg selisistat | Male | 6 | 30 ± 8.7 | 78.5 ± 8.7 | 24.8 ± 2.4 |
| D | 150 mg selisistat* | Male | 6 | 28 ± 13 | 84.5 ± 14 | 24.9 ± 3.6 |
| E | 300 mg selisistat | Male | 6 | 32 ± 14 | 80.6 ± 9.7 | 24.5 ± 2.2 |
| F | 600 mg selisistat | Male | 6 | 37 ± 11 | 84.4 ± 4.0 | 26.2 ± 2.4 |
| H | 300 mg selisistat | Female | 6 | 53 ± 6.8 | 66.6 ± 9.1 | 26.1 ± 2.3 |
| I, J, K | Placebo | Male | 6 | 29 ± 9.9 | 82.7 ± 11 | 27.2 ± 3.2 |
| L | Placebo | Female | 2 | 43, 48 | 67.1, 74.6 | 24.6, 28.8 |
| I | 100 mg selisistat OD | Male | 6 | 29 ± 8.7 | 79.3 ± 8.2 | 25.5 ± 3.7 |
| J | 300 mg selisistat OD | Male | 6 | 28 ± 8.3 | 67.7 ± 5.1 | 21.8 ± 2.3 |
| K | 100 mg selisistat BID | Male | 6 | 32 ± 6.2 | 83.6 ± 11 | 27.2 ± 3.0 |
| L | 100 mg selisistat BID | Female | 6 | 48 ± 15 | 68.2 ± 10 | 24.7 ± 3.0 |
Two-period crossover in the fasted and fed states. BMI, body mass index.
PK results
Single oral doses of 5 to 600 mg selisistat were rapidly absorbed by male subjects in the fasted condition, although the rate of absorption appeared to be dose-dependent with a median tmax of selisistat increasing from 1 h post-dose at 5 mg to 4 h post-dose at 600 mg (Figure 1). Elimination of selisistat occurred in a biphasic manner, with an apparent terminal plasma half-life that appeared to increase with dose (mean values ranging from 1.6 h at 5 mg to 6.1 h at 600 mg). The AUC(0,∞) of selisistat increased in a dose proportional manner over the 5 to 300 mg dose range, with a marked increase in supra-proportionality between the 300 and 600 mg dose levels, suggesting that one or more clearance mechanisms are approaching saturation at higher doses (Figure 2A and Table 2). The fraction of unchanged drug excreted in the urine with respect to dose was low for all dose levels in male subjects, with <0.02% being eliminated up to 24 h post-dose at each dose level. Following multiple dosing, the fraction of the dose excreted in the urine remained low, but increased with time, consistent with the plasma accumulation observed. Food had a minimal effect on the single dose pharmacokinetics of selisistat in male subjects. Following a high fat breakfast, the rate of absorption was delayed, whereas the extent of absorption was largely unchanged.
Figure 1.
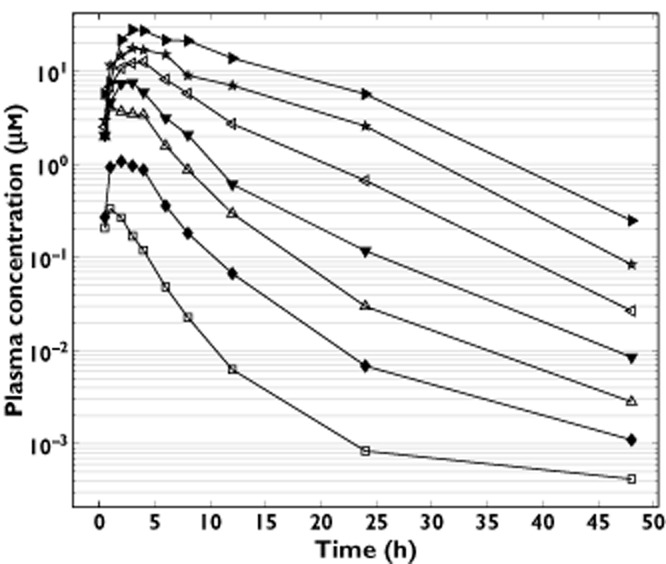
Mean selisistat plasma concentration vs. time following single doses between 5 and 600 mg.  , 5 mg;
, 5 mg;  , 25 mg;
, 25 mg;  , 75 mg;
, 75 mg;  , 150 mg;
, 150 mg;  , 300 mg;
, 300 mg;  , 600 mg;
, 600 mg;  , 300 mg; female
, 300 mg; female
Figure 2.
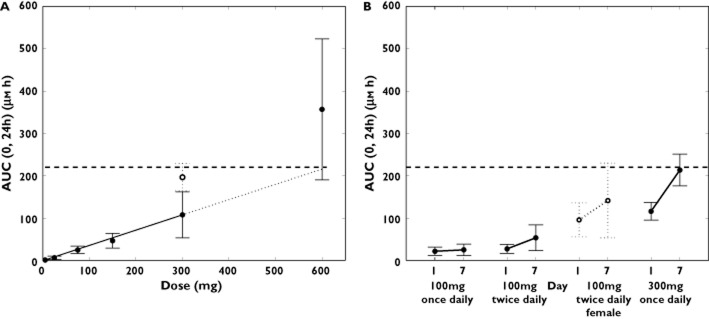
(A) Mean (± SD) selisistat AUC0–24 (μm h) following single doses between 5 and 600 mg in males (•) and females ( ). The dashed line represents the NOAEL exposure. (B) Mean (± SD) selisistat AUC(0,24 h) (μm h) following repeated doses between 100 and 300 mg once daily in males (•) and females (
). The dashed line represents the NOAEL exposure. (B) Mean (± SD) selisistat AUC(0,24 h) (μm h) following repeated doses between 100 and 300 mg once daily in males (•) and females ( ). The dashed line represents the NOAEL exposure
). The dashed line represents the NOAEL exposure
Table 2.
