

Article first published online 7 January 2015.
Supplemental Digital Content is Available in the Text.
Key Words: gene expression, genetic association, eQTL
Abstract
Background:
Many genetic risk loci have been identified for inflammatory bowel disease and colorectal cancer; however, identifying the causal genes for each association signal remains a challenge. Expression quantitative trait loci (eQTL) studies have identified common variants that induce differential gene expression and eQTLs can be cross-referenced with disease association signals for gene prioritization. However, the genetics of gene expression are highly tissue-specific, and further eQTL datasets from primary tissues are needed.
Methods:
We have conducted an eQTL discovery study using tissue extracted endoscopically from the terminal ileum and 4 colonic locations of non-inflamed bowel from 65 controls and patients with quiescent inflammatory bowel disease. A genome-wide cis-eQTL analysis was performed on >3,600,000 variants and 13,558 expressed probes.
Results:
We identified 1312 independent eQTLs associated with the differential expression of 1222 genes in rectal mucosa. One hundred seventy-one, 211, 168, and 102 independent eQTLs were identified in the sigmoid, descending colon, ascending colon, and terminal ileum, respectively. Twenty-six percent of genes with rectal eQTLs were novel and unique compared with 7 published eQTL datasets. Rectal eQTLs were significantly enriched for genes expressed in the colon. Examining 163 inflammatory bowel disease risk loci identified 11 tag single-nucleotide polymorphisms that were rectal eQTLs. A colorectal cancer locus at 11q23 contained a rectal eQTL for COLCA2, a protein implicated in colon cancer pathogenesis.
Conclusions:
This study defines a catalog of ileal and colonic eQTLs. Our data reaffirm the tissue specificity of eQTLs and support the notion that identification of functional variants in relevant tissue can be effective in fine-mapping genetic risk loci.
Genome-wide association studies have identified thousands of common genetic variants associated with many complex diseases and traits.1 Association studies in Crohn's disease (CD) and ulcerative colitis (UC), the most common forms of inflammatory bowel disease (IBD), have been particularly successful examples of the approach. Through standard commercial chipsets and, more recently, custom genotyping arrays, the International IBD Genetics Consortium has identified 163 distinct genomic loci associated with IBD.2 This body of findings has implicated novel genes and pathways in the pathogenesis of IBD, including intestinal barrier function, microbial defense, immune regulation and autophagy,3 and has provided genetic evidence supporting the role of defective innate immunity in CD.4
By design, the signals discovered in association studies are driven by common variants that are in high linkage disequilibrium (LD) with other common, and sometimes rare, polymorphisms within the same locus. Variants in high LD can span large genomic distances that are often multigenic, making it difficult to identify the causal gene(s) or mechanism(s) driving the association signal.
It is known, from previous genetics of gene expression studies, that some common disease-associated variants act by modifying noncoding regulatory elements inducing the differential expression of nearby (or distant) genes.5 If this were a sufficiently common mechanism, one approach to fine-mapping association results would be to identify specific variants, known collectively as expression quantitative trait loci (eQTLs) that regulate gene expression and cross-reference them with disease association signals. Although a number of large-scale eQTL studies have been performed to identify such putative functional variants, these studies have generally used easily extracted or otherwise already available tissues or cells, such as lymphoblastoid cell lines or peripheral blood leukocytes, as a way of generalizing functionality across tissue types.6–12 However, increasing evidence has demonstrated that eQTLs may be tissue-specific: variants affecting expression in one tissue can potentially have no effect or an opposite effect in another.13 In the context of interpreting signals in genome-wide association studies from particular diseases, it is therefore necessary to examine eQTL data from tissues of direct relevance to the disease under study, e.g., the bowel in the case of IBD and colorectal cancer. In addition, studying the variation in tissue- (or stimulus-) specific eQTLs can inform our understanding of the biology of such systems.14
We have conducted a comprehensive genetics of gene expression study using ileal and colonic tissue biopsies extracted endoscopically from 65 individuals. This dataset has allowed us to identify variants that affect gene expression in colonic tissue for the first time, with the further aim of better understanding the biological mechanisms underpinning genetic risk for IBD and colorectal cancer.
