

Abstract
Aims
The rare association of flupirtine with liver injury is most likely caused by reactive quinone diimines and their oxidative formation may be influenced by the activities of N-acetyltransferases (NAT) that conjugate the less toxic metabolite D13223, and by glucuronosyltransferases (UGT) and glutathione S-transferases (GST) that generate stable terminal glucuronides and mercapturic acid derivatives, respectively. The influence of genetic polymorphisms of NAT2, UGT1A1 and GSTP1 on generation of the terminal mercapturic acid derivatives and analgesic effects was evaluated to identify potential genetic risk factors for hepatotoxicity of flupirtine.
Methods
Metabolic disposition of flupirtine was measured after intravenous administration (100 mg), after swallowing an immediate-release (IR) tablet (100 mg) and after repeated administration of modified release (MR) tablets (400 mg once daily 8 days) in 36 selected healthy subjects. Analgesic effects were measured using pain models (delayed onset of muscle soreness, electric pain).
Results
Flupirtine IR was rapidly but incompletely absorbed (∼72%). Repeated administration of flupirtine MR showed lower bioavailability (∼60%). Approximately 12% of bioavailable flupirtine IR and 8% of bioavailable flupiritine MR was eliminated as mercapturic acid derivatives into the urine independent of the UGT1A1, NAT2 and GSTP1 genotype. Carriers of variant GSTP1 alleles showed lower bioavailability but increased intestinal secretion of flupirtine and increased efficiency in experimental pain. Flupirtine was not a substrate for ABCB1 and ABCC2.
Conclusions
Formation of mercapturic acid derivatives is a major elimination route for flupirtine in man. However, the theoretically toxic pathway is not influenced by the frequent polymorphisms of UGT1A1, NAT2 and GSTP1.
Keywords: ABCB1, ABCC2, drug-induced liver disease, flupirtine, pharmacogenetics, quinone diimines
What is Already Known about this Subject
Flupirtine may induce severe liver injury.
In vitro, hepatotoxic quinone diimines are formed by peroxidases that can be conjugated with glutathione by glutathione-S-transferases (GST).
Alternative metabolic pathways are conjugation by N-acetyltransferases (NAT) and UDP-glucuronosyltransferases (UGT) which may influence the theoretically hepatotoxic pathway in man.
What this Study Adds
Approximately 8–12% of bioavailable flupirtine in man is eliminated in the form of mercapturic acid derivatives by conjugation of glutathione with intermediate quinone diimines.
The hepatotoxic pathway is not influenced by genetic polymorphisms of UGT1A1, NAT2 and GSTP1.
The analgesic effect appears to be linked to the GSTP1 genotype.
Flupirtine is not a substrate in vitro for ABCB1 and ABCC2.
Introduction
Flupirtine was launched 1989 in the European market as an alternative to opioids and non-steroidal anti-inflammatory drugs (NSAIDs). The drug acts centrally on GABA(A) receptors and the selective neuronal Kv7 potassium channel 1–3, thus offering a mechanism-based therapy for pain relief and normalization of muscle tension 4,5. In patients with acute and chronic pain, flupirtine is clinically as efficient as weak opioids and NSAIDs 4–7. Neuroprotective properties are making the drug a possible candidate for the treatment of parkinsonism, dementia and other neurodegenerative diseases 8–11.
Flupirtine does not share the therapeutic limitations of NSAIDs (e.g. gastrointestinal bleeding) or opioids (e.g. constipation). Normally, it is well tolerated and a safe alternative in patients with hypersensitivity to NSAIDs 12. The spectrum of side effects includes a very rare, mild and transient elevation of liver enzymes, aggravation of pre-existing liver disease and, in single cases, severe and fatal liver failure 13–18. There is evidence that the liver injury may be initiated by metabolic formation of hepatotoxic metabolites 19.
Flupirtine is rapidly and almost completely absorbed from the gut, widely distributed and slowly eliminated (half-life ∼6–10 h) 20,21. Its pharmacokinetics are not influenced by dose 22. The metabolism of flupirtine is not yet completely understood. In addition to the formation of para-fluorohippuric acid 19,20, the drug undergoes hydrolytic cleavage of the carbamate group followed by subsequent acetylation via the human N-acetyltransferases (NAT) 1 and the polymorphic NAT2 generating the pharmacologically active metabolite D13223 4,19,20,23. Both the parent flupirtine and D13223 can be conjugated with glucuronic acid by UDP-glucuronosyltransferases (UGT) most likely by the isoforms UGT1A1, 1A3, 1A4 and 1A9, as shown in vitro with recombinant enzymes for retigabine, an anticonvulsant drug closely related in chemical structure to flupirtine 24,25. Recently, we established that flupirtine is an excellent substrate for peroxidases in vitro, which generate highly reactive quinone diimines. The reactive intermediates can be conjugated with glutathione either chemically or catalyzed by glutathione-S-transferases (GST) to form glutathione conjugates. The metabolite D13223 is oxidized to a much lesser extent 19. Quinone diimine intermediates might be expected to cause cellular toxicity as shown for the quinone imine intermediates formed with acetaminophen and diclofenac 26,27. There is some evidence that the polymorphic GSTP1 isoform might be involved in this pathway 28–30. According to our understanding of the balance between the metabolic toxification and detoxification of flupirtine (Figure 1), we reasoned that subjects who were slow acetylators of NAT2 and carriers of variant alleles of UGT1A1 and GSTP1 would be more prone to flupirtine-induced liver injury because the net generation of reactive diimines would be greater than in subjects bearing wild-type enzymes.
Figure 1.

Theoretical metabolism of flupirtine in man (modified from Methling et al. 2009 19). POD, peroxidase; NAT, N-acetyltransferase; UGT, UDP-glycuronosyltransferase; GSH, gluthathione; D13223, major N-actylated metabolite of flupirtine in man; M424 and M466, mercapturic acid derivatives of the quinone diimine
Our study was initiated in order to assess the clinical significance of genetic polymorphisms in affected detoxification pathways, by determining the formation of mercapturic acid derivatives of flupirtine-derived quinine diimines in healthy subjects genotyped for NAT2, UGT1A1 and GSTP1, and in order to quantify the analgesic effects of flupirtine by using two accepted pain models.
Methods
Subjects
The clinical study was performed in 36 German White subjects (25 males, 11 females, age 20–32 years, body weight 51.5–102 kg, height 162–197 cm, body mass index 19.4–26.7 kg m−2). The subjects gave written informed consent prior to inclusion. They were selected from our clientele of genetically well characterized healthy subjects according to their combined NAT2, UGT1A1 and GSTP1 genotype as described below. All subjects were in good health as ascertained by means of history, physical examination, routine clinical chemistry and haematological screening. All subjects were negative in screenings for hepatitis B and C viruses, human immunodeficiency virus, drugs and pregnancy. They all took no drugs before the study except hormonal contraceptives (females). Ten subjects were smokers of less than 10 cigarettes day−1 and 30 subjects occasionally drank alcohol.
The groups of healthy subjects consisted of nine slow acetylators (SA) and nine rapid acetylators (RA) of NAT2, who were wild-type for UGT1A1 (UGT+) and GSTP1 (GST+). Among a further group of 18 SA, six subjects carried two UGT1A1*7 alleles (UGT−) but were GST+, six subjects were UGT+ and carriers of a least two deficient GSTP1-alleles (c.DNA 313A>G, p.105I>V; c.DNA 341C>T, p.114A>V; GST−) and a further six subjects were SA, UGT− and GST−. The sample sizes of the study groups were arbitrarily defined, however, primarily depending on the availability of volunteers with the predefined genotypes among our clientele of about 2000 subjects who had to be genotyped.