Summary of pharmacokinetic parameters in male and female subjects following ascending single doses of selisistat
| Parameter | Dose of selisistat (male, fasted state) | Dose of selisistat (Female, fasted state) | ||||||
|---|---|---|---|---|---|---|---|---|
| 5 mg (n = 6) | 25 mg (n = 6) | 75 mg (n = 6) | 150 mg‡ (n = 6) | 150 mg (n = 6) | 300 mg (n = 6) | 600 mg (n = 6) | 300 mg (n = 6) | |
| AUC(0,tlast) (μm h) | 1.02 ± 0.56 | 4.86 ± 3.89 | 22.5 ± 8.20 | 42.5 ± 16.9 | 39.6 ± 13.3 | 97.8 ± 57.4 | 317 ± 166 | 206 ± 29.9 |
| (56.0) | (71.1) | (34.5) | (44.7) | (38.6) | (49.0) | (68.4) | (13.8) | |
| AUC(0,24 h) (μm h) | 1.02 ± 0.56 | 4.85 ± 3.80 | 22.4 ± 8.21 | 41.7 ± 16.3 | 39.4 ± 13.1 | 94.3 ± 52.5 | 283 ± 143 | 188 ± 30.6 |
| (55.6) | (70.4) | (34.6) | (43.6) | (38.2) | (47.0) | (65.1) | (15.2) | |
| AUC(0,∞) (μm h) | 1.00 ± 0.63 | 4.87 ± 3.88 | 22.6 ± 8.30 | 42.5 ± 16.9 | 39.6 ± 13.3 | 98.0 ± 57.6 | 319 ± 168 | 206 ± 29.9 |
| (63.1)* | (71.1) | (34.7) | (44.7) | (38.6) | (49.1) | (68.6) | (13.8) | |
| Cmax (μm) | 0.33 ± 0.083 | 1.05 ± 0.43 | 4.83 ± 1.20 | 7.94 ± 1.61 | 6.74 ± 1.75 | 13.1 ± 4.48 | 26.6 ± 10.44 | 21.3 ± 6.9 |
| (25.8) | (42.3) | (24.1) | (22.5) | (26.1) | (32.0) | (45.8) | (34.3) | |
| tmax† (h) | 1.01 | 2.00 | 1.06 | 2.56 | 3.67 | 3.50 | 4.00 | 3.00 |
| (1.0, 2.0) | (1.0, 3.0) | (1.0, 4.0) | (1.0, 3.0) | (3.0, 4.0) | (2.0, 4.0) | (3.0, 4.0) | (2.0, 6.0) | |
| t1/2 (h) | 1.63 ± 0.69 | 3.03 ± 0.84 | 4.26 ± 1.56 | 4.94 ± 1.48 | 3.86 ± 1.52 | 4.91 ± 0.77 | 6.12 ± 1.38 | 6.43 ± 1.17 |
| (41.4)* | (25.6) | (38.1) | (33.3) | (39.3) | (15.8) | (23.2) | (20.7) | |
Geometric mean ± SD (CV%) data are presented; n = number of subjects studied; *n = 5; †Median (min–max); ‡Fed state.
The multiple oral dose pharmacokinetics of selisistat showed no dose or time dependency in tmax or apparent terminal half-life. At each dose level, the morning trough selisistat plasma concentrations for individual subjects showed that steady-state was generally achieved by day 4. Consistent with the single dose finding, a supra-proportional increase in steady-state AUC(0,τ) was observed across the 100 mg once daily to 300 mg once daily range (Figure 2B), whilst the steady-state Cmax increased in a dose-proportional manner. Furthermore, the steady-state AUC(0,τ) was approximately two-fold higher for the 100 mg twice daily dose level as compared with the 100 mg once daily dose level (Table 3).
Table 3.
Summary of the pharmacokinetic parameters in male and female subjects following multiple doses of selisistat
| Parameter | 100 mg selisistat once daily (Male) | 300 mg selisistat once daily (Male) | 100 mg selisistat twice daily (Male) | 100 mg selisistat twice daily (Female) | ||||
|---|---|---|---|---|---|---|---|---|
| Day 1 (n = 6) | Day 7 (n = 6) | Day 1 (n = 6) | Day 7 (n = 6) | Day 1 (n = 6) | Day 7 (n = 6) | Day 1 (n = 6) | Day 7 (n = 6) | |
| AUC(0,τ) (μm h) | 19.2 ± 9.2 | 21.5 ± 12.8 | 108 ± 19.2 | 199 ± 34.7 | 21.5 ± 7.2 | 39.4 ± 22.0 | 49.2 ± 15.9 | 88.3 ± 56.4 |
| (41.0) | (46.8) | (18.3) | (17.0) | (35.5) | (59.1) | (39.8) | (68.2) | |
| AUC(0,∞) (μm h) | 19.3 ± 9.3 | 119 ± 19.7 | 22.1 ± 7.9 | 58.7 ± 23.6 | ||||
| (41.1) | (16.4)* | (37.5) | (50.5) | |||||
| Cmax (μm) | 4.3 ± 1.7 | 4.7 ± 2.2 | 15.3 ± 6.1 | 22.5 ± 9.7 | 5.2 ± 1.0 | 7.3 ± 1.8 | 8.8 ± 2.4 | 13.7 ± 6.2 |
| (36.2) | (38.2) | (41.0) | (45.7) | (21.4) | (24.6) | (28.0) | (41.4) | |
| tmax† (h) | 3.0 | 2.4 | 4.0 | 3.5 | 2.0 | 2.1 | 2.2 | 3.8 |
| (2.00, 4.00) | (1.00, 4.12) | (3.00, 6.00) | (1.02, 6.00) | (1.00, 3.02) | (1.00, 4.00) | (1.00, 3.02) | (3.02, 4.00) | |
| t1/2 (h) | 2.43 ± 0.30 | 4.61 ± 1.26 | 4.76 ± 2.04 | 3.72 ± 0.47 | 1.85 ± 0.51 | 4.55 ± 0.58 | 3.73 ± 1.55 | 4.85 ± 0.70 |
| (12.2) | (28.7) | (42.7)* | (11.9) | (26) | (12.7) | (48.5) | (14.4) | |
| RAobs | 1.12 ± 0.13 | 1.84 ± 0.27 | 1.83 ± 0.48 | 1.79 ± 0.59 | ||||
| (12.1) | (15.9) | (25.7) | (30.0) | |||||
Geometric mean (CV%) data are presented; n = number of subjects studied; *n = 5; †Median (min–max). RAobs, observed accumulation ratio.
