

Abstract
The composition of the oral microbiome differs from one intraoral site to another, reflecting in part the host response and immune capacity at each site. By focusing on two major oral infections, periodontal disease and caries, new principles of disease emerge. Periodontal disease affects the soft tissues and bone that support the teeth. Caries is a unique infection of the dental hard tissues. The initiation of both diseases is marked by an increase in the complexity of the microbiome. In periodontitis, pathobionts and keystone pathogens such as Porphyromonas gingivalis appear in greater proportion than in health. As a keystone pathogen, P. gingivalis impairs host immune responses and appears necessary but not sufficient to cause periodontitis. Historically, dental caries had been causally linked to Streptococcus mutans. Contemporary microbiome studies now indicate that singular pathogens are not obvious in either caries or periodontitis. Both diseases appear to result from a perturbation among relatively minor constituents in local microbial communities resulting in dysbiosis. Emergent consortia of the minor members of the respective microbiomes act synergistically to stress the ability of the host to respond and protect. In periodontal disease, host protection first occurs at the level of innate gingival epithelial immunity. Secretory IgA antibody and other salivary antimicrobial systems also act against periodontopathic and cariogenic consortia. When the gingival immune response is impaired, periodontal tissue pathology results when matrix metalloproteinases are released from neutrophils and T cells mediate alveolar bone loss. In caries, several species are acidogenic and aciduric and appear to work synergistically to promote demineralization of the enamel and dentin. Whereas technically possible, particularly for caries, vaccines are unlikely to be commercialized in the near future because of the low morbidity of caries and periodontitis.
4 keywords for indexing: Periodontitis, Caries, Microbiome, Pathogenesis
Introduction
The digestive system begins with the oral cavity where food and microorganisms are introduced, mixed with salivary proteins and digestive enzymes, swallowed, and enter the lower gastrointestinal (GI) tract to be further digested. In the oral environment, several unique ecological niches can be mapped where microorganisms establish in consortial communities. Failing to establish in oral communities, some environmental microorganisms simply transit to the lower GI tract.
Despite continuous shedding of superficial epithelial layers, the oral mucosae are persistently colonized by microorganisms growing in unique ecological niches. One distinctive feature of the oral cavity is the surface of the tooth or tooth enamel (Fig 1). This non-shedding surface supports the growth and maturation of a complex microbial biofilm. The nutrient foundations of the microbiota surviving on mucosae or within the tooth biofilm are the proteins and glycoproteins of saliva, and the carbohydrates, proteins and lipids of dietary food. Since the teeth are anchored to the jaws, but grow out of the gums or gingivae, serum proteins that exude at the gingival sulcus (the junction of the tooth and the gingiva; Fig. 2) are an additional source of nutrients in specific ecological niches. In this review, we will discuss the composition of the microbiota that has shed from oral surface niches into saliva, biofilm communities on the tooth enamel, and within gingival sulcus. The current literature reveals that contrary to what occurs in the GI tract, initiation of oral infectious diseases and disease status are associated with increased diversity and richness of the microbiota. Oral health is associated with low diversity and richness within the microbial community. This review also highlights the host response to the oral microbiomes in specific niches by consideration of the immunopathogenesis of periodontal disease and the immune defenses against caries.
Figure 1.

Anatomy and ecological niches of the oral cavity.
Figure 2. Microbial diversity and richness in periodontal health and disease.

Microbial communities with lower diversity and richness harbor keystone pathogens, symbionts and pathobionts at very low frequency and in proportions adequate to ensure health. When environmental perturbations occur in the periodontal tissues (e.g., trauma or idiopathic growth of a keystone pathogen) or the host is genetically susceptible, keystone pathogens elicit inflammation that changes the nutrient foundation of the ecological niche (i.e. periodontal pocket). The altered nutrient foundation promotes the proportional expansion of pathobionts relative to symbionts, promoting inflammation that ultimately leads to connective tissue and bone destruction. Diversity and richness are higher.
The CORE oral microbiome today
Many basic observations of the composition of the oral microbiome were formulated well before ribosomal RNA-based systematics. Unachievable with classical methods, however, the power and scope of molecular taxonomy have resulted in the discovery of new phylotypes and, more importantly, a high level of bacterial community analysis that had been hypothesized [1].
Today, one of the most important databases of taxa present in the oral cavity is the Human Oral Microbiome Database (HOMD) (http://www.homd.org/). HOMD stores 34,753 filtered cloned sequences representing a wide variety of healthy and diseased sites throughout the oral cavity such as the dorsum of the tongue, lateral sides of the tongue, buccal fold where the gingiva folds into the cheek, surface of the cheeks (buccal mucosa), hard palate, soft palate, labial gingiva, tonsils, and supragingival and subgingival plaques from tooth surfaces (Fig. 1). [2,3]. As a system open to the environment, accounting for 100% of the organisms found in the oral cavity can potentially include all of the organisms in nature. Classified as operational taxonomic units (OTUs), the organisms of the oral cavity will differ among individuals, reflecting diet, sampling times of day, and geographical locations. From a data-driven perspective, the HOMD 34,753 clones yielded 1179 OTUs; 875 OTUs comprise 99% of the clones. The remaining 1% or 347 clones represent 304 OTUs, comprising organisms rarely retrieved from the oral cavity. As of March 2014, HOMD stores 688 taxa, of which 343 are named species, 101 are unnamed cultivated microorganisms and 243 are uncultured phylotypes [4]. The relative abundance of the oral microbiome at the genus level stored in HOMD is presented in Figure 3 (reviewed in [5]).