The study was approved by the Ethics Committee of the University Medicine Greifswald and by the German Federal Institute for Drugs and Medical Devices (BfArM; no. 4033937). Planning and performance of the study followed the regulations of the German Medicines Act (AMG) and the recommendations of the ICH-GCP guidance (CPMP/ICH/135/95, 1997). The study was registered at EudraCT 2007-007483-17 and ClinicalTrials.gov (NCT01676246).
Study protocol
The study was performed with an initial randomized, controlled, two period, single dose, crossover part followed by a second, multiple dose part. In the first crossover part, 100 mg flupirtine in 50 ml solution were infused within 30 min (Katadolon® inject, AWD.pharma, Dresden, Germany) and a single oral dose of 100 mg in immediate release (IR) dosage form (flupirtine IR, Katadolon® hard capsules, AWD.pharma, Dresden, Germany) was given randomly (block randomization) with at least 7 days for wash-out. In the second part of the study, the subjects were treated with 400 mg flupirtine in modified release (MR) dosage form once daily for 8 days (flupirtine MR, Katadolon® S long, AWD.pharma, Dresden, Germany). This formulation consisted of 100 mg IR flupirtine combined with 300 mg extended release flupirtine. The chronic treatment started at least 7 days after the second administration in the first part of the study.
After overnight fasting, flupirtine solution was infused by using Alaris® GH pumps (CareFusion, Rolle, Switzerland). The flupirtine IR and flupirtine MR was swallowed with 240 ml table water. Compliance of oral dosing was supervised by mouth inspection. Standard meals were provided 5, 8 and 11 h after administration of the study medication. Drinking of table water was strictly standardized for 4 h after administration. Venous blood was sampled via a forearm vein before and 0.25 (i.v. administration), 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 16, 24 h (repeated dosing) and 36 h after administration (single dosing). Urine was sampled for 3 days after single dose administration and for 24 h after last administration of the repeated dose part of the study. Faeces were sampled for 5 days after single dose administration and on study days 5–9 during the repeated dose part. Plasma and aliquots of urine and faeces were stored at −20°C minimum.
Measurement of pain
Delayed onset of muscle soreness (DOMS) and electric pain threshold and tolerance were measured before and 2, 4, 6 and 8 h after intravenous flupirtine and flupirtine IR as described by Tegeder et al. 31. Electric pain threshold was also measured before, daily during repeated dosing and 2, 4, 6, 8, 10, 12 and 16 h after the last administration of flupirtine MR. DOMS was induced by unaccustomed exercise incorporating eccentric muscle contractions in calf muscles. The exercise protocol consisted of two sets of 50 concentric/eccentric contractions of the calf muscles of one leg with a rest of 5 min in-between. The exercise was performed 23–25 h prior to medication. For pain measurements, muscle pain was stimulated by standing on the tiptoes of one leg for 30 s, which requires constriction of the respective calf muscles. The other leg was lifted while the subjects held on to a table to keep their balance. The pain intensity during this stimulation was rated by means of a 10 cm visual analogue scale (VAS). The VAS extended from ‘no pain’ to ‘intolerable pain’ with a precision of 1 mm. The stimulation was repeated with the other leg. The sequence of leg stimulations was randomly defined.
Electric pain was induced by 5 Hz sine waves increasing in intensity by 0.2 mA s−1 from 0 to 20 mA, which predominantly activate C-fibres. The electric current was applied via two gold electrodes placed on the medial and lateral side of the distal joint of the left hand middle finger as the default-testing site (NEUROMETER® CPT, Neurotron Inc., Baltimore, MD, USA). During testing, subjects kept a button continuously pressed until they found the pain intolerable and interrupted the current by releasing the button. The electrical current at which this occurred was defined as pain tolerance. Each pain tolerance value was the median of five subsequent measurements obtained at intervals of 1 min. This pain model has been demonstrated previously to be suitable for quantifying analgesic effects of strong opioids 32–35.
Drug and metabolite assays
Flupirtine and D13223 were quantified by using validated LC-MS/MS methods (API2000 ABSciex combined with an Agilent 1100 LC-system, both Darmstadt, Germany, and the analytical column XTerra®MS C18 3.5 μm, 2.1 × 100 mm, Waters, Ireland) 36. In serum, urine and faeces, flupirtine and D-13223 were assayed with diphenhydramine as internal standard after liquid−liquid extraction with methyl-tert-butylether. The mercapturic acid metabolites M424 and M466 in urine were measured with sulfadimethoxine as internal standard after protein precipitation with acetonitrile (equipment: API3000, ABSciex, combined with a Perkin Elmer series 200 LC-system, Darmstadt, Germany; analytical column: Synergi Fusion-RP, 2.5 μm, 2.0 × 50 mm, Phenomex, Darmstadt, Germany).
The methods were validated for flupirtine and the main metabolites for the ranges of 0.5–500 ng ml−1 in serum, 0.02–5.0 μg ml−1 in urine and 0.005–5.0 μg ml−1 in faeces. The urine assay for M424 and M466 was validated for concentrations of 0.005–5.0 μg ml−1. The analytics were performed in our laboratory certified according to the international recommendations for drug analysis in pharmacokinetic studies 37,38 and according to good laboratory practice (GLP-certificate, Mecklenburg-Vorpommern, Germany, 12.03.2008). Between-day and within-day accuracy (expressed as relative error) for the assay of flupirtine and D13223 in serum ranged from −8.7 to 6.5% and from −3.4 to 5.7%, respectively, of the nominal concentrations. Between-day and within-day precision (expressed as relative SD) was 3.6 to 13.8% for flupirtine and 4.1 to 11.8% for D13223 of the respective mean concentrations. The quality parameters of the assays for urine were the following: accuracy, flupirtine −4.3% to 11.4%, D13223 −5.6 to 5.1%, M424 −3.1% to 3.7%, M466 −4.4% to 3.8%; precision, flupirtine 2.0 to14.1%, D13223 2.9 to 13.5%, M424 0.9 to 10.0%, M466 0.9 to 8.5%. The respective data for faeces were: accuracy, flupirtine −4.2 to 12.5%, D13223 −8.4% to 12.1% and precision, flupirtine 3.8 to 10.0%, D13223 1.8 to 11.0%.
Serum concentrations of 4-β-hydroxycholesterol (4β-OH-C) were assayed using GC-MS for an isotope dilution method with [26.26.26.27.27.27-2H6] 4β-OH-C as an internal standard as described previously 39. The lower limit of quantification was 3.0 ng ml−1 for serum. Between-day and within-day precision was 2.1 and 2.7%, respectively, of the mean values, and between-day and within-day accuracy was between 2.9 and 3.3% of the nominal values.
Permeability of flupirtine and D13223 in MDCK2 cells transfected with ABCB1 and ABCC2
ABCB1 (GenBank accession no. NM_000927.4) was amplified by using reverse transcribed RNA from human placenta (forward primer 5′-GCGATGGATCTTGAAGGGGACC-3′ and reverse primer 5′-TTCACTGGCGCTTTGTTC-3′). The coding sequence of ABCB1 was cloned into the retroviral expression vector pQCXIH (Takara Bio Europe/Clontech, Saint-Germain-en-Laye, France). MDCK2 cells were infected according to the instructions of the manufacturer and were selected using 375 μg ml−1 hygromycin. Control cells were infected with the empty vector (vector control, VC). Resistant cell clones were characterized by Western blot analysis and immunofluorescence staining as described previously 40. The mouse monoclonal antibody anti-ABCB1 (C219) was obtained from Alexis Biochemicals, (Gruenberg, Germany). As secondary antibodies, the horseradish peroxidase-conjugated antibody rabbit-anti-mouse (Acris, Herford, Germany) and the goat-anti-mouse antibody conjugated to Alexa Fluor-488 (Life Technologies, Darmstadt, Germany) were used, respectively. Immunofluorescence microscopy was performed with the Zeiss Axio Observer.D1 (Carl Zeiss MicroImaging, Jena, Germany). The functionality of the ABCB1-transfected cells was verified by an in vitro accumulation assay using calcein-AM 41. Stable transfected MDCK2-ABCC2 cells were generated and characterized as described previously 42.