In the single dose phase, between-subject variability (%CV) in terms of AUC(0,∞) and Cmax was 35–71% and 23–46%, respectively. Across all dose levels, the pooled between-subject variability for AUC(0,∞) and Cmax was 56% and 33%, respectively. In the multiple dose phase, between-subject variability (%CV) was 17–59% in males and 28–68% in females. Systemic exposure following both single and multiple dosing was higher in females than in males. AUC(0,∞), AUC(0,τ) and Cmax values were 1.1-fold, 2.2–2.3-fold and 1.7–1.9-fold higher in females than inmale subjects. There were no differences in systemic exposure or pharmacokinetic parameter estimates between Caucasian and non-Caucasians subjects.
Transcriptional profiling
Microarray data were analyzed to identify genes showing differential expression in treated vs. placebo subjects following normalization to pre-dose data, resulting in two types of effects, genes that showed modulation in the placebo group but not in the selisistat-treated subjects and another group with the converse situation. The whole array comprised approximately 55 000 probe sets and was analyzed with repeated measure anova to obtain 8200 probe sets with an interaction P value less than 0.05. Additional clustering to theoretical profiles of interest led to the selection of approximately 2000 probe sets. The final selection of 23 differentially regulated genes was performed by adding a final filter to the fold-change after treatment with respect to baseline and selecting those genes changing at least 1.3-fold (Table 4). These genes were further investigated by the more sensitive real-time quantitative polymerase chain reaction (RT-qPCR) method in the same blood samples, confirming the transcriptional modulation for nine out of the 23 genes.
Table 4.
Genes differentially modulated by selisistat in healthy volunteers
| Microarray results | |||
|---|---|---|---|
| Affymetrix ID | Symbol | Gene name | RT-qPCR results |
| 1553395_a_at | CD200R1 | CD200 receptor 1 | Discarded |
| 220384_at | TXNDC3 | thioredoxin domain containing 3 | Validated |
| 224310_s_at | BCL11B | B-cell CLL/lymphoma 11B | Discarded |
| 222562_s_at | TNKS2 | tankyrase | Discarded |
| 200603_at | PRKAR1A | protein kinase cAMP-dep. Regulatory, I alpha | Validated |
| 209160_at | AKR1C3 | aldo-keto reductase 1-C3 | Validated |
| 209137_s_at | USP10 | ubiquitin specific peptidase10 | Validated |
| 206643_at | HAL | histidine ammonia-lyase | Discarded |
| 200907_s_at | PALLD | palladin, cytoskeletal associated protein | Validated |
| 235146_at | TMCC3 | transmembrane and coiled coil domains 3 | Validated |
| 223080_at | GLS | glutaminase | Not tested (a) |
| 214321_at | NOV | ? | Not tested (a) |
| 206942_s_at | PMCH | pro-melanin-concentrating hormone | Discarded |
| 228624_at | TMEM144 | transmembrane protein 144 | Discarded |
| 240757_at | CLASP1 | cytoplasmic linker associated protein 1 | Not tested (b) |
| 216102_at | PHLDB1 | pleckstrin homology-like domain, B-1 | Not tested (b) |
| 202323_s_at | ACBD3 | acyl-CoA binding domain containing 3 | Validated |
| 204893_s_at | ZFYVE9 | zinc finger, FYVE domain containing 9 | Discarded |
| 219529_at | CLIC3 | chloride intracellular channel 3 | Validated |
| 217863_at | PIAS1 | protein inhibitor of activated STAT1 | not tested (b) |
| 202067_s_at | LDLR | low density lipoprotein receptor | Validated |
| 223849_s_at | MOV10 | Moloney leukemia virus 10 homolog | Discarded |
| 1555766_a_at | GNG2 | guanine nucleotide binding protein, gamma 2 | Discarded |
The table reports the selected probe set and the corresponding gene name. The RT-qPCR column reports genes for which the modulation observed in the microarray was confirmed as ‘Validated’. Five genes were not tested because the Affymetrix ID recognized ambigous targets, indicated in table with (a) or a low annotation grade, indicated with (b).