METHODS
Sample Collection
Seventy-two patients undergoing routine colonoscopy at University College London Hospital (UCLH) were recruited as part of a parallel study investigating mucosal IBD transcriptomics.15 Ethical approval was obtained from the Joint UCL/UCLH Committees on the Ethics of Human Research. The study cohort consisted of 33 healthy controls, 16 patients with CD, and 23 patients with UC. Mucosal pinch biopsies were obtained endoscopically from macroscopically normal tissue in subsets of these patients from the terminal ileum (n = 19) and 4 colonic locations: the ascending colon (n = 23), the descending colon (n = 29), the sigmoid colon (n = 23), and the rectum (n = 66). At each site, 2 biopsies were taken: one for RNA extraction and another for histological assessment. At the time of sample collection, all patients with IBD were clinically quiescent with a Harvey–Bradshaw score of less than 3 (CD) or a partial Mayo activity score of 2 or less (UC).16,17 Healthy controls were under investigation of gastrointestinal symptoms but had both macroscopically and microscopically normal tissue at colonoscopy and histopathological examination, respectively. RNA was extracted from biopsies at each of these locations and the Illumina HT12v4 or WG6v3 microarray chipsets were used to measure relative expression levels. In all, 160 microarray measurements were made from 72 individuals in 5 intestinal locations. Genomic DNA was isolated from peripheral blood or saliva. Further details regarding the sample acquisition and processing can be found in our previous publication.15
Genotyping, Quality Control, and Imputation
All 72 individuals were genotyped at 730,525 single-nucleotide polymorphisms (SNPs) on the Illumina Human OmniExpress platform. Standard technical quality control was performed using PLINK to remove potential sources of technical and genetic bias.18 Samples were tested for familial relatedness, significant genetic missingness, and outlying genome-wide heterozygosity rates. Principal component analysis was performed using EIGENSTRAT to remove significant population outliers as compared with HapMap individuals.19 Under these criteria, 70 samples remained after individual-level quality control.
After outlier sample removal, all SNPs with missingness >3%, Hardy–Weinberg equilibrium chi-square P-values <1 × 10−6, and minor allele frequency <5% were removed resulting in a dataset of 590,471 SNPs. All 70 samples were imputed using Impute2 with the 1000 Genomes Phase I integrated variant set release as the primary reference panel.20,21 Only variants with an INFO score >0.8, probability threshold >0.9, minor allele frequency >0.05, missingness >0.03, and Hardy–Weinberg equilibrium P-values >1 × 10−6 were retained. After applying these very stringent filters, 3,656,633 SNPs and indels were available in the final eQTL analysis.
Measuring Gene Expression
Gene expression was measured on either Illumina WGv3 or HT12v4 microarray platforms. Data were log2 transformed following cubic spline normalization with background subtraction. Samples measured on each platform were treated as a single batch, and quality control was performed on each batch independently to remove principal component analysis outliers. Probes common between the HT12v4 and WG6v3 arrays (n = 39,425) were selected. Only probes that reached a detection P-value threshold of <0.01 in at least 2 individuals across all bowel locations on each platform separately were taken forward (n = 14,491). We adjusted for both experimental and chip-level batch effects using the empirical Bayes method ComBat.22
A number of Illumina probes are known to have common genetic variants within them, which affect binding affinity resulting in them appearing incorrectly as eQTLs.23 To reduce the occurrence of these technical false positives, 934 probes containing common 1000 Genomes SNPs and indels (minor allele frequency >1%) were removed from our analysis. The remaining probe sets were mapped and annotated using the BioMart and BioConductor package illuminaHumanv4.24,25 Ambiguous probes (probes that did not map to at least one Entrez Gene or a known genomic location) were also removed, leaving 13,558 probes.