ABCB1- and ABCC2-mediated transport of flupirtine or D13223 was measured using the Transwell® technique. In brief, 0.4 or 0.2 ml 100 μm flupirtine and D13223, respectively, were added to the basal or apical donor compartment in the presence or absence of the ABCB1 inhibitor PSC-833 (Novartis, Nuremberg, Germany) and the ABCC2-inhibitor MK-571 (Sigma-Aldrich, Taufkirchen, Germany) (100 μm), respectively. Every 30 min, the incubation buffer was removed from the receiver compartment and replaced by fresh preheated buffer for a total time of 3 h. The concentrations of flupirtine and D13223 in the receiver compartment were measured using the LC-MS/MS method described above.
Pharmacokinetic and statistical evaluation
The maximum concentration (Cmax) and the time to Cmax (tmax) were obtained directly from the measured concentration−time curves after single dose administration and at steady-state, respectively. The area under the concentration–time curve (AUC) and the area under the moment curve (AUMC) were calculated from the time of administration until the last quantified concentration by the trapezoidal formula (AUC(0–t)) and extrapolated to infinity (AUC(0–∞)). The terminal elimination rate constant (λz) was derived from the terminal slope by log-linear regression analysis and the apparent elimination half-life (t1/2) was calculated by ln2/λz. Distribution volume at steady-state after intravenous dosing (Vss) was calculated as dose × AUMC/AUC2. Bioavailability (F) was derived from AUC × CL/doseoral. Renal clearance (CLR) was derived from cumulative urinary excretion (Ae urine) of flupirtine over AUC(0–∞) (single dose) and AUC(0–24 h) (steady-state), respectively, after intravenous and oral administration. Intestinal clearance (CLI) was assessed by using the cumulative faecal excretion (Ae faeces) over AUC. Metabolic clearances (CLM) were assessed by the amounts of D13223 and the mercapturic acid derivatives (MERC) M424 and M466, respectively, over AUC(0–∞) after intravenous administration.
Arithmetic means ± SD are given. Statistical differences between samples were evaluated using the Wilcoxon and Mann–Whitney test as appropriate with P < 0.05 as the level of statistical significance. The sample size of n = 18 (Table 1) was sufficient to confirm the significant differences in AUC of flupirtine and Ae urine of the mercapturic acid derivatives with a power of >90%. In Table 2, the AUC difference after multiple dose administration in 24 subjects with GST+ and 12 subjects with GST− was identified with a power of ∼35%. All statistical and pharmacokinetic calculations were performed with the SPSS Statistics version 2.0 (IBM Corporation, Armonk, USA) and SAS statistical package (SAS 8.02, SAS Institute Inc., Carry, USA). The power considerations were done using the nQuery Advisor 7.0 (Statistical Solutions, Cork, Ireland).
Table 1.
Pharmacokinetic characteristics of flupirtine after intravenous infusion, single dosing of flupirtine IR and chronic administration of flupirtine MR in 18 healthy subjects. The steady-state data for flupirtine MR were normalized to a 100 mg dose
| 100 mg intravenously | 100 mg orally, single dose | 400 mg, orally, multiple dose | ||
|---|---|---|---|---|
| AUC/AUC(0–24 h) (FPT) | (μg ml−1 h) | 10.8 ± 4.69 | 6.96 ± 2.16 | 5.77 ± 1.84†‡ |
| AUC/AUC(0–24 h) (D13223) | (μg ml−1 h) | 7.67 ± 2.56 | 5.92 ± 2.67 | 4.01 ± 1.22†‡ |
| Cmax | (μg ml−1) | 1.73 ± 0.782 | 0.857 ± 0.274 | 0.483 ± 0.168†‡ |
| F | (%) | – | 71.9 ± 28.7 | 59.9 ± 25.6 |
| tmax | (h) | – | 1.69 ± 0.893 | 1.92 ± 0.974 |
| t1/2 | (h) | 9.06 ± 1.45 | 8.94 ± 1.24 | 22.2 ± 12.7†‡ |
| Vss | (l kg−1) | 1.61 ± 0.762 | – | – |
| CLR | (ml min−1) | 15.2 ± 8.04 | 14.8 ± 9.25 | 14.1 ± 8.20 |
| CLI | (ml min−1) | 0.382 ± 0.225 | – | – |
| CLM, D13223, urine | (ml min−1) | 15.3 ± 6.82 | – | – |
| CLM, D13223, faeces | (ml min−1) | 0.125 ± 0.087 | – | – |
| CLM, D13223, urine+faeces | (ml min−1) | 15.5 ± 6.86 | – | – |
| CLM,MERC,urine | (ml min−1) | 12.6 ± 5.21 | – | – |
| Aeurine, FLU free | (mg) | 8.48 ± 3.65 | 5.51 ± 2.59 | 4.68 ± 2.75†‡ |
| Aeurine, D13223 free | (mg) | 6.68 ± 2.22 | 4.70 ± 1.39 | 3.98 ± 1.75†‡ |
| Aeurine, M424 | (mg) | 6.51 ± 1.58 | 5.52 ± 1.98 | 2.95 ± 0.904†‡ |
| Aeurine, M466 | (mg) | 3.73 ± 0.819 | 3.25 ± 1.09 | 2.38 ± 0.559†‡ |
| Aeurine, MERC | (mg) | 10.2 ± 2.22 | 8.77 ± 2.99 | 5.32 ± 1.43†‡ |
| Aefaeces, FLU free | (mg) | 0.232 ± 0.164 | n.d. | 6.15 ± 3.24† |
| Aefaeces, D13223 free | (mg) | 0.072 ± 0.056 | n.d. | 0.102 ± 0.052 |
| Aeall/doseurine+faeces | (%) | 25.7 ± 5.48 | n.d. | 20.2 ± 5.97‡ |
†P < 0.05 compared with 100 mg intravenously; ‡P < 0.05 compared with 100 mg orally (Wilcoxon); n.d. = not determined; MR = modified release.
Table 2.
Pharmacokinetic characteristics of flupirtine after intravenous infusion and chronic administration of flupirtine MR in rapid metabolizers (GST+) and poor metabolizers (GST-) of the glutathione-S-transferase P1
| 100 mg intravenously | 400 mg orally, multiple dose | ||||
|---|---|---|---|---|---|
| GST+ (n = 24) | GST- (n = 12) | GST+ (n = 24) | GST- (n = 12) | ||
| AUC/AUC(0–24 h) (FPT) | (μg ml−1 h) | 10.5 ± 4.12 | 10.2 ± 4.19 | 22.5 ± 6.59 | 18.3 ± 7.79* |
| AUC/AUC(0–24 h) (D13223) | (μg ml−1 h) | 7.37 ± 2.42 | 7.61 ± 5.37 | 15.9 ± 4.35 | 15.1 ± 8.87 |
| F | (%) | – | – | 58.3 ± 22.6 | 45.7 ± 11.8 |
| t1/2 | (h) | 9.00 ± 1.29 | 8.61 ± 1.26 | 27.1 ± 25.0 | 21.1 ± 10.5 |
| CLR | (ml min−1) | 13.7 ± 7.89 | 13.2 ± 7.11 | 12.6 ± 7.63 | 14.4 ± 9.06 |
| Aeurine, FLU free | (mg) | 7.72 ± 3.77 | 7.03 ± 2.89 | 16.5 ± 10.3 | 14.0 ± 8.06 |
| Aeurine, D13223 free | (mg) | 6.48 ± 2.22 | 6.94 ± 2.63 | 14.8 ± 6.81 | 12.8 ± 7.49 |
| Aeurine, MERC | (mg) | 10.6 ± 2.84 | 11.3 ± 3.29 | 21.8 ± 6.77 | 22.4 ± 6.74 |
| Aefaeces, FLU free | (mg) | 0.249 ± 0.187 | 0.468 ± 0.374* | 26.7 ± 14.8 | 38.6 ± 18.2* |
| Aefaeces, D13223 free | (mg) | 0.078 ± 0.059 | 0.119 ± 0.068* | 0.381 ± 0.193 | 0.463 ± 0.252 |
| Aeall/Doseurine+faeces | (%) | 25.1 ± 6.32 | 25.9 ± 6.67 | 20.1 ± 5.57 | 22.1 ± 5.62 |
*P < 0.05 compared with GST+ (Mann–Whitney); n.d. = not determined; MR = modified release.