Safety
There were no serious adverse events reported during the study and no subjects were withdrawn due to adverse events. Single oral doses of selisistat were considered to be safe and well tolerated by healthy male subjects when administered at doses up to 600 mg, and by female subjects when administered at a dose of 300 mg selisistat (Table 5). Multiple oral doses of selisistat were also considered to be safe and well tolerated by healthy male subjects at doses up to 300 mg once daily for 7 days and by healthy female subjects when administered doses of 100 mg twice daily for 7 days. There was a low incidence of drug related adverse events in male subjects, with no increase in the number of subjects experiencing adverse events with increasing dose of selisistat. The incidence of adverse events did not exceed that observed in the placebo group. No increase in the number of adverse events reported was observed following administration of multiple doses of selisistat compared with single doses (Table 6). The majority of adverse events reported by male and female subjects were mild in severity and resolved without treatment. Only one adverse event graded as severe in intensity occurred during the study. One 18-year-old male subject experienced an episode of postural syncope 1 h and 18 min after dosing at 150 mg. This event was considered possibly related to the study drug by the investigator. Dietary state had no effect on adverse events. Following single oral doses of selisistat, the most frequent drug-related adverse event was headache, experienced by 12% of male subjects and 83% of female subjects. Following multiple oral doses of selisistat, the incidence of adverse events was low in male subjects. In female subjects, three out of six subjects reported at least one incident of gastrointestinal complaint. Overall, adverse events were more frequently reported in females than in males on drug and on placebo (Tables 5 and 6).
Table 5.
Treatment emergent adverse events for all causalities (single dose)
| System organ class | Number of subjects (%) with adverse events [number of adverse events] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | Dose of selisistat | ||||||||||
| Male (fasted) (n = 12) | Male (fed) (n = 2) | Female (fasted) (n = 2) | Male 5 mg (fasted) (n = 6) | Male 25 mg (fasted) (n = 6) | Male 75 mg (fasted) (n = 6) | Male 150 mg (fasted) (n = 6) | Male 150 mg (fed) (n = 6) | Male 300 mg (fasted) (n = 6) | Female 300 mg (fasted) (n = 6) | 600 mg (fasted) (n=6) | |
| Nervous system disorders | |||||||||||
| Headache | 2 (17%) [2] | 1 (50%) [1] | 1 (17%) [1] | 1 (17%) [1] | 2 (33%) [2] | 1 (17%) [1] | 5 (83%) [5] | 1 (17%) [1] | |||
| Dizziness | 1 (17%) [1] | ||||||||||
| Dizziness postural | 1 (17%) [1] | ||||||||||
| Somnolence | 1 (17%) [1] | ||||||||||
| Syncope | 1 (17%) [1] | ||||||||||
| Total | 2 (17%) [2] | 1 (50%) [1] | 1 (17%) [1] | 2 (33%) [3] | 2 (33%) [3] | 1 (17%) [2] | 5 (83%) [5] | 1 (17%) [1] | |||
| Musculoskeletal and connective tissue disorders | |||||||||||
| Pain in extremity | 1 (8%) [1] | 1 (50%) [1] | 1 (17%) [1] | ||||||||
| Arthralgia | 1 (17%) [1] | ||||||||||
| Total | 1 (8%) [1] | 1 (50%) [1] | 1 (17%) [1] | 1 (17%) [1] | |||||||
| Respiratory, thoracic and mediastinal disorders | |||||||||||
| Oropharyngeal pain | 1 (8%) [1] | 1 (17%) [1] | |||||||||
| Total | 1 (8%) [1] | 1 (17%) [1] | |||||||||
| Skin and subcutaneous disorders | |||||||||||
| Dermatitis Acneiform | 1 (8%) [1] | ||||||||||
| Rash | 1 (17%) [1] | ||||||||||
| Total | 1 (8%) [1] | 1 (17%) [1] | |||||||||
| Cardiac disorders | |||||||||||
| Palpitations | 1 (17%) [1] | ||||||||||
| Total | 1 (17%) [1] | ||||||||||
| General disorders and administration site conditions | |||||||||||
| Fatigue | 1 (17%) [1] | ||||||||||
| Total | 1 (17%) [1] | ||||||||||
| Infections and infestations | |||||||||||
| Nasopharyngitis | 1 (17%) [1] | ||||||||||
| Total | 1 (17%) [1] | ||||||||||
| Gastrointestinal disorders | |||||||||||
| Nausea | 1 (17%) [1] | ||||||||||
| Total | 1 (17%) [1] | ||||||||||
| Overall total | 5 (42%) [5] | 0 (0%) [0] | 1 (50%) [1] | 1 (17%) [1] | 0 (0%) [0] | 2 (33%) [5] | 3 (50%) [5] | 2 (33%) [4] | 0 (0%) [0] | 5 (83%) [7] | 1 (17%) [1] |
n = number of subjects studied.
Table 6.