Regression Framework
We used a standard additive linear model for eQTL identification, regressing normalized probe intensities on genotypes. Because of limited sample size, only cis effects were considered, and pairwise associations were limited to each probe and all SNPs situated one mega-base (Mb) upstream and downstream of its start and end positions. To correct for multiple testing, false discovery rates (FDR) were estimated using the Benjamini and Hochberg26 procedure for each probe-SNP pair. The genome-wide analyses, including multiple-testing correction, were performed using the Matrix eQTL R package.27
To test the robustness of our additive model, we evaluated our eQTL results for genomic inflation by accounting for potential covariates of gene expression such as age, sex, disease status, population-level principal components, and additional clinical metadata. We also tested for differential effects of genotypes across diseases (CD and UC) by including fixed effects and interaction terms for each condition in our linear model. Our core results remained robust under various regression specifications, and genomic inflation did not vary across models despite the inclusion of potential confounders. In addition, we were unable to identify any genotype-disease interaction effects at genome-wide significance. Because of the reduction of power from the inclusion of all possible covariates and the robustness of our results across models with and without covariates, we chose to report the results from the direct pairwise regressions between probes and variants without dividing the cohort by disease status. Because of the variability in gene expression between bowel locations, genome-wide regressions of each location were conducted separately and were compared at the summary level. Given that the study was powered most effectively to detect eQTLs with the rectal tissue dataset with over twice the number of samples than in any other location, this was used as the primary discovery dataset.
RESULTS
Analysis Summary
Genome-wide cis-eQTL analysis of the 65 rectal samples revealed 56,186 significant probe-variant pairs at an FDR threshold of 5% (P = 9.1 × 10−5) as shown in the Manhattan plot in Figure 1. In total, 48,323 unique variants were associated with 1375 microarray probes. To distinguish independent signals, LD pruning was performed to cluster SNPs with an r2 correlation of 0.1 or greater. After pruning, 1312 independent loci were found to be associated with the expression of 1375 probes, which represented 1222 unique and identifiable genes (see Table, Supplemental Digital Content 1, http://links.lww.com/IBD/A662). Example plots for the 10 most significant eQTLs are shown in Figure 2. Two hundred forty-two of the 1312 independent eQTLs spanned more than 1 probe, and of these, 131 mapped to more than 1 Entrez gene (Table 1), suggesting that some SNPs may regulate the expression of multiple genes. Two hundred ninety-eight experiment-wide significant loci (P < 10−7, FDR 1.7 × 10−4) contained a mean of 11.3 directly genotyped SNPs supporting the signal.
FIGURE 1.
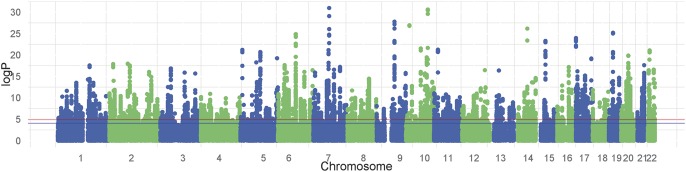
Manhattan plot showing unadjusted P-values of all tested cis-eQTLs in rectal tissue. The blue line indicates significance at FDR 5% (reached by 56,186 probe-variant pairs), and the red line indicates significance at FDR 1%.
FIGURE 2.
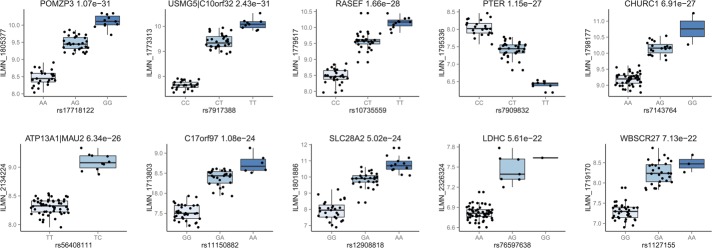
Box plots for the 10 best eQTLs identified in the rectal dataset sorted by P-value. Adjusted Illumina expression intensities are plotted against SNP genotypes. The name of the gene and unadjusted P-values are indicated for each association.