Results
Pharmacokinetics and metabolism
Flupirtine IR was rapidly but incompletely absorbed after oral administration. The drug reached maximum concentrations approximately 2 h after oral administration. Absolute bioavailability was approximately 70%. After administration of flupirtine MR (steady-state conditions), maximum concentrations were also reached after approximately 2 h. However, there was a distinct shoulder in the elimination slope 3–6 h after oral dosing, which was most likely caused by the delayed dissolution of the MR part of the dosage form (Figure 2). The AUC(0–24 h) of 400 mg flupirtine MR was increased compared with the AUC(0–∞) of 100 mg flupirtine IR by a factor of 3.47 ± 1.05 and Cmax by a factor of 2.38 ± 0.77. Therefore, the dose-corrected AUC(0–24 h) and Cmax after repeated dose administration of flupirtine MR were significantly lower than AUC(0–∞) and Cmax after single dose administration of 100 mg flupirtine IR. Absolute bioavailability of flupirtine MR at steady-state tended to be lower than after single dose administration of flupirtine IR (59.9 ± 25.6% vs. 71.9 ± 28.7%, P = 0.058).
Figure 2.
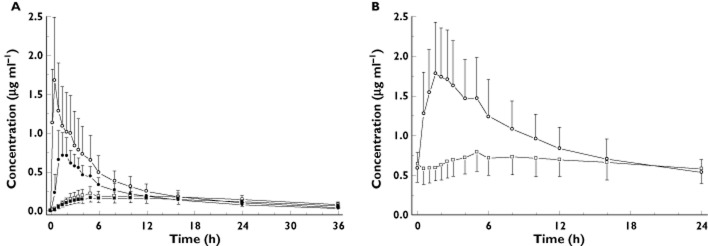
Concentration–time curves of flupirtine and its acetylated metabolite D13223 after intravenous flupirtine infusion and single dosing of flupirtine IR (A) and after chronic administration of flupirtine MR in 24 healthy subjects (B).  , flupirtine i.v.;
, flupirtine i.v.;  , flupirtine IR;
, flupirtine IR;  , D13223 i.v.;
, D13223 i.v.;  , D13223 (IR);
, D13223 (IR);  , flupirtine MR;
, flupirtine MR;  , D13223
, D13223
Flupirtine was widely distributed with a Vss of >1.0 l kg−1. The elimination t1/2s of intravenous flupirtine and flupirtine IR were both approximately 9 h. In contrast, the apparent elimination half-life of the 400 mg flupirtine MR tablet was prolonged by more than two-fold (∼22 h), indicating absorption delayed (flip-flop) drug elimination.
The major metabolite in human serum was the acetylated D13223 which, independent of the mode of administration, reached nearly the same exposure (AUC) as the parent drug. Renal clearance contributed less than 10% to total body clearance and was not influenced by the dosing schedule. Metabolic clearances as determined by formation of the acetylated D13223 and mercapturic acid derivatives were similar in value to renal clearance. A negligible proportion of serum was cleared by intestinal elimination of the parent drug. Up to ∼25% of the intravenous dose and of the bioavailable oral single dose were eliminated via the urine, ∼15% as unchanged compound and D13223 and ∼12% as mercapturic acid derivatives. It should be noted, that <1% of the dose was eliminated via faeces. After repeated oral administration of flupirtine MR, ∼23% and ∼10% of the bioavailable dose were recovered in urine and faeces, respectively. Compared with oral single dose administration of flupirtine IR, the amounts of flupirtine, D13223 and mercapturic acid derivatives were significantly lower after repeated treatment with flupirtine MR. The amount of mercapturic acid derivatives excreted accounted for ∼8% of the bioavailable dose of flupirtine MR at steady-state (Table 1).
The acetylator status of the subjects (RA vs. SA) was without statistical significance for all pharmacokinetic parameters. Furthermore, the *7/*7 promoter polymorphism of UGT1A1 did not influence metabolic disposition of flupirtine (data not shown).
As flupirtine disposition was not influenced by polymorphisms of NAT2 and UGT1A1, in a second evaluation step we analyzed the pharmacokinetic parameters amongst different GST-genotypes (Table 2, Figure 3). Carriers of variant GST alleles had significantly lower AUC of the parent drug after repeated treatment. Bioavailability tended to be lower, however, without reaching the level of statistical significance (45.7 ± 11.8% vs. 58.3 ± 22.6%; Mann-Whitney P = 0.081). Likewise, the half-life, the volume of distribution, renal clearance and excretion of flupirtine and of the metabolites D13223, M424 and M466 into urine were not influenced by the GST polymorphism. The amount of flupirtine eliminated via the faeces was significantly increased (P < 0.05) after both intravenous (0.457 ± 0.365 mg vs. 0.249 ± 0.187 mg) and repeated oral administration (38.6 ± 18.2 mg vs. 26.7 ± 14.8 mg).
Figure 3.

Concentration–time curves of flupirtine (A) and the acetylated metabolite D13223 (B) after intravenous flupirtine infusion, single dosing of flupirtine IR and chronic administration of flupirtine MR in 12 healthy carriers of variant alleles of the glutathione-S-transferase P1 (GST-) and 24 carriers of wild-type alleles (GST+).  , GST+;
, GST+;  , GST−
, GST−
Analgesic effects
Independent of the route of administration, 100 mg flupirtine was effective in carriers of the reference GST alleles (GST+) in reducing joint pain induced by electric current and muscle hyperalgesia induced by a series of concentric and eccentric contractions of the calf muscle (DOMS model; delayed onset of muscle soreness, Figure 4). DOMS was not measured during repeated treatment.
Figure 4.