Figure 3.

Proportions of oral microorganisms in the Human Oral Microbiome Database (HOMD) [5].
Representing a minimally redundant collection of OTUs regularly found in the oral cavity (the “core” microbiome), CORE (http://microbiome.osu.edu/) is another sequence database complementary to HOMD (Fig. 4) [6]. The CORE database contains 636 phylotypes; each phylum was defined to contain DNA sequences that are 98% similar. At this level of similarity, patterns of variation due to sequence artifacts could be distinguished from heterogeneity among paralogous operons and the co-existence of similar but differentiated taxa [7]. Of the 636 phylotypes, 365 presently lack a cultured member and none of which is a singleton sequence. Singleton sequences are unique with no overlap with other sequences to generate the contig, or with so many overlaps that cannot be assembled to be assigned to any particular phylum. Interestingly, 1000 pyrosequencing reads were compared from subgingival samples of 24 patients, sequences were more effectively assigned to existing taxa by both CORE and HOMD than the Ribosome Database Project or GenBank [6].
Figure 4. CORE microbiome of oral cavity.

The tree was generated with RAxML BlackBox Web server [154] and viewed in ITOL [155]. Genera are color-coded by phyla, except for the Firmicutes and Proteobacteria, which are shown at the level of class (Adapted from [6]).
Oral microbiome modulated by dietary habits
The members of the oral microbiome in health or disease appear to select depending on the availability of nutrients (reviewed in [8]). Diet of the human host appears to shape the symbiosis of the microbiota residing on the mucosae and on the non-shedding surfaces of teeth. A simple example is the use of refined carbohydrates in the diet of modern humans. Sucrose contributes to increased risk of caries.
Shifts in the diet of early human ancestors to modern foods have contributed to subtle changes in the oral microbial community. The oral microbiome of Neolithic hunter-gatherers appeared surprisingly stable over the millennia to medieval times in the proportional composition of different phyla [9]. Although small changes were detectable in the composition of the microbiota between hunter/gatherers, early agriculturist humans, and medieval humans, shifting dietary habits was generally associated with a decrease in microbial diversity. Interestingly, periodontal disease-associated taxa, including Porphyromonas gingivalis and members of the Tannerella and Treponema genera, were more prevalent in the farming than hunter-gatherer populations. The decrease in microbial diversity culminated in dramatic shifts in the proportional composition of the microbial communities of humans of the industrial revolution. In contrast, from medieval to modern times, the oral microbiome associated with the gingival sulcus and periodontal disease also appeared to change little in the face of significant dietary changes and the advent of antibiotics [10]. From early humans to modern times, subtle trends include a decrease in non-pathogenic clostridia taxa and members of the Ruminociccaceae family, and an increased frequency of caries-associated Veillonellaceae, Lachnospiraceae, and Actinomycetales. Notably, the frequency of S. mutans is significantly higher in modern samples than in preindustrial agricultural samples [9]. It is reasonable to point out, however, that DNA sequences buried in ancient calculus sampled to compare to modern viable microbiomes may not be representative of the loose plaque biofilm directly facing the epithelium of the periodontal pocket or sites on the tooth surface that are susceptible to caries.
Clearly, the environment and perhaps host genetics modify the oral microbiome. In South American Amerindians living in a remote village of the Amazon forest, who are less exposed to selective pressures of modern diet and are genetically less diverse than multiracial and multiethnic urban societies, show a more restricted oral mucosal microbiome than urban people. Despite the lower number of genera identified, the Amerindians harbor an increased frequency of previously unclassified Proteobacteria [11]. Similarly, remote Eskimo tribes showed low prevalence of periodontal disease and caries [12] until modern diets were introduced [13], whereas Sri Lankan tea workers with diets essentially identical to early ancestors and in the absence of conventional oral hygiene measures had little caries but showed a range of incidence and severity of periodontitis as people aged in a landmark longitudinal study [14]. In the Sri Lankan population, the severity of periodontitis appeared to reflect the presence of putative pathogens in the subgingival microflora [15]. Since the Sri Lankans employed no oral hygiene measures and enjoyed similar diets, host genetic polymorphisms within racially and ethnically similar populations may account for differences in acquisition or outgrowth of pathogens and the occurrence of disease.