TABLE 1.
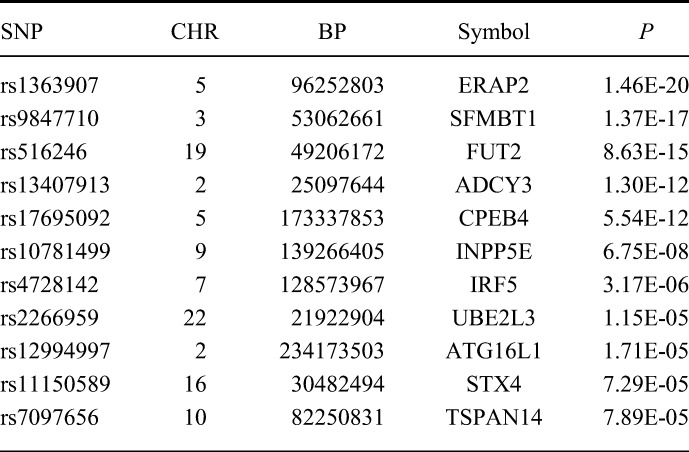
Rectal eQTLs Found Within Loci Associated with IBD. P-value is Unadjusted
Similar analyses revealed 171, 211, 168, and 102 independent eQTLs in the sigmoid colon, descending colon, ascending colon, and terminal ileum, respectively at an FDR of 5% (see Table, Supplemental Digital Contents 2–5, http://links.lww.com/IBD/A663; http://links.lww.com/IBD/A664; http://links.lww.com/IBD/A665; and http://links.lww.com/IBD/A666). The difference in eQTL discovery between the locations tested is driven by variability in sample size, and consequently, statistical power in association testing. Despite the difference in power, the list of most significantly associated genes (FDR 1%) in different intestinal tissues overlapped substantially. The percentage overlap between significant SNP-probe pairs identified in the rectum and each of the other sites were 88.4%, 94.9%, 87.3%, and 75.0% for the sigmoid colon, descending colon, ascending colon, and terminal ileum, respectively. However, 74.0% to 80.6% of genes with eQTLs only in the rectum showed detectable expression in over 90% of non-rectal tissue samples. Thus, the nonoverlap of eQTLs between intestinal tissues within our study was more likely to be driven by the statistical power rather than tissue-specific gene expression.
Comparison with Other eQTL Datasets
The eQTLs identified in the rectal dataset was compared with 7 published eQTL datasets in lymphoblastoid cell lines, fibroblasts, monocytes, T cells, and liver.6–12 Approximately, 3 quarters (74%) of all genes with eQTLs identified in our rectal dataset also had eQTLs in at least one of the existing eQTL datasets. The 26% of genes with eQTLs that were unique to the rectum amounted to 318 genes at FDR 5% and 48 genes at the more stringent P < 10−8. The precise gene overlap between our study and each individual eQTL dataset varied from 9% to 45.4%, influenced by the variation in study sample size and assay type (microarray or RNAseq) in addition to true tissue-specific differences.
eQTL Coregulation Between Tissues
Our observation that a single SNP sometimes regulated the expression of more than 1 gene led us to test whether rectal eQTLs regulated the same or different genes in other published studies in a tissue-specific manner. SNP identifiers and corresponding Entrez gene identifiers between rectal eQTLs and the 216,768 published eQTLs were compared to identify loci that regulated multiple transcripts across tissues. Of the 48,323 unclumped rectal eQTLs (preremoval of variants in LD), 13,356 SNPs were also found to be an eQTL in another study. Of these 13,356 SNPs, 4802 were associated with differential expression of an alternative gene to the one found in our study (see Table, Supplemental Digital Content 6, http://links.lww.com/IBD/A667).