Treatment-emergent adverse events for all causalities (multiple dose)
| Number of subjects (%) with adverse events [number of adverse events] | ||||||
|---|---|---|---|---|---|---|
| System organ class | Placebo | Dose of selisistat | ||||
| Male (n = 6) | Female (n = 6) | Male 100 mg once daily (n = 6) | Male 300 mg once daily (n = 6) | Male 100 mg twice daily (n = 6) | Female 100 mg twice daily (n = 6) | |
| Gastrointestinal disorders | ||||||
| Diarrhoea | 1 (50%) [1] | 1 (17%) [1] | 1 (17%) [1] | |||
| Abdominal pain upper | 1 (50%) [1] | 1 (17%) [1] | ||||
| Constipation | 1 (50%) [1] | 1 (17%) [1] | ||||
| Nausea | 1 (50%) [1] | 1 (17%) [1] | ||||
| Flatulence | 1 (17%) [1] | |||||
| Abdominal distension | 1 (17%) [1] | |||||
| Total | 2 (100%) [4] | 1 (17%) [1] | 1 (17%) [1] | 3 (50%) [5] | ||
| Nervous system disorders | ||||||
| Headache | 1 (17%) [2] | 1 (17%) [1] | ||||
| Dizziness | 1 (17%) [1] | |||||
| Total | 1 (17%) [2] | 1 (17%) [1] | 1 (17%) [1] | |||
| Musculoskeletal and connective tissue disorders | ||||||
| Neck pain | 1 (17%) [1] | |||||
| Back pain | 1 (17%) [1] | 1 (17%) [1] | ||||
| Total | 1 (17%) [1] | 1 (17%) [1] | 1 (17%) [1] | |||
| Skin and subcutaneous tissue disorders | ||||||
| Rash | 1 (17%) [1] | 1 (17%) [1] | 1 (17%) [1] | |||
| Total | 1 (17%) [1] | 1 (17%) [1] | 1 (17%) [1] | |||
| Ear and labyrinth disorders | ||||||
| Hearing impaired | 1 (17%) [1] | |||||
| Total | 1 (17%) [1] | |||||
| Infections and infestations | ||||||
| Nasopharyngitis | 1 (17%) [1] | 1 (17%) [1] | ||||
| Total | 1 (17%) [1] | 1 (17%) [1] | ||||
| Injury, poisoning and procedural complications | ||||||
| Arthropod bite | 1 (17%) [1] | |||||
| Total | 1 (17%) [1] | |||||
| Metabolism and nutrition disorders | ||||||
| Decreased appetite | 1 (17%) [1] | |||||
| Total | 1 (17%) [1] | |||||
| Renal and urinary disorders | ||||||
| Micturition urgency | 1 (17%) [1] | |||||
| Total | 1 (17%) [1] | |||||
| Respiratory, thoracic and mediastinal disorders | ||||||
| Epistaxis | 1 (17%) [1] | |||||
| Total | 1 (17%) [1] | |||||
| General disorders and administration site conditions | ||||||
| Fatigue | 1 (17%) [1] | |||||
| Total | 1 (17%) [1] | |||||
| Overall total | 3 (50%) [6] | 2 (100%) [6] | 2 (33%) [2] | 2 (33%) [4] | 3 (50%) [3] | 6 (100%) [8] |
n = number of subjects studied.
There were no dose- or treatment-related trends in terms of clinical laboratory evaluations, including liver function tests, haematological parameters, vital signs or cardiac function. Specifically, no treatment or dose-related trends in parameters recorded on 12-lead safety ECGs were noted and there were no clinically relevant findings in the ECG morphology at any dose level of selisistat. There were no subjects with a QTc interval >480 ms or an increase from baseline >60 ms as assessed from the 12-lead safety ECGs. There were no clinically significant findings in physical examinations, postural control or neurological examinations and no changes in sway platform performance.
Concentration−effect modelling of ECG parameters
The variability of the QTc data measured as the standard deviation of the between-subject ΔQTcF was low, 5.3 ms and 6.8 ms in the single ascending dose (SAD) and multiple ascending dose (MAD) parts, respectively 15. The change from baseline QTcF across dose groups in part 1, in which the highest plasma concentrations were achieved, is shown in Table 7. The pattern across time points and dose groups did not suggest a dose-dependent effect of selisistat on the QTc interval. No significant concentration-dependent effect on ΔΔQTcF was seen after single doses from 5 mg to 600 mg of selisistat within the observed plasma concentration range. A linear model with an intercept provided an acceptable fit of the data and the estimated population intercept and slope were 0.9 ms (90% CI −0.2, 2.0) and −0.00026 ms per ng ml−1 (90% CI −0.00063, 0.00010), respectively (Figure 3A). The analysis of data from the MAD part provided similar results with an intercept of 2.8 ms (90% CI −0.16, 5.71) and an estimated slope of −0.00011 ms per ng ml−1 (90% CI −0.00087, 0.00066; Figure 3B). The ΔΔQTcF effect at the observed geometric Cmax of 26.6 μm after a single dose of 600 mg using this model can be predicted to −0.9 ms (90% CI −3.3, 1.4). A ΔΔQTcF effect of approximately 2.8 ms (90% CI −0.1, 5.6) can be predicted for the observed Cmax level of 22.5 μm after 7 days of dosing of 300 mg once daily. For plasma concentrations exceeding the mean Cmax level, e.g. 30 μm, a QTcF effect of 3.7 ms (90% CI −0.1, 7.5) can be predicted using the same model. The upper bound of the 90% CI of the projected ΔΔQTcF effect was below 10 ms for all plasma concentrations observed in both the SAD and the MAD part of the study (Figure 3A, B).
Table 7.