During the past few hundred years, the human mouth appears to have become a substantially less biodiverse ecosystem. Since higher phylogenetic diversity is associated with greater ecosystem resilience [16,17], the decreased diversity of the modern oral environment may be associated with less resistance to perturbations and greater susceptibility to insertion of pathobionts or even true pathogens in the microbial community [9]. Whereas this hypothesis has yet to be tested, the microbiota of different ecological niches within the oral cavity may reflect greater or less stability over time.
Characteristics of different oral ecological niches
In defining health and disease or the microbial communities that prelude the establishment of caries or periodontal disease, it is critical to define the characteristic microbiota within specific ecological niches. In the broadest terms, the oral cavity harbors at least five communities: the teeth, which are non-shedding surfaces; the saliva; the dorsal and lateral surfaces of the tongue; and the gingival sulcus and the periodontal pocket; and the remaining epithelial surfaces of the oral mucosae [2,18]
Salivary microbiome
Saliva has no indigenous microbiota. The bacteria in saliva are those shed from biofilms on oral tissues. All epithelial surfaces desquamate, releasing associated bacteria into the bathing saliva. The salivary microbiome consists disproportionally of microorganisms from the tongue biofilm. The papillate surfaces of the tongue harbor a microbiota skewed towards anaerobic genera such as Prevotella and Veillonella, whereas the ventral surface bears a microbiota rich in streptococci and Gemella [19]. Consistent with the contribution of the shedding biofilm from the tongue, the salivary microbiome has been reported to contain a number of genera with the most prevalent or autochthonous being Prevotella and Streptococcus (Fig. 5) [20]. Genera present at <1% prevalence are considered transients or allochthonous microorganisms.
Figure 5. Proportions of different genera recovered from whole saliva of healthy adults.

Saliva was collected from a group of 71 healthy individuals by mouthrinse with 10 mL UV-irradiated sterile saline for 30 sec and stored at −80°C. The asterisks (*) denote the best classification possible as adapted from a table in [20].
Saliva and salivary constituents
The tissues and surface biofilms of the oral cavity are constantly immersed in saliva. The myriad proteins and glycoproteins in saliva provide lubrication for mastication and gustatory sensation, and both support and antagonize biofilm formation [21]. In conditions of nutrient deprivation, bacteria in the oral milieu show consortial behavior to facilitate metabolism of specific salivary glycoproteins as a nutrient source [22]. A salivary film conditions the enamel of the tooth crown. In the salivary film, salivary proteins (glycoproteins) are available to interact with microbial adhesins of pioneer colonizers as modeled in vitro, facilitating the initiation of biofilm formation on the tooth surface [23–25]. Expression of adhesins is dynamic and responsive to the adhesion status of the pioneer colonizer [26]. As the salivary film transitions from the crown into the gingival sulcus, the composition changes and the proportion of serum proteins increases due to the proximity with the gingival crevicular fluid (GCF) (Fig. 2) [27]. With the adsorption of serum proteins, the composition of the dental biofilm also changes from predominantly pioneer Streptococci and Actinomyces spp. to an increased proportion of putative periodontal pathogens.
Salivary proteins (glycoproteins) that affect oral biofilm formation include secretory IgA [28], mucins [29], agglutinin (GP340) [30], and proline-rich proteins [31,32]. These proteins can promote microbial adhesion because the salivary film and its constituent proteins coat the teeth and mucosal membranes [33]. Also present as fluid-phase salivary constituents, however, the constituent proteins also appear to promote desorption, agglutination and microbial clearance by swallowing of saliva. Indeed, saliva is swallowed at the rate of ~1 mL per minute. The constituent antimicrobial proteins and peptides of whole saliva include cystatins [34] and histatins [35,36], lysozyme, lactoferrin and lactoperoxidase [37], defensins [38], cathelicidin [39], and calprotectin (S100A8/A9) [40]. The antimicrobial proteins/peptides all likely limit the overgrowth of many species in the dental biofilm.
Microbiota on teeth
Unlike the shedding surfaces of the oral epithelia, the tooth surfaces are the only “non-shedding” surfaces in the oral cavity. The non-shedding surfaces facilitate a stable anchoring location for long-term biofilm development. As a substrate for biofilm formation, the surfaces of the teeth are more complex than the hydroxyapatite mineral of the enamel forming the crowns and the cementum, which coats the roots. The tooth enamel in the mouth is coated with a salivary film, whereas the roots can be coated with an admix of salivary and serum proteins. The protein-rich films are the actual sites of initial adhesion of the pioneer microbial colonizers (reviewed in [33,41]. Pioneer streptococcal adhesins initially bind the salivary film of the enamel via “catch-bond” or shear-enhancement interactions, requiring sheer-induced conformational changes in the adhesins [42]. As the biofilm matures, the community becomes more complex. During maturation, the changing architecture of the dental plaque biofilm reflects the forces of interspecies coaggregation more than new interactions between early tooth colonizers with the salivary film (Fig. 6) [43].