If we use a very conservative measure of independence by looking at eQTL coregulation only at the tag SNPs in our data, 67 of the 1312 independent eQTLs in the rectal dataset were associated with differential expression of a gene different than the one in our study. This coregulation could be attributed to a range of design factors: existing studies may have limited power to detect multiple associations, annotation between studies is inconsistent and often cannot be compared, and probe sets in older microarray chips may have assayed only a small number of non-overlapping transcripts. However, complex regulation was still observed between our study and a well-powered study identifying eQTLs in the monocytes of 1490 unrelated individuals.12 This supports the notion that tissue-specific effects of variants on expression drive some aspects of regulation and that the same variant could be associated with expression of different genes across tissues.
Comparison with Known Ileal eQTLs
A recent eQTL study examining postsurgical ileal biopsy specimens identified over 15,000 cis- and trans-eQTLs.28 Interestingly, the overlap in rectal eQTLs identified in this study and those from the larger existing ileal study were not substantial. At FDR of 1%, 244 of 671 genes (36.4%) with rectal eQTLs also have ileal eQTLs, decreasing to 27.8% of genes at 5% FDR (see Table, Supplemental Digital Content 7, http://links.lww.com/IBD/A668). Thus, 881 of 1222 genes with eQTLs in our study did not have significant evidence of any eQTLs in the ileal biopsy study. We also assessed how many genetic loci acted in both tissues in precisely the same manner, i.e., the same SNP allele correlated with increased expression of the same gene. Of the 48,323 unclumped rectal eQTLs, 2004 SNPs also were found to be significantly associated with gene expression in the ileal study at FDR 5% (see Table, Supplemental Digital Content 7, http://links.lww.com/IBD/A668). However, only 1389 SNPs acted on the same gene, whereas 615 SNPs seemed to regulate mutually exclusive genes within the same region.
Gene Ontology Enrichment
We performed hypergeometic tests to look for enrichment of particular gene ontology categories, pathways, and tissue types in our rectal eQTL gene list compared with all Illumina HT12v4 probes. The most significant gene ontology terms were related to basic cellular functions, including a range of metabolic processes and transport mechanisms (see Table, Supplemental Digital Content 8, http://links.lww.com/IBD/A669). As expected, enrichment analysis using Uniprot expression gene lists as baseline showed a 1.58-fold enrichment of genes expressed in the colon, the largest enrichment among all tissue types (P = 4.16 × 10−7). We did not find any significant enrichment in biological processes linked to IBD, such as autophagy. Similar enrichment results were found when using the genes identified from the ileal eQTL study.28 As expected, the largest fold enrichment of tissue type for these data was for the small intestine (1.8-fold, P = 7.6 × 10−5). Genes expressed in the colon were not significantly enriched in the ileal dataset.
IBD and Colorectal Cancer Risk Loci Found to be rectal eQTLs
In a recent meta-analysis of genome-wide association studies, 163 independent loci were found to be associated with IBD.2 We tested if any of these IBD loci were also eQTLs in the rectum as a way of suggesting a mechanism of action for these variants. Of the 163 IBD tag SNPs, 132 were typed and tested for differential expression in our analysis. Eleven IBD-associated regions had tag SNPs that were associated with the differential expression of 11 genes (Table 1). Six of these loci (ERAP2, SFMBT1, FUT2, ADCY3, INPP5E, and UBE2L3) were previously identified as eQTLs in the ileum.28 Of the remaining 5 IBD loci with rectal eQTLs that were not found to be eQTLs in ileal tissues, 4 were found to be associated with differential expression of the same genes (CPEB4, IRF5, ATG16L1, and TSPAN14) in other tissues.6–12 Variant rs11150589 is a novel eQTL and has not previously been associated with STX4 expression (Figure 3A).
FIGURE 3.