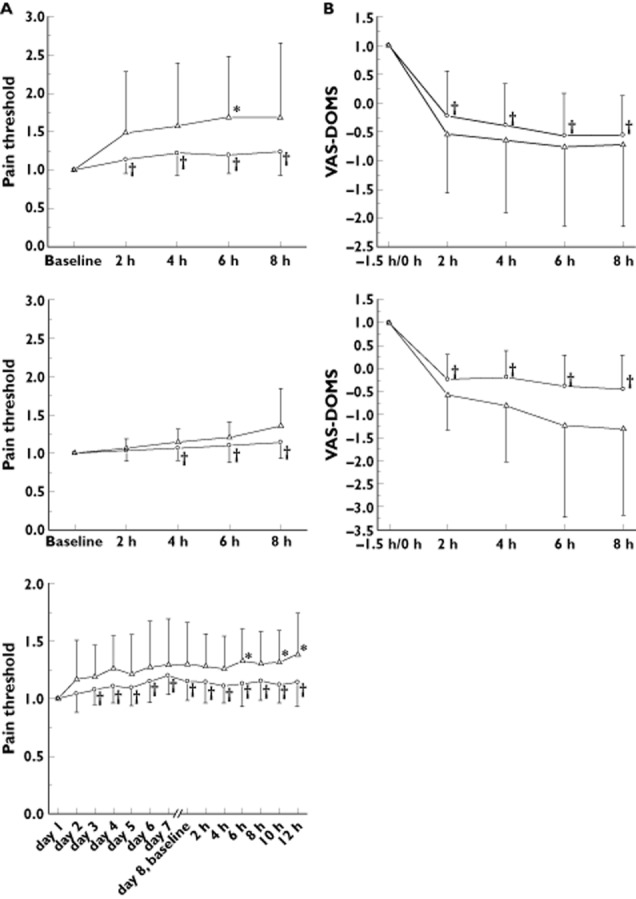
Effects of intravenous flupirtine (100 mg), single dosing of flupirtine IR (100 mg) and chronic administration of flupirtine MR (400 mg) on electric pain threshold (A) and delayed onset of muscle soreness (DOMS, B) in 12 healthy carriers of variant alleles of the glutathione-S-transferase P1 (GST-) and 24 carriers of wild-type alleles (GST+). Higher pain threshold (electric pain) or lower values on a visual analogue scale (VAS-DOMS) refer to stronger analgesic effect of flupirtine
Safety
Generally, flupirtine was safe and well tolerated in healthy subjects. Adverse events probably related to flupirtine after intravenous administration and single dose flupirtine IR were tiredness and somnolence (19 reports), dizziness (three reports), disturbance of accommodation (one report) and dry mouth (one report). After repeated treatment for 8 days, medication related adverse events were not observed and the liver function tests were within normal range amongst all subjects (before vs. after repeated dose treatment: ALAT 0.41 ± 0.19 μkatal l−1 vs. 0.39 ± 0.17 μkatal l−1; ASAT 0.34 ± 0.12 μkatal l−1 vs. 0.31 ± 0.08 μkatal l−1; γ-GT 0.32 ± 0.19 μkatal l−1 vs. 0.30 ± 0.16 μkatal l−1). Repeated dose treatment with flupirtine-ER also resulted in no changes of the plasma concentration ratio of 4-β-hydroxycholesterol over cholesterol (0.34 ± 0.07 vs. 0.32 ± 0.07), an accepted in vivo measure for pregnane-X receptor- (PXR-) type enzyme induction 39,43.
Permeability in ABCB1-MDCK2 and ABC2-MDCK2 cells
Flupirtine and its metabolite D13223 showed almost similar apical and basolateral transport in ABCB1-transfected, ABCC2-transfected and vector-transfected MDCK2 cells. PSC-833 and MK-571 exerted no significant effects on the permeability of flupirtine and D13223 (data not shown). Thus, flupirtine and D13223 are not substrates of the efflux transporter P-glycoprotein (ABCB1) and MRP2 (ABCC2).
Discussion
Approximately 8–12% of bioavailable flupirtine appears in the urine in the form of mercapturic acid derivatives after single and repeated dose administration in healthy subjects. Formation of mercapturic acid derivatives in man was predicted from the results of our recent in vitro experimental studies showing that the drug is readily oxidized by peroxidases 19. The oxidation products are likely to be quinine diimines, which can dimerise and trimerise or be trapped by bionucleophiles, such as glutathione, in addition reactions. Further metabolism of the GSH conjugates would be expected to yield the mercapturic acid derivatives M242 and M466 (Figure 1). Another major pathway to generate diimines and, in turn, stable addition end products is hydrolytic cleavage of the carbamate group of flupirtine by lesterases. The putative descarboethoxy product is efficiently acetylated to D13223 by human NAT1 and NAT2 19. Serum exposure and amount of D13223 excreted via urine are similar in extent to the parent flupirtine. The acetylated D13223, however, is less reactive towards peroxidases than flupirtine and the quinone diimines are relatively stable and can be detected in vitro as compared with the extreme reactivity of flupirtine diimines, which polymerize and/or conjugate more rapidly.
Metabolic activation of intermediate quinone imines that bind to native protein (haptenization) has been comprehensively described for paracetamol (acetaminophen). These mechanisms may also be the rationale for understanding flupirtine-induced hepatotoxicity 27. Since 2007, flupirtine-induced liver injury has been in the focus of an ongoing debate in Germany in response to an increasing prescription rate and the number of hepatobiliary adverse events 44. The incidence of flupirtine-related hepatobiliary adverse events was estimated to be about 8 in 100 000 patients according to a recent re-evaluation of 226 unselected, spontaneously reported cases in the BfArM database 13.
According to current theories to explain the pathogenesis of drug induced liver injury (DILI), a parent drug or more frequently its reactive metabolite(s) cause direct cell stress, directly impair mitochondrial function and/or indirectly trigger specific immune reactions. The initial injury may cause mitochondrial dysfunction followed by apoptosis or necrosis of liver cells. Genetic factors and environmental influences may influence the initial trigger reactions (e.g. drug metabolizing enzymes and transporters, HLA system) and downstream processes (e.g. cytokines regulating inflammation and immune response) and in turn the individual risk for DILI 45–47. A common feature of flupirtine-induced liver injury is the dose-independent behaviour, a latency period of several weeks to months and absence of correlations between severity and cumulative dose 16,18,48. An in depth clinico-pathological evaluation of flupirtine hepatotoxicity reported extensive perivenular (zone 3) necrosis that was associated with ceroid pigment-laden macrophages and a mild to moderate lymphocytic infiltrate. Zone 3 hepatocytes appear to be particularly vulnerable to necrosis, possibly due to a reduced ability to replenish depleted glutathione stores in the detoxification of reactive metabolites as reported for paracetamol. Furthermore, while centrolobular necrosis is commonly seen with several intrinsic hepatotoxins, the presence of ceroid-laden macrophages is a feature in liver injury related to halogenated hydrocarbons or dihydralazine 49,50. Immune-mediated processes might also be involved in flupirtine-induced liver injury as a plasma cell rich hepatitis occurred in one patient after re-exposure 18. The putative toxic-allergic mechanism is also in line with observations on rapid recurrence or worsening of DILI symptoms after unintended re-challenge with flupirtine 13.
Whatever downstream mechanisms may contribute to flupirtine-induced liver injury, the initial key point mechanism may likely be metabolic activation of flupirtine by peroxidases to quinone diimines. Therefore, flupirtine could damage human liver cells by pathogenetic processes similar to other hepatotoxic drugs such as paracetamol or diclofenac. However, unlike paracetamol and diclofenac, flupirtine is not a good substrate for human CYP450 29. The common mechanism of metabolic activation to putative toxic benzoquinone imines and diimines from the parent compounds (paracetamol and flupirtine, respectively) and/or intermediate metabolites (5-hydroxydiclofenac) could lead to hepatotoxicity in susceptible subjects 51–53. We do not think that the metabolite of flupirtine, D13223, is also responsible because it is only a poor substrate for peroxidases and we did not see any evidence for mercapturic acid metabolites of D13223.