Mean change from baseline QTcF across dose groups and post-dosing time points in part 1
| Time (h) | Mean | SE | 90% CI | Mean | SE | 90% CI | ||
|---|---|---|---|---|---|---|---|---|
| Lower | Upper | Lower | Upper | |||||
| Placebo | Group A (5 mg) | |||||||
| 0.5 | −2.79 | 1.45 | −5.35 | −0.22 | −2.50 | 0.92 | −4.36 | −0.64 |
| 1 | −4.07 | 1.71 | −7.10 | −1.04 | −2.17 | 1.38 | −4.94 | 0.61 |
| 2 | −2.64 | 1.12 | −4.62 | −0.66 | 0.17 | 1.40 | −2.66 | 2.99 |
| 3 | −2.57 | 1.46 | −5.16 | 0.01 | 1.50 | 1.18 | −0.87 | 3.87 |
| 4 | −3.79 | 1.04 | −5.63 | −1.94 | 1.17 | 2.39 | −3.64 | 5.98 |
| 6 | −2.00 | 1.64 | −4.91 | 0.91 | 2.50 | 3.59 | −4.74 | 9.74 |
| 8 | −2.71 | 1.62 | −5.58 | 0.15 | 0.17 | 2.60 | −5.07 | 5.41 |
| 12 | −0.57 | 2.13 | −4.35 | 3.21 | 0.00 | 3.02 | −6.09 | 6.09 |
| 24 | 0.43 | 1.74 | −2.65 | 3.51 | 0.00 | 2.45 | −4.94 | 4.94 |
| Time (h) | Group B (25 mg) | Group C (75 mg) | ||||||
|---|---|---|---|---|---|---|---|---|
| 0.5 | −4.83 | 2.15 | −9.17 | −0.50 | −4.17 | 1.35 | −6.89 | −1.44 |
| 1 | −2.67 | 2.30 | −7.31 | 1.98 | −3.50 | 1.48 | −6.48 | −0.52 |
| 2 | −2.00 | 1.88 | −5.79 | 1.79 | −3.00 | 2.46 | −7.96 | 1.96 |
| 3 | −2.17 | 2.50 | −7.20 | 2.86 | −3.17 | 1.58 | −6.35 | 0.02 |
| 4 | 1.50 | 2.11 | −2.75 | 5.75 | −1.00 | 2.45 | −5.94 | 3.94 |
| 6 | 6.50 | 3.04 | 0.37 | 12.63 | −4.50 | 2.31 | −9.15 | 0.15 |
| 8 | −1.67 | 2.69 | −7.09 | 3.76 | −3.00 | 1.65 | −6.33 | 0.33 |
| 12 | 2.67 | 2.70 | −2.78 | 8.12 | −0.67 | 2.29 | −5.28 | 3.95 |
| 24 | 3.83 | 6.23 | −8.71 | 16.38 | −0.33 | 1.52 | −3.40 | 2.73 |
| Time (h) | Group D (150 mg fasted) | Group D (150 mg fed) | ||||||
|---|---|---|---|---|---|---|---|---|
| 0.5 | −2.50 | 0.81 | −4.12 | −0.88 | 2.33 | 1.78 | −1.26 | 5.93 |
| 1 | −5.00 | 1.00 | −7.02 | −2.98 | −3.00 | 2.05 | −7.13 | 1.13 |
| 2 | −3.67 | 1.50 | −6.69 | −0.65 | −4.00 | 2.07 | −8.16 | 0.16 |
| 3 | −0.33 | 1.17 | −2.70 | 2.03 | −7.17 | 1.08 | −9.34 | −5.00 |
| 4 | −1.33 | 1.12 | −3.58 | 0.91 | −6.17 | 1.68 | −9.56 | −2.78 |
| 6 | −0.33 | 2.49 | −5.34 | 4.68 | 0.00 | 2.58 | −5.20 | 5.20 |
| 8 | −1.50 | 3.13 | −7.80 | 4.80 | −2.50 | 2.91 | −8.36 | 3.36 |
| 12 | 0.00 | 2.46 | −4.96 | 4.96 | 0.83 | 2.40 | −4.00 | 5.67 |
| 24 | −1.00 | 1.90 | −4.82 | 2.82 | −3.83 | 2.24 | −8.35 | 0.68 |
| Time (h) | Group E (300 mg) | Group F (600 mg) | ||||||
|---|---|---|---|---|---|---|---|---|
| 0.5 | −2.00 | 0.86 | −3.73 | −0.27 | −4.00 | 2.28 | −8.60 | 0.60 |
| 1 | −2.17 | 1.96 | −6.11 | 1.78 | −4.50 | 1.18 | −6.87 | −2.13 |
| 2 | −2.67 | 0.56 | −3.79 | −1.54 | −4.83 | 1.74 | −8.34 | −1.33 |
| 3 | 0.17 | 1.25 | −2.35 | 2.68 | −4.67 | 0.61 | −5.91 | −3.43 |
| 4 | −2.17 | 0.40 | −2.98 | −1.36 | −5.83 | 1.19 | −8.24 | −3.43 |
| 6 | 1.00 | 1.90 | −2.82 | 4.82 | −7.50 | 3.17 | −13.89 | −1.11 |
| 8 | 2.83 | 2.90 | −3.02 | 8.68 | −7.50 | 2.51 | −12.56 | −2.44 |
| 12 | 0.83 | 1.87 | −2.93 | 4.60 | −3.50 | 2.75 | −9.05 | 2.05 |
| 24 | −0.83 | 2.33 | −5.53 | 3.86 | −4.00 | 1.41 | −6.85 | −1.15 |
Figure 3.
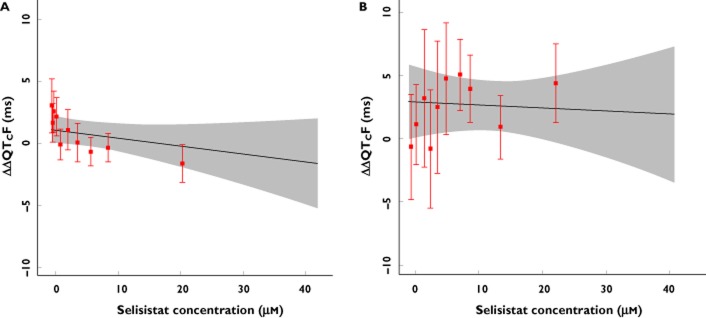
(A, B) Projected and observed QTc effect after single doses of selisistat across plasma concentrations. The model based estimate (solid black line) with 90% CI (grey shaded area) is shown across the range of plasma concentrations observed in the SAD (A) and MAD (B) study. The horizontal red line shows the plasma concentration divided into deciles and the vertical, red bars show the observed ΔΔQTcF with 90% CI within each plasma concentration decile (plotted at the median concentration of each decile). As shown by the upper bound of the 90% CI, a drug-induced effect on the QTc interval exceeding 10 ms could be excluded at all observed plasma concentrations after single or multiple dose administration
Discussion
This was the first time selisistat was administered to humans. Since selisistat represents a first-in-class compound, precautions were taken in the study to assess safety across all critical organ systems; cardiovascular and vital signs monitoring included continuous 24 h Holter monitoring with serial extraction of replicate 12-lead ECGs during both parts of the study. In addition, and given the intended patient population, extensive characterization of any neurological effects of selisistat treatment was included as regards the motor, cerebellar, sensory and vestibular systems. The compound was found to be safe and well tolerated by healthy male and female subjects when administered as a single dose up to 600 mg and repeated doses up to 300 mg day−1 and the overall safety assessment did not present with any concerns regarding future studies with the compound in the intended HD patient population.