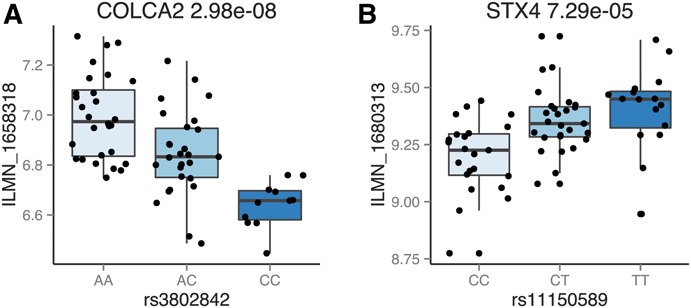
Box plots for COLCA2 (A) and STX4 (B). In (A), the C risk genotype is associated with decreased COLCA2 expression.
Similarly, a colorectal cancer risk locus at 11q23 tagged by rs3802842 was found to be a rectal eQTL associated with differential expression of colorectal cancer associated 2 (COLCA2)29–31 (Figure 3B). COLCA2 has recently been implicated in the pathogenesis of colon cancer, and the same risk variant has been reported to affect COLCA2 expression in candidate or locus-specific studies.32 The C risk allele in rs3802842 significantly downregulates the expression of the COLCA2 transcript in our data, suggesting that COLCA2 expression may be related to colonic tumor formation.
DISCUSSION
In this study, we have identified 1312 independent QTLs that were associated with the expression of 1222 unique and identifiable genes in rectal tissue at FDR of 5%. This study has reaffirmed the tissue-specific nature of some eQTLs: 318 of genes (approximately 26%) in our study had not been associated with eQTLs in previous studies. Of the 163 IBD risk associations,2 11 tag SNPs were found to be eQTLs in colonic tissues. A colorectal cancer risk locus at 11q23 was identified as a genome-wide significant rectal eQTL associated with COLCA2 expression.29–31 These results show that tissue-specific characterization of functional variants can be important to understand the biological mode of action for risk loci discovered in association studies.
Consistent with previous reports, our rectal eQTLs show incomplete overlap with published eQTLs in other cell types, even in closely related tissues such as ileum. We believe that 3 factors contribute to this observation, and each is an important consideration for researchers using published eQTL lists. First, the statistical power (driven by sample size) of different eQTL studies contributes enormously to estimates of overlap.33 Our sample size is modest, especially in the non-rectal samples, so it is hard to draw conclusions about previously reported eQTLs that we are not well powered to find. Second, technical differences, including method of sampling, possible disease state or inflammation, and type of expression assay used (microarray or RNAseq) can result in differential detectability of specific eQTLs in different datasets. Finally, it is clear that both tissue- and context- (e.g., stimulated or resting immune cells) specific effects are widespread, and some, such as the COLCA2 eQTL reported here, may be essential to dissecting disease associations.
Out study furthers the understanding of the relationship between genetic variation and gene expression across the body by contributing new data sampled from biopsies of the large bowel. These mucosal pinch biopsies are composed, however, of multiple cell types including epithelial cells and those of the lamina propria such as immune cells, and thus do not provide a uniform picture of gene expression in these cell types. As previously commented, the study of individual sorted primary cell types isolated from complex tissues may help to reduce this heterogeneity and enable the precise cellular origin of the eQTLs discovered here to be ascertained.28,34 It is also not clear the extent to which disease and treatment history affect subsequent expression profiles; studies of large numbers of samples from multiple bowel locations in both patients and healthy individuals will be required.
Supplementary Material
ACKNOWLEDGMENTS
Author contributions: Contributed equally: T. Singh and A. P. Levine; Jointly supervised the work: A. W. Segal and J. C. Barrett; Study conception and design: T. Singh, A. P. Levine, P. J. Smith, A. M. Smith, A. W. Segal, and J. C. Barrett; Recruitment, sample acquisition and processing: P. J. Smith; Data preparation and analyses: T. Singh and A. P. Levine; Drafting of the manuscript: T. Singh, A. P. Levine, A. M. Smith, and J. C. Barrett; Obtained funding: A. W. Segal and P. J. Smith; Study supervision: A. M. Smith, A. W. Segal, and J. C. Barrett.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.ibdjournal.org).