As metabolic activation can be influenced by genetic variability of the enzymes involved, we hypothesized that the risk of flupirtine-induced liver injury may be influenced by genetic polymorphisms of NAT2, UGTs and GSTs controlling the generation of reactive diimine intermediates in the liver. In our study, however, the appearance of terminal mercapturic acid derivatives of flupirtine was only related to the bioavailable flupirtine dose and was not influenced by treatment duration (single dose, repeated dose) or by genetic polymorphisms. It must be pointed out that the net formation of toxic quinone diimines and, in turn, the risk for flupirtine-induced liver injury might also be influenced by genetic variability of human myeloperoxidase, NAT1 and esterases or by induction of hepatic peroxidases, inhibition and/or deficiency of GST (e.g. pre-existing liver diseases) or depletion of hepatic glutathione (e.g. alcohol, malnourishment, glutathione depleting co-medication) 47. Some of these factors are already considered as contraindications for the prescribing of flupirtine. In this context, it should be re-evaluated whether flupirtine can be prescribed in combinations with paracetamol and diclofenac since the quinone diimines and quinone imines will consume GSH by the same detoxification pathway. Furthermore, it should be evaluated in the future whether subjects with genetic polymorphisms of downstream mechanisms for DILI (e.g. HLA-system, pro-inflammatory cytokines) are more prone to flupirtine hepatotoxicity, e.g. by genome-wide association studies.
Limitations and problems of the study
Regarding our recent in vitro study 19, the hydrolysis product of flupirtine is conjugated by both human N-acetyltransferases (NAT1 and NAT2) with nearly equal efficacy. It is unknown to what extent the polymorphic NAT2 contributes to net acetylation of the parent drug and whether N-acetylation via NAT2 is the rate-limiting step. Possibly, the NAT1 compensates for deficient N-acetylation in subjects being SA of NAT2. It must be also considered, that about 20% of the flupirtine dose is hydrolyzed and excreted as para-fluorohippuric acid, a condensation product with glycine 19,20. To our knowledge, however, it is unknown how its formation via 5-fluorobenzoic acid is catalyzed and regulated.
As a further limitation, the rationale for the pharmacogenetic approach was derived from previously available data on the disposition of the anticonvulsant retigabine, which is a chemical analogue of flupirtine metabolized by recombinant UGT1A1, 1A3, 1A4 and 1A9 24,25. In the meantime, we have shown that flupirtine is a competitive inhibitor of UGT2B7 and 2B15/17 but their polymorphisms were not considered in our pharmacogenetic study 54. We have focused our study on UGT1A1 as it is the only glucuronosyltransferase with functional relevant polymorphisms that occur with clinically relevant frequency.
A third limitation is that there is a lack of data on the mechanisms of glutathione conjugation to the flupirtine-derived quinone diimines in man. We have shown that the addition of GSH to the quinone diimine can take place in the absence of GST in vitro 19 but do not yet know if the reaction can be catalyzed by GST. We included subjects with deficient GSTP1 alleles in our study because of some evidence in the literature that GSTP1 is involved in the detoxification of the paracetamol-derived N-acetyl-p-benzoquinoneimine (NAPQI) and in the pathogenesis of paracetamol-induced liver injury and bladder cancer 28–30. With our pharmacogenetic approach, we confirmed that detoxification of the haptenizing quinone diimines was not significantly influenced by genetic variability of the GSTP1. However, other GST isoforms might be included in the detoxification of the diimines or the mercapturic acid derivatives were formed spontaneously independent of the GST pathway. Interestingly, GSTP1 must be involved in some way in flupirtine disposition because less of the drug was absorbed in subjects with at least two variant GSTP1 alleles. In such subjects, a significantly increased amount of flupirtine was eliminated via faeces both after intravenous infusion and after repeated oral administration. This means that intestinal secretion of flupirtine must be higher in carriers of two variant GSTP1 alleles. We do not yet have an explanation for this observation, but, it should be noted that glutathione is involved in the membrane (co)transport of many drugs, e.g. by ABCC2 55.
Flupirtine and D13223 are not substrates of the efflux transporters ABCB1 and ABCC2 which can be regulated by the nuclear pregnane-X receptor (PXR) as CYP enzymes or UGTs. In this context, it must be noted that repeated treatment with flupirtine MR resulted in lower bioavailability and lower urinary excretion of the parent drug, D13223 and mercapturic acid derivatives but increased renal excretion of the glucuronide. However, repeated treatment with flupirtine in our healthy subjects did not change the plasma concentration ratio of 4β-OHC over cholesterol, which is an accepted measure for PXR-type enzyme induction 39,43, i.e. PXR-type enzyme induction of any of the variables involved in flupirtine disposition cannot explain the changes in absorption after repeated dose administration.
Finally, we confirm that flupirtine exerts analgesic effects in healthy subjects. Interestingly, the analgesic effect of flupirtine appears to be linked to the GSTP1 genotype. In both experimental models (electric pain and DOMS 31), flupirtine was apparently more efficient in carriers of variant GSTP1 alleles.
Conclusions
In conclusion, approximately 8–12% of bioavailable flupirtine in healthy subjects is eliminated in the form of mercapturic acid derivatives by conjugation of GSH with intermediate quinone diimines. The latter are considered to be reactive metabolites with the potential to cause liver injury. However, the theoretically toxic pathway is not influenced by frequent polymorphisms of UGT1A1, NAT2 and GSTP1. The complex background of flupirtine induced liver injury should be evaluated by future pharmacogenomics analyses.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf and declare CM, ES, KM, MK, AN, DR PJB and JB had no support from any organization for the submitted work.
WS received grants from AWD.pharma GmbH & Co. KG, Radebeul, which was integrated into Teva Germany, Ulm, Germany and from the German Federal Ministry of Education and Research (grant 01 GG 9845/5, 01 ZZ 0103) for the submitted work. There are no other relationships or activities that could appear to have influenced the submitted work.
BT was an employee of AWD.pharma GmbH & Co. KG, Radebeul, which was integrated into Teva Germany, Ulm, Germany in the previous 3 years and has no other relationships or activities that could appear to have influenced the submitted work.
The authors are indebted to Andrea Werzner, Danilo Wegner, Gitta Schumacher and Sabine Bade for their excellent technical assistance.
This work was supported by an institutional grant from AWD.pharma GmbH & Co. KG, Radebeul, which was integrated into Teva Germany, Ulm, Germany and the German Federal Ministry of Education and Research (grant 01 GG 9845/5, 01 ZZ 0103).
Contributors
The authors have contributed to the manuscript in the following manner:
Werner Siegmund initiated the clinical study, served as principal investigator and wrote the draft and final version of the paper.
Christiane Modess and Ali Nassif conducted the clinical study in healthy subjects and edited the manuscript for all clinical study issues.
Eberhard Scheuch and Karen Methling developed and validated the quantitative analytical method and performed the assays of flupirtine and its metabolites and edited the analytical part of the manuscript.
Markus Keiser performed the in vitro experiments using ABCB1- and ABCC2-transfected cells and edited the respective sections in the manuscript.
Dieter Rosskopf was included in recruiting by genotyping of our clientele of healthy subjects including development of the respective screening methods and editing the respective parts of the manuscript.
Patrick J. Bednarski, Jürgen Borlak and Bernd Terhaag participated by evaluation and discussion of the study data and by editing all parts of the manuscript.
Adrea Werzner assisted in the clinical study by nursing.
Gitta Schumacher and Sabine Bade assisted in the drug analysis as laboratory technicians.
Danilo Wegner assisted by data management, pharmacokinetic and statistical evaluation.