Figure 6. Selective coaggregation and relative abundance of microorganisms in supra/subgingival dental plaque of humans.
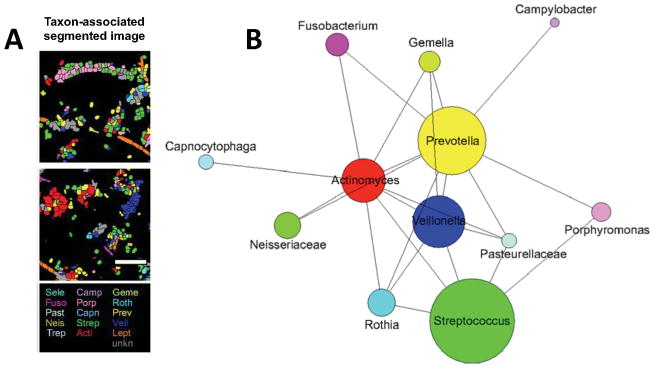
(A) Spatial analysis of human dental plaque using Combinatorial Labeling and Spectral Imaging - Fluorescence In Situ Hybridization (CLASI-FISH) strategy reveals specific interactions between certain microorganisms and not with others. (B) The 2D plot reveals the relative abundance (diameter of circles) and intertaxon associations observed between labeled taxons. A line connecting two taxa indicates that cells of the lower-abundance taxon of any pair were observed to associate with cells of the higher-abundance taxon with >3% frequency and more frequently than would be expected from random associations. (Adapted with permission from Valm A.M. et al. PNAS 2011;108:4152–4157.)
The dental plaque microbial community that forms on the enamel salivary film (supragingival; above the gum line) differs from the subgingival (below the gum line) community, which forms on the proteinaceous film that coats the cementum of the root. As the growth of the community extends along the root and away from the salivary environment, the film contains more serum and less saliva. The environment becomes more anaerobic and increasing shielded from foodstuffs and extremes in pH and temperature. Surface sheer is reduced. Consequently the subgingival and supragingival communities differ in the proportion of facultative bacteria.
The microbiota of the tooth surface (crown) has a composition slightly different than the saliva with Streptococcus ssp. and Veillonella being the most prevalent (Fig. 7). These frequency distributions among the different taxa however should be considered in light of the method of analysis. Cloning and sequencing of 16s rRNA genes and pyrosequencing of the different V (variable) regions of 16s rRNA genes have different levels of bias in amplifying such variable rRNA sequences [44].
Figure 7. Proportions of different genera recovered from dental plaque of healthy adults.

Supragingival plaque from a group of 98 healthy individuals by sampling cheek-side dental surfaces using a sterile, DNA-free wooden toothpick and stored at −80°C. The asterisks (*) denote the best classification possible as adapted from a table in [20].
Ecological niches on the crown of the tooth
The tooth crown can be further divided in five distinct areas and ecological niches, each characterized by specific caries risks: the occlusal or chewing surface; the approximal surface or contact point between teeth; the supragingival surface; the buccal or cheek-contacting surface; and the lingual surface approximating the tongue (Fig. 1).
Occlusal and approximal surfaces are the most susceptible to caries. These ecological niches harbor microbial communities that are acidogenic, producing organic acids, and/or aciduric and able to withstand an acid environment. Plaque community diversity is greater on the approximal and lingual (tongue) surfaces of molar teeth, and less diverse on buccal (cheek) and anterior or front teeth [18,45]. The approximal surfaces are protected from regular toothbrushing and more susceptible to caries development. Plaque stagnates in these sites; the resident microbiota is bathed, however, in a serum-like exudate. This exudate, gingival crevicular fluid, is dissimilar from saliva and further contributes to the proportional composition of the microbiota. The microbiota in the approximal area differs from other flat surfaces of the crown or the chewing surfaces where the enamel forms pits and fissures. Therefore the composition of the tooth microbiota is influenced not only by the location of the tooth within the mouth and the proximity to salivary flow from nearby ducts, but also by the anatomy and physiology of the tooth surface and the surrounding gingiva (Fig. 8).
Figure 8. Diversity and richness of microbial communities directly relate to caries risk.

Microbial communities of the tooth surface and irregularities in the enamel differ with respect to diversity and richness. Surfaces and sites with highest diversity and richness mature at ecological niches most susceptible to caries. When caries is established, the acid environment reduces the diversity and richness of the local microbiota.
Hence, the undisturbed and more established ecological niches of the tooth display more diverse communities [18,45]. Since more established and undisturbed microbial communities are more susceptible to caries, these findings seem to contradict the earlier hypothesis established for the intestine, where a less diverse community is more permissive to the insertion of new pathogenic taxa [17].