Supported by the Medical Research Council and Wellcome Trust. A. P. Levine is funded by the Irwin Joffe Memorial Fellowship.
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Visscher PM, Brown MA, McCarthy MI, et al. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marks DJB, Rahman FZ, Sewell GW, Segal AW. Crohn's disease: an immune deficiency state. Clin Rev Allergy Immunol. 2010;38:20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cookson W, Liang L, Abecasis G, et al. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dimas A, Deutsch S, Stranger B. Common regulatory variation impacts gene expression in a cell type–dependent manner. Science. 2009;325:1246–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Innocenti F, Cooper GM, Stanaway IB, et al. Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS Genet. 2011;7:e1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown CD, Mangravite LM, Engelhardt BE. Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eQTLs. PLoS Genet. 2013;9:e1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schadt EE, Molony C, Chudin E, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veyrieras JB, Kudaravalli S, Kim SY, et al. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeller T, Wild P, Szymczak S, et al. Genetics and beyond—the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grundberg E, Small KS, Hedman AK, et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raj T, Rothamel K, Mostafavi S, et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science. 2014;344:519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith PJ, Levine AP, Dunne J, et al. Mucosal transcriptomics implicates under expression of BRINP3 in the pathogenesis of ulcerative colitis. Inflamm Bowel Dis. 2014;20:1802–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harvey RF, Bradshaw MJ. Measuring Crohn's disease activity. Lancet. 1980;1:1134–1135. [DOI] [PubMed] [Google Scholar]
- 17.Lewis JD, Chuai S, Nessel L, et al. Use of the noninvasive components of the Mayo score to assess clinical response in ulcerative colitis. Inflamm Bowel Dis. 2008;14:1660–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 20.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. [DOI] [PubMed] [Google Scholar]
- 23.Ramasamy A, Trabzuni D, Gibbs JR, et al. Resolving the polymorphism-in-probe problem is critical for correct interpretation of expression QTL studies. Nucleic Acids Res. 2013;1–12. 10.1093/nar/gkt069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Durinck S, Moreau Y, Kasprzyk A, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. [DOI] [PubMed] [Google Scholar]
- 25.Dunning M, Lynch A, Eldridge M. IlluminaHumanv4 db: Illumina HumanHT12v4 Annotation Data (chip illuminaHumanv4). R package version 1.22.1.
- 26.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a Practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995. Available at: http://www.jstor.org/stable/10.2307/2346101. [Google Scholar]
- 27.Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabakchiev B, Silverberg MS. Expression quantitative trait loci analysis identifies associations between genotype and gene expression in human intestine. Gastroenterology. 2013;144:1488–1496, 1496.e1–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tenesa A, Farrington SM, Prendergast JGD, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet. 2008;40:631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters U, Hutter CM, Hsu L, et al. Meta-analysis of new genome-wide association studies of colorectal cancer risk. Hum Genet. 2013;131:217–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peters U, Jiao S, Schumacher FR, et al. Identification of genetic susceptibility loci for colorectal tumors in a genome-wide meta-analysis. Gastroenterology. 2013;144:799–807.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peltekova VD, Lemire M, Qazi AM, et al. Identification of genes expressed by immune cells of the colon that are regulated by colorectal cancer-associated variants. Int J Cancer. 2013;134:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding J, Gudjonsson JE, Liang L, et al. Gene expression in skin and lymphoblastoid cells: refined statistical method reveals extensive overlap in cis-eQTL signals. Am J Hum Genet. 2010;87:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raine T, Liu JZ, Anderson CA, et al. Generation of primary human intestinal T cell transcriptomes reveals differential expression at genetic risk loci for immune-mediated disease. Gut. [published online ahead of print May 5, 2014]. 10.1136/gutjnl-2013-306657. [DOI] [PMC free article] [PubMed] [Google Scholar]