References
- Klinger F, Geier P, Dorostkar MM, Chandaka GK, Yousuf A, Salzer I, Kubista H, Boehm S. Concomitant facilitation of GABA(A) receptors and K(V) 7 channels by the non-opioid analgesic flupirtine. Br J Pharmacol. 2012;166:1631–1642. doi: 10.1111/j.1476-5381.2011.01821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhuber J, Bleich S, Wiltfang J, Maler M, Parsons CG. Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltage independent potassium channels. Rapid communication. J Neural Transm. 1999;106:857–867. doi: 10.1007/s007020050206. [DOI] [PubMed] [Google Scholar]
- Raffa RB, Pergolizzi JV., Jr The evolving understanding of the analgesic mechanism of action of flupirtine. J Clin Pharm Ther. 2012;37:4–6. doi: 10.1111/j.1365-2710.2010.01233.x. [DOI] [PubMed] [Google Scholar]
- Friedel HA, Fitton A. Flupirtine. A review of its pharmacological properties and therapeutic efficacy in pain states. Drugs. 1993;45:548–569. doi: 10.2165/00003495-199345040-00007. [DOI] [PubMed] [Google Scholar]
- Harish S, Bhuvana K, Bengalorkar GM, Kumar T. Flupirtine: clinical pharmacology. J Anaesthesiol Clin Pharmacol. 2012;28:172–177. doi: 10.4103/0970-9185.94833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devulder J. Flupirtine in pain management: pharmacological properties and clinical use. CNS Drugs. 2010;24:867–881. doi: 10.2165/11536230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Klawe C, Maschke M. Flupirtine: pharmacology and clinical applications of a nonopioid analgesic and potentially neuroprotective compound. Expert Opin Pharmacother. 2009;10:1495–1500. doi: 10.1517/14656560902988528. [DOI] [PubMed] [Google Scholar]
- Uberall MA, Mueller-Schwefe GH, Terhaag B. Efficacy and safety of flupirtine modified release for the management of moderate to severe chronic low back pain: results of SUPREME, a prospective randomized, double-blind, placebo- and active-controlled parallel-group phase IV study. Curr Med Res Opin. 2012;28:1617–1634. doi: 10.1185/03007995.2012.726216. [DOI] [PubMed] [Google Scholar]
- Otto M, Cepek L, Ratzka P, Doehlinger S, Boekhoff I, Wiltfang J, Irle E, Pergande G, Ellers-Lenz B, Windl O, Kretzschmar HA, Poser S, Prange H. Efficacy of flupirtine on cognitive function in patients with CJD: a double-blind study. Neurology. 2004;62:714–718. doi: 10.1212/01.wnl.0000113764.35026.ef. [DOI] [PubMed] [Google Scholar]
- Sattler MB, Williams SK, Neusch C, Otto M, Pehlke JR, Bahr M, Diem R. Flupirtine as neuroprotective add-on therapy in autoimmune optic neuritis. Am J Pathol. 2008;173:1496–1507. doi: 10.2353/ajpath.2008.080491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder HC, Muller WE. Neuroprotective effect of flupirtine in prion disease. Drugs Today (Barc) 2002;38:49–58. doi: 10.1358/dot.2002.38.1.660505. [DOI] [PubMed] [Google Scholar]
- Treudler R, Pohle K, Simon JC. Flupirtine is a safe alternative drug in patients with hypersensitivity to NSAIDs. Eur J Clin Pharmacol. 2011;67:961–963. doi: 10.1007/s00228-011-1022-7. [DOI] [PubMed] [Google Scholar]
- Anderson N, Borlak J. Correlation versus causation? Pharmacovigilance of the analgesic flupirtine exemplifies the need for refined spontaneous ADR reporting. PLoS ONE. 2011;6:e25221. doi: 10.1371/journal.pone.0025221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein F, Glanemann M, Rudolph B, Seehofer D, Neuhaus P. Flupirtine-induced hepatic failure requiring orthotopic liver transplant. Exp Clin Transplant. 2011;9:270–272. [PubMed] [Google Scholar]
- Li C, Ni J, Wang Z, Li M, Gasparic M, Terhaag B, Uberall MA. Analgesic efficacy and tolerability of flupirtine vs. tramadol in patients with subacute low back pain: a double-blind multicentre trial*. Curr Med Res Opin. 2008;24:3523–3530. doi: 10.1185/03007990802579769. [DOI] [PubMed] [Google Scholar]
- Michel MC, Radziszewski P, Falconer C, Marschall-Kehrel D, Blot K. Unexpected frequent hepatotoxicity of a prescription drug, flupirtine, marketed for about 30 years. Br J Clin Pharmacol. 2012;73:821–825. doi: 10.1111/j.1365-2125.2011.04138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell-Jackson P, Williams R. Use of flupirtine maleate as an analgesic in patients with liver disease. Br J Clin Pract. 1985;39:63–66. [PubMed] [Google Scholar]
- Puls F, Agne C, Klein F, Koch M, Rifai K, Manns MP, Borlak J, Kreipe HH. Pathology of flupirtine-induced liver injury: a histological and clinical study of six cases. Virchows Arch. 2011;458:709–716. doi: 10.1007/s00428-011-1087-9. [DOI] [PubMed] [Google Scholar]
- Methling K, Reszka P, Lalk M, Vrana O, Scheuch E, Siegmund W, Terhaag B, Bednarski PJ. Investigation of the in vitro metabolism of the analgesic flupirtine. Drug Metab Dispos. 2009;37:479–493. doi: 10.1124/dmd.108.024364. [DOI] [PubMed] [Google Scholar]
- Hlavica P, Niebch G. [Pharmacokinetics and biotransformation of the analgesic flupirtine in humans] Arzneimittelforschung. 1985;35:67–74. [PubMed] [Google Scholar]
- Schneider E, Niebch G, Hermann R, Nowak H, Ruus P, Borbe HO. Absolute bioavailability of flupirtine capsules, drops and suppositories in man. Eur J Clin Pharmacol. 1995;49:A161. [Google Scholar]
- Niebch G, Borbe HO, Hummel T, Kobal G. Dose-proportional plasma levels of the analgesic flupirtine maleate in man. Application of a new HPLC assay. Arzneimittelforschung. 1992;42:1343–1345. [PubMed] [Google Scholar]
- Obermeier K, Niebch G, Thiemer K. [Pharmacokinetics and biotransformation of the analgesic flupirtine in the rat and dog] Arzneimittelforschung. 1985;35:60–67. [PubMed] [Google Scholar]
- Borlak J, Gasparic A, Locher M, Schupke H, Hermann R. N-Glucuronidation of the antiepileptic drug retigabine: results from studies with human volunteers, heterologously expressed human UGTs, human liver, kidney, and liver microsomal membranes of Crigler-Najjar type II. Metabolism. 2006;55:711–721. doi: 10.1016/j.metabol.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Hempel R, Schupke H, McNeilly PJ, Heinecke K, Kronbach C, Grunwald C, Zimmermann G, Griesinger C, Engel J, Kronbach T. Metabolism of retigabine (D-23129), a novel anticonvulsant. Drug Metab Dispos. 1999;27:613–622. [PubMed] [Google Scholar]
- Boelsterli UA. Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicol Appl Pharmacol. 2003;192:307–322. doi: 10.1016/s0041-008x(03)00368-5. [DOI] [PubMed] [Google Scholar]
- Park BK, Kitteringham NR, Maggs JL, Pirmohamed M, Williams DP. The role of metabolic activation in drug-induced hepatotoxicity. Annu Rev Pharmacol Toxicol. 2005;45:177–202. doi: 10.1146/annurev.pharmtox.45.120403.100058. [DOI] [PubMed] [Google Scholar]