The single dose pharmacokinetics of selisistat were found to be essentially linear in the dose interval up to 300 mg, while systemic exposure increased in a more than proportional manner to dose from 300 to 600 mg, suggesting saturation of one or more clearance mechanisms. Since the pre-defined stopping criterion for systemic exposure was reached at the 600 mg dose level, no further dose levels were explored. Inspection of the trough concentrations following repeated doses of selisistat suggested that pharmacokinetic steady-state was reached within 4 days of dosing, although the observed steady-state exposure appeared higher than predicted from the single dose data. A two compartment model with first order input and elimination, lag time and log-transformed squared error fit was found to describe the fasted single dose data (5–600 mg) with satisfactory goodness-of-fit parameters, and confirms the observation that food appears to affect only the rate of absorption (data not shown). Females appeared to show a higher systemic exposure, and while this difference may be due to the uneven number of male and female subjects, any possible gender differences will be the subject of future studies in HD patients. Data from cells transfected with mutant huntingtin, transgenic Drosophila and R6/2 mice suggest a therapeutically relevant concentration range from 100 to 400 nm. In the R6/2 model, the minimally effective dose level resulted in a steady-state average plasma concentration of 370 nm, corresponding to a daily dose of between 25 and 75 mg in humans based on the results of this study.
The microarray and subsequent RT-qPCR data suggest that selisistat treatment is associated with a specific transcriptional signature in circulating blood cells. These genes are involved in mechanisms of signal transduction and transmembrane transport (TMCC3, LDLR, CLIC3, PRKAR1A, USP10), in metabolic processes related to cholesterol, lipid and steroid homeostasis (LDLR, AKR1C3, ACBD3, TXNDC3, USP10) and redox processes (AKR1C3, TXNDC3). While this transcriptional signature may itself be useful as a target engagement marker, it also generates further hypotheses regarding the molecular mechanism of action of selisistat in HD, similarly to the work performed in the transgenic R6/2 HD mouse model 17 and also the possibility to develop target engagement or disease progression markers based on the relative gene products.
Cardiovascular safety pharmacology assessments with selisistat included an in vitro assay to evaluate potential effects on cardiac potassium channels (IKr) and in vivo evaluation for cardiovascular effects in beagle dogs using telemetry. A concentration-dependent inhibition of the IKr channels occurred starting from the concentration of 10 μm, with a calculated IC50 of 43 μm. However, no meaningful changes were seen on blood pressure, body temperature, heart rate and ECG intervals at single oral doses of 25, 50 and 100 mg kg−1, with associated Cmax plasma concentrations (min–max) of 31–67, 43–72 and 53–130 μm, respectively. In addition, no effect on ECGs was seen in repeat dose toxicity testing 18. Interestingly, by implementing a relatively intense ECG schedule and combining a highly precise QT measurement technique with concentration−effect modelling, a QT effect exceeding 10 ms could be excluded. Using the concentration−effect model, the ΔΔQTcF effect of selisistat at high plasma concentrations, reaching up to 30 μg ml−1, can be estimated to be a few milliseconds (Figure 3A, B), with an upper bound of the 90% CI clearly below the level of regulatory concern (10 ms). In our view, and as suggested by others 19–21, the study thereby illustrates the potential for a standard clinical pharmacology study to replace the ICH E14 required thorough QT study 22. It can be argued that the lack of a positive control makes it difficult to claim the absence of an effect, but there is little in these data to suggest that selisistat causes QT prolongation at plasma concentrations thought to be pharmacologically relevant. Based on non-clinical data, it can be assumed that the therapeutic dose range reaches no higher than 100 mg, which is associated with Cmax levels of less than 8 μm.
Biotransformation of selisistat proceeds via hydroxylation and oxidative deamination followed by glucuronic acid conjugation across all species studied (mouse, rat, dog, human). A phenotyping study in vitro showed that CYP3A4 and CYP1A2 were the major isoforms involved in the formation of the hydroxylated metabolites while CYP2D6 and CYP2C19 play a minor role. CYP2C9 was shown not to be involved in the metabolism of the compound. The effect of direct inhibition was measured by incubating selisistat with human liver microsomes at concentrations reflecting expected human plasma concentrations. For CYP 2C8, 2D6, 2E1 and 3A4 (midazolam and testosterone sites), IC50 values were estimated to be higher than 100 μm. For CYP 1A2, 2C9 and 2C19, calculated IC50 values of 8.7, 62.4 and 72.2 μm were determined, respectively 17. Selisistat is therefore unlikely to exhibit clinically significant CYP-mediated drug–drug interactions with compounds metabolized by CYP 2C8, 2C9, 2C19, 2D6, 2E1 and 3A4 at the expected plasma concentrations, whereas interactions with compounds primarily or exclusively metabolized by CYP 1A2 cannot be excluded at pharmacologically relevant plasma concentrations.
In conclusion, in this first-in-human study, selisistat was shown to be safe and well tolerated by healthy male and female subjects when administered as a single dose up to 600 mg and repeated doses up to 300 mg day−1, and was associated with a low rate of adverse events at dose levels thought to exceed the therapeutically relevant levels. By combining serial recording of 12-lead ECGs paired with PK blood sampling and then analyzing the data using concentration−effect modelling, a QTc effect exceeding the level of regulatory concern (10 ms) could be excluded after single and multiple doses of selisistat.