Gingival sulcus and periodontal pocket
The gingival sulcus and periodontal pocket form unique ecological niches for microbial colonization. The gingival sulcus or crevice is a space between the enamel of the tooth crown and the epithelium of the visible gingiva that folds to create an epithelial-lined crease around the tooth (Fig 2). Superficially the sulcus or pocket opens into the oral cavity, whereas the base of the trough is bounded by a thin, modified epithelium (junctional epithelium), which ends where the connective tissue bundles attach to the cementum surface of the root [46]. The sulcular and junctional epithelia are immunologically active sensors of the proximal dental plaque biofilm, expressing Toll-like receptors and other pathogen recognition receptors (PRR) on the lining stratified squamous, keratin-17-positive epithelial cells [47]. The keratinocytes of the gingival epithelium connect to one another through desmosomes and cell adhesion molecules such as CEACAM1 [46]. The junctional and sulcular epithelia release chemotactic molecules such as IL-8 (CXCL8) MCP-1 [48], CCL28, secretory leukocyte protease inhibitor (SLPI) [47,49] and RANTES [50]. These cytokines and chemokines signal neutrophils and monocytes to transmigrate through the epithelium into the sulcus or pocket [51]. Microbe-associated molecular patterns (MAMPs) from the biofilm activate the epithelium, increasing the proliferative rate, expression of adhesion molecules [47], and production of IL-1β [52] and antimicrobial proteins and peptides such as calprotectin and defensins [53] (reviewed in [54]). This mixture of immune and inflammatory mediators and white cells is contained in the gingival crevicular fluid (Fig. 2 and 8) (reviewed in [55]), which brings serum proteins such as IgG and albumin and cytokines such as IL-1β, IL-6, TNFα into the microbial biofilm and oral environments.
The gingival crevicular epithelium [56,57] and PMNs [58] express antimicrobial proteins including S100A8/A9. In the extracellular environment, S100A8/A9 appears antimicrobial when incorporated in neutrophil extracellular traps (NETs) [58,59]. When present in the cytoplasm of the mucosal epithelial cells, S100A8/A9 contributes to innate intracellular immunity, protecting against invasive bacterial pathogens [60,61]. Employing S100A8/A9, therefore, the gingival crevicular epithelium forms a barrier against invasive bacteria using innate intracellular immunity and extracellular NETs.
The microbiota of the dental plaque biofilm drives the inflammatory process. In susceptible individuals, inflammation in the gingival tissues activates neutral proteases, elastases [62], collagenases [63] and metalloproteinases [64] destroying the epithelial and connective tissue attachments to the tooth. In an apparent attempt to heal, the junctional epithelium responds to the damage by migrating toward the apex of the tooth. Some workers suggest that apical migration of the junctional epithelium is stimulated by proteolytic activity from degranulating neutrophils within the gingival crevice [65]. As the sulcus deepens due to the loss of connective tissue attachment to the root of the tooth a periodontal pocket forms. An anaerobic environment characterizes deep periodontal pockets with a pH range from 6 to 8 [66]. Ultimately, the microbial biofilm in the gingival sulcus and periodontal pocket elicits inflammation in the surrounding connective tissue. Characteristic of periodontitis, this chronic inflammatory process, ultimately drives the destruction of the alveolar bone that supports the tooth in the socket (Fig. 2) [67,68].
Periodontal microbiome and immunity
Microbiota of periodontal health
The microbiota of the healthy gingival sulcus or crevice has recently been defined by pyrosequencing of V1-2 and V4 regions of 16S rRNA genes [45,69]. In healthy gingival sulci (less than 4 mm deep), the phylum Proteobacteria, particularly the gammaproteobacteriae of genus Acinetobacter, Haemophilus and Moraxella, were most prevalent. Within the phylum Firmicutes, the class Bacilli comprising genus Streptococcus, Granulicatella and Gemella were also health-associated (Fig. 9 and 10). These genera can be considered symbionts, which also return to periodontal pockets in high proportion after periodontal treatments [70].
Figure 9. Microbiome of the periodontal pocket in health and disease.

Subgingival samples were collected form pockets < 4 mm (healthy) or > 5mm (diseased) in depth. After the removal of supragingival plaque and drying the target sites, samples were collected by insertion of four medium paper points for 10 s into three sites. Deep and shallow sites were sampled separately in subjects with periodontitis. (Adapted with permission from Griffen AL. et al. ISME J. 2012 Jun;6(6):1176–85)
Figure 10. Frequency distribution of periodontal microorganisms in periodontitis patients with ≥ 4mm pockets and in healthy individuals.

Data show differences between health and disease at level of phylum, genus and species. Pie charts indicate the number of taxa that were significantly different. The graphs show levels for genera that were ≥ 0.1% different and species that were ≥ 0.2% different in health and periodontitis samples. Taxa were sorted according to magnitude of change. P ≤ 0.05 after FDR correction for all taxa shown. (Adapted with permission from Griffen AL. et al. ISME J. 2012 Jun;6(6):1176–85)
In the gingival sulcus, the conditions of ecological niche are driven from steady state by host and microbial perturbations. Through the crevicular fluid, the host dispenses isotype-switched immunoglobulins [71] specific for components of the microbiota [72,73]. Perturbations in the microbial community are also caused by growth of microorganisms such as P. gingivalis, which can alter the nutrient foundation of the niche and disrupt the equilibrium between symbionts and pathobionts (Fig. 2) (reviewed by [74]). The community is also susceptible to modulatory effects of bacteriophagic activity [75]. Hence, both host-induced species-specific suppression and interspecies microbial competition can increase the pathogenic potential of the complex, mixed-species dental plaque community.