- Coles B, Wilson I, Wardman P, Hinson JA, Nelson SD, Ketterer B. The spontaneous and enzymatic reaction of N-acetyl-p-benzoquinonimine with glutathione: a stopped-flow kinetic study. Arch Biochem Biophys. 1988;264:253–260. doi: 10.1016/0003-9861(88)90592-9. [DOI] [PubMed] [Google Scholar]
- Fortuny J, Kogevinas M, Garcia-Closas M, Real FX, Tardon A, Garcia-Closas R, Serra C, Carrato A, Lloreta J, Rothman N, Villanueva C, Dosemeci M, Malats N, Silverman D. Use of analgesics and nonsteroidal anti-inflammatory drugs, genetic predisposition, and bladder cancer risk in Spain. Cancer Epidemiol Biomarkers Prev. 2006;15:1696–1702. doi: 10.1158/1055-9965.EPI-06-0038. [DOI] [PubMed] [Google Scholar]
- Henderson CJ, Wolf CR, Kitteringham N, Powell H, Otto D, Park BK. Increased resistance to acetaminophen hepatotoxicity in mice lacking glutathione S-transferase Pi. Proc Natl Acad Sci USA. 2000;97:12741–12745. doi: 10.1073/pnas.220176997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegeder I, Meier S, Burian M, Schmidt H, Geisslinger G, Lotsch J. Peripheral opioid analgesia in experimental human pain models. Brain. 2003;126:1092–1102. doi: 10.1093/brain/awg115. [DOI] [PubMed] [Google Scholar]
- Angst MS, Drover DR, Lotsch J, Ramaswamy B, Naidu S, Wada DR, Stanski DR. Pharmacodynamics of orally administered sustained- release hydromorphone in humans. Anesthesiology. 2001;94:63–73. doi: 10.1097/00000542-200101000-00014. [DOI] [PubMed] [Google Scholar]
- Lotsch J, Angst MS. The mu-opioid agonist remifentanil attenuates hyperalgesia evoked by blunt and punctuated stimuli with different potency: a pharmacological evaluation of the freeze lesion in humans. Pain. 2003;102:151–161. doi: 10.1016/s0304-3959(02)00349-4. [DOI] [PubMed] [Google Scholar]
- Oertel BG, Schmidt R, Schneider A, Geisslinger G, Lotsch J. The mu-opioid receptor gene polymorphism 118A>G depletes alfentanil-induced analgesia and protects against respiratory depression in homozygous carriers. Pharmacogenet Genomics. 2006;16:625–636. doi: 10.1097/01.fpc.0000220566.90466.a2. [DOI] [PubMed] [Google Scholar]
- Skarke C, Darimont J, Schmidt H, Geisslinger G, Lotsch J. Analgesic effects of morphine and morphine-6-glucuronide in a transcutaneous electrical pain model in healthy volunteers. Clin Pharmacol Ther. 2003;73:107–121. doi: 10.1067/mcp.2003.5. [DOI] [PubMed] [Google Scholar]
- Scheuch E, Methling K, Bednarski PJ, Siegmund W. Quantitative LC-MS/MS determination of flupirtine, its N-acetylated and two mercapturic acid derivatives in man. J Pharm Biomed Anal. 2014 doi: 10.1016/j.jpba.2014.09.010. (in press) [DOI] [PubMed] [Google Scholar]
- Shah VP, Midha KK, Dighe S, McGilveray IJ, Skelly JP, Yacobi A, Layloff T, Viswanathan CT, Cook CE, McDowall RD. Analytical methods validation: bioavailability, bioequivalence and pharmacokinetic studies. Conference report. Eur J Drug Metab Pharmacokinet. 1991;16:249–255. doi: 10.1007/BF03189968. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration (FDA) Bioanalytical method validation. 2001. Guidance for Industry.
- Tomalik-Scharte D, Lutjohann D, Doroshyenko O, Frank D, Jetter A, Fuhr U. Plasma 4beta-hydroxycholesterol: an endogenous CYP3A metric? Clin Pharmacol Ther. 2009;86:147–153. doi: 10.1038/clpt.2009.72. [DOI] [PubMed] [Google Scholar]
- Leonhardt M, Keiser M, Oswald S, Kuhn J, Jia J, Grube M, Kroemer HK, Siegmund W, Weitschies W. Hepatic uptake of the magnetic resonance imaging contrast agent Gd-EOB-DTPA: role of human organic anion transporters. Drug Metab Dispos. 2010;38:1024–1028. doi: 10.1124/dmd.110.032862. [DOI] [PubMed] [Google Scholar]
- Hanke U, May K, Rozehnal V, Nagel S, Siegmund W, Weitschies W. Commonly used nonionic surfactants interact differently with the human efflux transporters ABCB1 (p-glycoprotein) and ABCC2 (MRP2) Eur J Pharm Biopharm. 2010;76:260–268. doi: 10.1016/j.ejpb.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Jia J, Puls D, Oswald S, Jedlitschky G, Kuhn JP, Weitschies W, Hosten N, Siegmund W, Keiser M. Characterization of the intestinal and hepatic uptake/efflux transport of the magnetic resonance imaging contrast agent gadolinium-ethoxylbenzyl-diethylenetriamine-pentaacetic acid. Invest Radiol. 2013;49:78–86. doi: 10.1097/RLI.0b013e3182a70043. [DOI] [PubMed] [Google Scholar]
- Tirona RG. Molecular mechanisms of drug transporter regulation. Handb Exp Pharmacol. 2011;201:373–402. doi: 10.1007/978-3-642-14541-4_10. [DOI] [PubMed] [Google Scholar]
- Schwabe U, Paffrath D. Arzneiverordnungs-Report 2011. Heidelberg, Germany: Springer Medizin Verlag; 2012. [Google Scholar]
- Alfirevic A, Pirmohamed M. Predictive genetic testing for drug-induced liver injury: considerations of clinical utility. Clin Pharmacol Ther. 2012;92:376–380. doi: 10.1038/clpt.2012.107. [DOI] [PubMed] [Google Scholar]
- Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov. 2005;4:489–499. doi: 10.1038/nrd1750. [DOI] [PubMed] [Google Scholar]
- Russmann S, Jetter A, Kullak-Ublick GA. Pharmacogenetics of drug-induced liver injury. Hepatology. 2010;52:748–761. doi: 10.1002/hep.23720. [DOI] [PubMed] [Google Scholar]
- Pirmohamed M, Breckenridge AM, Kitteringham NR, Park BK. Adverse drug reactions. BMJ. 1998;316:1295–1298. doi: 10.1136/bmj.316.7140.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin SB, Goodman ZD, Ishak KG, Zimmerman HJ, Irey NS. The morphologic spectrum of halothane-induced hepatic injury: analysis of 77 cases. Hepatology. 1985;5:1163–1171. doi: 10.1002/hep.1840050617. [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Kakar S. Histological patterns in drug-induced liver disease. J Clin Pathol. 2009;62:481–492. doi: 10.1136/jcp.2008.058248. [DOI] [PubMed] [Google Scholar]
- Aithal GP, Ramsay L, Daly AK, Sonchit N, Leathart JB, Alexander G, Kenna JG, Caldwell J, Day CP. Hepatic adducts, circulating antibodies, and cytokine polymorphisms in patients with diclofenac hepatotoxicity. Hepatology. 2004;39:1430–1440. doi: 10.1002/hep.20205. [DOI] [PubMed] [Google Scholar]
- Tang W. The metabolism of diclofenac–enzymology and toxicology perspectives. Curr Drug Metab. 2003;4:319–329. doi: 10.2174/1389200033489398. [DOI] [PubMed] [Google Scholar]
- Thummel KE, Lee CA, Kunze KL, Nelson SD, Slattery JT. Oxidation of acetaminophen to N-acetyl-p-aminobenzoquinone imine by human CYP3A4. Biochem Pharmacol. 1993;45:1563–1569. doi: 10.1016/0006-2952(93)90295-8. [DOI] [PubMed] [Google Scholar]
- Miners JO, McKinnon RA, Mackenzie PI. Genetic polymorphisms of UDP-glucuronosyltransferases and their functional significance. Toxicology. 2002;181-182:453–456. doi: 10.1016/s0300-483x(02)00449-3. [DOI] [PubMed] [Google Scholar]
- Nies AT, Keppler D. The apical conjugate efflux pump ABCC2 (MRP2) Pflugers Arch. 2007;453:643–659. doi: 10.1007/s00424-006-0109-y. [DOI] [PubMed] [Google Scholar]