Competing Interests
This study was funded by Siena Biotech SpA, ClinicalTrial.gov Identifier NCT01521832. All authors have completed the Unified Competing Interest form and declare that CA, DD, LM and GP are employees of Siena Biotech SpA with no other relationships or activities that could appear to have influenced the submitted work. GW was an employee of Siena Biotech during the period of planning and conduction of the study and subsequently a paid contractor of Siena Biotech. JC, BD and MZ were paid contractors of Siena Biotech SpA.
The authors would like to thank the staff at Springfield House, Covance Clinical Research Ltd, for the conduct of the study. The authors are grateful to Dr Elisa Mori for the statistical analyses of the RT-qPCR data.
References
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Walker FO. Huntington's disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- Gutekunst C, Norflus F, Hersch S. The neuropathology of Huntington's disease. In: Bates G, Harper P, Jones L, editors. Huntington's Disease. New York: Oxford University Press; 2002. pp. 251–275. [Google Scholar]
- Bonelli RM, Wenning GK, Kapfhammer HP. Huntington's disease: present treatments and future therapeutic modalities. Int Clin Psychopharmacol. 2004;19:51–62. doi: 10.1097/00004850-200403000-00001. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Jeong H, Then F, Melia TJ, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallos J, Bodai L, Lukacsovich T, Purcell JM, Steffan JS, Thompson LM, Marsh JL. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington's disease. Hum Mol Genet. 2008;17:3767–3775. doi: 10.1093/hmg/ddn273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadri-Vakili G, Cha JH. Histone deacetylase inhibitors: a novel therapeutic approach to Huntington's disease. Curr Alzheimer Res. 2007;3:403–408. doi: 10.2174/156720506778249407. [DOI] [PubMed] [Google Scholar]
- Hughes RE. Polyglutamine disease: acetyltransferases awry. Curr Biol. 2002;12:R141–R143. doi: 10.1016/s0960-9822(02)00709-1. [DOI] [PubMed] [Google Scholar]
- Butler R, Bates GP. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat Rev Neurosci. 2006;7:784–796. doi: 10.1038/nrn1989. [DOI] [PubMed] [Google Scholar]
- Bruserud Ø, Stapnes C, Ersvaer E, Gjertsen BT, Ryningen A. Histone deacetylase inhibitors in cancer treatment: a review of the clinical toxicity and the modulation of gene expression in cancer cells. Curr Pharm Biotechnol. 2007;8:388–400. doi: 10.2174/138920107783018417. [DOI] [PubMed] [Google Scholar]
- Napper AD, Hixon J, McDonagh T, Keavey K, Pons JP, Barker J, Yau WT, Amouzegh P, Flegg A, Hamelin E, Thomas RJ, Kates M, Jones S, Navia MA, Saunders JO, DiStefano PS, Curtis R. Discovery of indoles as potent and selective inhibitors of the deacetylase SirT1. J Med Chem. 2005;48:8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- Smith MR, Syed A, Lukacsovich T, Purcell J, Barbaro BA, Worthge SA, Wei SR, Pollio G, Magnoni L, Scali C, Massai L, Franceschini D, Camarri M, Gianfriddo M, Diodato E, Thomas R, Gokce O, Tabrizi S, Caricasole A, Landwehrmeyer B, Menalled L, Ramboz S, Luthi Carter R, Westerberg G, Marsh JL. Sirtuin 1 inhibition alleviates pathology in multiple animal and cell models of Huntington's disease. Hum Mol Genet. 2014;23:2995–3007. doi: 10.1093/hmg/ddu010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamanti D, Lahiri N, Tarditi A, Magnonia M, Fondelli C, Morena E, Malusa F, Pollio G, Diodato E, Tripepi G, Tabrizi SJ, Caricasole A, Mori E. Reference genes selection for transcriptional profiling in blood of HD patients and R6/2 mice. J Huntingtons Dis. 2013;2:185–200. doi: 10.3233/JHD-120042. [DOI] [PubMed] [Google Scholar]
- Darpo B, Fossa AA, Couderc JP, Zhou M, Schreyer A, Ticktin M, Zapesochny A. Improving the precision of QT measurements. Cardiol J. 2011;18:401–410. [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Research. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamanti D, Mori E, Incarnato D, Malusa F, Fondelli C, Magnoni L, Pollio G. Whole gene expression profile in blood reveals multiple pathways deregulation in R6/2 mouse model. Biomark Res. 2013;1:1–14. doi: 10.1186/2050-7771-1-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siena Biotech SpA. Data on file.
- Darpo B, Garnett C. Early QT assessment – how can our confidence in the data be improved? Br J Clin Pharmacol. 2012;76:642–648. doi: 10.1111/bcp.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatagi S, Carrothers TJ, Kuwabara-Wagg J, Khariton T. Is a thorough QTc study necessary? The role of modeling and simulation in evaluating the QTc prolongation potential of drugs. J Clin Pharmacol. 2009;49:1284–1296. doi: 10.1177/0091270009341184. [DOI] [PubMed] [Google Scholar]
- Darpo B, Sarapa N, Garnett C, Benson C, Dota C, Ferber G, Jarugula V, Johannesen L, Keirns J, Krudys K, Ortemann-Renon C, Riley S, Rogers-Subramaniam D, Stockbridge N. The IQ-CSRC prospective clinical Phase 1 study: ‘can early QT assessment using exposure response analysis replace the thorough QT study?’. Ann Noninvasive Electrocardiol. 2014;19:70–81. doi: 10.1111/anec.12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2005. ICH harmonized tripartite guideline E14. The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. Available at http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf (last accessed September 2014)