Microbiota of periodontal disease
Since the 1950s, the microbiota of the periodontal pocket has been studied with culture methods. Investigators sought to define the microbial species critical for the initiation and progression of the disease. Defined historically as microorganisms of the “red complex” by Sigmund Socransky, P. gingivalis, Tannerella forsythia (formerly Bacteroides forsythus) and Treponema denticola were considered the cultivable microorganisms most associated with disease as defined by deep periodontal pockets [76]. Although present in low numbers in healthy subjects [77,78], these species were considered to be responsible for initiation and progression of disease. After periodontal treatment, the red complex microorganisms disappear (below the limits of detection). When inflammation and deep pockets reappear, the red complex is again prominent. A cluster of species with less stringent association with disease was defined as the “orange complex” and includes Prevotella spp., Fusobacterium spp. and Parvimonas micra (formerly Peptostreptococcus micros). The red and orange clusters and their association with periodontitis were originally characterized by culture studies and more recently confirmed using DNA-DNA hybridization.
Newly identified microorganisms have also been associated with progression of periodontal disease using more contemporary sequencing technology. In earlier studies using cloning and Sanger sequencing [78–80], the complexity of the dental plaque ecosystem could not be ascertained because the expense of sequencing limited the genetic information that could be obtained. The power to comprehensively study bacterial community composition through determination of thousands of sequences per sample was facilitated by the advent of 454 pyrosequencing of V regions of 16S rRNA genes [18,20]. When V1-2, V4-6 and V7-9 regions are assessed, differences are observed at all phylogenetic levels between health- and periodontitis-associated bacterial communities (Fig. 9 and 10) [44,69,81].
Using pyrosequencing, the microbiota highly associated with diseased pockets greater than 4 mm in depth included the phyla Spirochaetes genus Treponema, Synergistetes genus Sinergistes [82], and Bacteroidetes such as genera Porphyromonas, Prevotella and Tannerella. The class of Fusobacteria genera Fusobacterium and Leptotrichia was also highly associated with disease. As the level of disease increased as measured by deeper pockets, the class Negativicutes genera Seleomonas and Megasphera appeared most prevalent [69,79,83,84] and Clostridia became prominent including the genera Filifactor, Lachnospiraceae and Peptostreptococcus. In deeper diseased sites, also associated was the class Erysipelotrichia genera Erysipelothrix, Solobacterium and Bulleidia [69,83].
New sequencing technologies also facilitated novel associations between periodontitis and previously uncultivable or previously underappreciated species, including the Gram-positive Filifactor alocis [69] and Peptostreptococcus stomatis, and species from the genera Prevotella, Synergistes [82], Megasphaera, Selenomonas, and Desulfobulbus [3,79]. Many of these newly recognized organisms correlate with disease as strongly as the classical red complex bacteria (Fig. 10). Clearly, periodontitis is a polymicrobial infection resulting from the expansion of pathobionts within the microbial community. The expansion appears to be initiated by low prevalence microorganisms capable of modulating the nutrient foundation of the community through induction of inflammation, extravasation of blood born nutrients and serum proteins (Fig. 2). The presence of Koch’s postulate-fulfilling “true” pathogens is not apparent (Fig. 10).
Generally microorganisms associated with pockets deeper than 4 mm are reduced to very low or even undetectable levels after periodontal treatment. Scaling and root planing (“deep cleaning”) or periodontal surgery, disrupt the microbiota of the periodontal pocket by mechanically scraping the biofilm off the root surface to a point that the microbial richness and biodiversity are significantly decreased [70]. Antibiotic treatments alone or in combination with scaling and root planing disrupt the relative proportions of the taxa within the community. However, when the selective pressure is lifted, the community tends to return to equilibrium without persistence of antibiotic resistance in the various taxa [85].
Other domains of life
Bacteria are not the only microorganisms present in the periodontal pocket. Members of the Archaea domain have also been described in the subgingival biofilm (reviewed in [86]). Methanobrevibacter oralis phylotypes SBGA-1 and SGBA-2 are detected in a subset of severe periodontitis patients who harbor low levels of Treponema spp [87]. Treponema spp. and Methanobrevibacter spp. are potential syntrophic competitors. Both are hydrogenotrophic and synthesize organic acids (acetogenic) using CO2 and H2 in anaerobic conditions [88]. Potential users of H2 derived from the fermentation processes of other anaerobic bacteria in the subgingival plaque biofilm in deep pockets of severe periodontitis patients include methanogenic archaea (mcrA gene) and sulfate-reducing bacteria (dsrAB gene) such as Desulfovibrio and Desulfubulbus [69,89].
Viruses have also been hypothesized to contribute to the microbiome of the periodontal pocket. The hypothesis posits that subgingival bacteria and viruses infecting the adjacent periodontal tissues would form a pathogenic consortium. Epstein-Barr virus type 1 (EBV-1) infects periodontal B-lymphocytes and human cytomegalovirus (HCMV) infects periodontal monocytes/macrophages and T-lymphocytes. EBV-1, HCMV and other herpesviruses are present more frequently in periodontitis lesions and acute necrotizing ulcerative gingivitis-lesions than in gingivitis or periodontally healthy sites [90]. Although plausible, this hypothesis of viral consortial or cooperative pathogenesis in periodontal disease has never been tested definitively.
Challenges sampling the periodontal pocket
The periodontal plaque biofilm harbors different constituents in the loose superficial plaque and in plaque most adherent to the root surface [91]. Given the small space, applying precise sampling methods is a challenge. Depending on the sampling method, different clades of microorganisms from the “same site” can be identified because of inadvertent sampling of proximal ecological communities [92]. Given the anatomy, the crude sampling tools for the periodontal pocket such as curettes and paper points yielded similar results. Sub-sites or communities could not be resolved. Nonetheless the subgingival biofilm is characterized by specific aggregation patterns or co-colonization between different genera [43]. It is important to note that in diseased sites the diversity and richness of the community was significantly greater than in healthy sites [69,81]. The microbiota in the subgingival diseased pocket at the time of sampling may reflect both a proportional shift among genera secondary to initiation of disease and also a shift that foreshadows the progression of disease.
Cellularity and pathogenesis of periodontal disease
The immunological response elicited by the microbial biofilm is clearly complex. Innate immunity maintains tissue homeostasis and prevents periodontal tissue destruction. Neutropenia, agranulocytosis, neutrophil adhesion and chemotaxis deficiencies and diseases affecting degranulation of lysosomal contents are characterized by severe periodontitis [93]. Phagocytic antigen presenting cells such as dendritic cells (DCs) and Langerhans cells direct the phenotype of T helper (Th) cells. The cellular infiltrate in human gingivitis is primarily composed of Th cells [94]. During an undefined transition, the immune response in the gingiva switches from the recruitment and activation of neutrophils to clear pathogenic bacteria to a chronic infiltrate of T, B cells and plasma cells [50,95,96]. The chronic infiltrate of immune cells induces destruction of connective tissue, vascular proliferation and alveolar bone destruction characteristic of periodontitis.
In murine models of periodontitis, CD4+ T cells are central to the development of alveolar bone destruction [65,97,98]. Although memory B cells and T cells release RANK-ligand to contribute to osteoclastic activation, Th cells release an ill-defined set of cytokines immediately before initiation of alveolar bone destruction [99]. Current research seeks to characterize P. gingivalis-specific CD4+ T cells locally in the marginal gingiva, define the cellularity that predicts disease initiation and progression, and determine the kinetics of clonal expansion and cytokine expression. Using new immunological tools, epitope-specific CD4+ T cell phenotypes have been recognized that are specific for virulence factors of keystone pathogens such as P. gingivalis [100,101]. Also critical to the understanding of the pathogenesis of periodontal disease, the epithelial innate immune cells such as DCs and Langerhans cells appear to drive differentiation of Th phenotypes against keystone pathogens [102].
Polymicrobial synergy and dysbiosis hypothesis
Periodontal disease is a polymicrobial infection. It is likely that members of the microbiome show consortial behavior to initiate and cause progression of periodontitis. For example, conventional rodents (not germ-free) develop inflammation and periodontal bone destruction when silk ligatures are placed around their teeth [103]. In contrast, germ-free rodents are not susceptible to periodontal destruction even when placing silk ligature around teeth [103]. In rodents and humans, periodontitis appears to be a disease in which host immunity activates by outgrowth of multiple bacterial species within the biofilm community. Although the direct pathogenic effects of the bacteria remain unclear in vivo, evidence suggests that collateral damage from the host response to the infection ultimately destroys the periodontal structures that support the tooth.
To explain the occurrence of periodontitis without one or more emergent pathogens, the dysbiosis hypothesis has been recently proposed [104]. The dysbiosis hypothesis maintains that the transition from periodontal health to disease reflects changes in abundance of low-abundance species in the bacterial community of the periodontal pocket. This shift in the composition of the microbial community leads to alterations in the host-microbe crosstalk sufficient to mediate destructive inflammation and bone loss [74] (Fig. 2). One microorganism potentially responsible for the initiation of the dysbiosis phenomenon is the Gram-negative asaccharolytic bacterium P. gingivalis. P. gingivalis appears to thrive at low frequency within the microbial community [69,80], but is consistently associated with the onset of periodontal disease, progression, and treatment failure [76,105].
As a minor member of the plaque biofilm but nonetheless a keystone pathogen, the virulence factors of P. gingivalis appear to manipulate and depress the host response [74] rather than induce inflammation and bone destruction [106]. Monocolonization of mice with P. gingivalis does not induce periodontal bone destruction after silk ligatures were placed on the teeth unless the commensal microbiota was present [107]. P. gingivalis, therefore, appeared necessary but not sufficient to induce bone destruction in a murine model of periodontal disease [104].
Indeed, P. gingivalis impairs host defenses in ways that facilitate the growth and development of the entire microbial community. The ability of P. gingivalis to modify the nutritional foundation for the microbial community promotes significant shifts in the composition of the community. These features define P. gingivalis as a keystone pathogen, a microorganism that can change the environment to alter proportions of other microorganisms within the ecological niche. The consequent disruption of the proportional relationship between strict symbionts and pathobionts triggers the destructive cascade leading to activation of inflammation and subsequent bone destruction [108] (Fig. 2).
In general, the diversity and richness of the plaque microbiome associated with periodontal disease is greater than in health. Whereas studies of the oral microbiome must be interpreted in view of the technical sampling challenges, the increased diversity in disease reflects the etiology. In contrast to the gastrointestinal microbiota, for example, in which a singular pathogen emerges in a microbiota of less complexity, the microbiota in periodontal disease shifts to include a higher proportion of pathobionts and keystone pathogens. No singular pathogen emerges and the disease-associated microbiome is of greater complexity than in health (Fig. 10).
Virulence strategies of P. gingivalis
An asaccharolytic microorganism, P. gingivalis expresses a set of proteases that degrade proteins to use as an energy source and also potentiate inflammation. The activation of inflammation is part of a basic survival strategy increasing the protein-rich gingival crevicular fluid bathing the gingival sulcus while dampening killing mechanisms in phagocytic cells [109]. P. gingivalis produces Lys-and Arg-proteases (Kgp, RgpA and RgpB gingipains), which can activate complement C1q independently of antibody [110] and use the C5 convertase-like enzymatic activity of RgpA and RgpB to generate C5a anaphylotoxin [111]. Interfering with complement-toll-like receptor cross talk and phagocytosis facilitates survival of P. gingivalis whereas the products of protein hydrolysis provide a nutrient source in the complex antimicrobial environment of the gingival sulcus.
P. gingivalis lipoprotein is another important virulence factor. TLR2 recognition of the lipoprotein induces a weak cAMP response. Activation of either CXCR4 or C5aR fails to induce cAMP. Strikingly, however, cAMP production is synergistically increased when P. gingivalis-stimulated TLR2 cooperates with activated C5aR and CXCR4 in lipid rafts. Combined activation greatly increases cAMP-dependent protein kinase (PKA) signaling, inactivating glycogen synthase kinase-3β and impairing inducible nitric oxide synthase (iNOS)-dependent killing of bacteria in vitro and in vivo [112].
P. gingivalis also expresses an atypical lipopolysaccharide (LPS) (4-acyl monophosphate lipid A). In contrast to agonist E. coli LPS, the atypical P. gingivalis LPS functions as a TLR4 antagonist [113,114]. Using this immunological subterfuge, P. gingivalis further reduces TLR-dependent iNOS production and iNOS-dependent killing.
P. gingivalis employs other subterfuge mechanisms to internalize into phagocytic cells and yet be protected against killing mechanisms. Binding of P gingivalis fimbriae (FimA) to phagocyte CD14-TLR2/TLR1 elicit an inside-out signal, which promotes internalization of P. gingivalis. Normally committed to the uptake of apoptotic cells, the physiological role of CR3 is essentially hijacked to internalize P. gingivalis [115,116]. Binding of CR3 activates ERK1 and ERK2 signaling in human monocytes, thereby inhibiting TLR2-induced IL-12 production and possibly Th1 differentiation [117]. After phagocytosis, P. gingivalis can sequester from phagolysosomes by diverting into the autophagic pathway.
Other pathogenic microorganisms
Emerging in aggressive juvenile forms of periodontitis, Aggregatibacter actinomycetemcomitans (formerly Actinobacillus actinomycetemcomitans) and Treponema spp. are considered true pathogens. A. actinomycetemcomitans is always below the levels of detection in healthy sites whereas the Treponema spp. are also considered pathobionts. Recently, a murine commensal microorganism similar to A. actinomycetemcomitans (NI1060, a Gram-negative member of the Pasteurellaceae family) has been associated with severe alveolar bone loss in a murine silk ligature model of periodontitis [118]. The bone destruction was dependent on activation of the nucleotide-binding oligomerization domain receptor 1 (NOD1). NOD1 is an innate immune receptor that mediates neutrophil recruitment by inducing the secretion of chemokines from non-hematopoietic cells. Mice lacking NOD1 show decreased chemokine CXCL1 secretion from the gingival epithelium in a ligature-induced model of periodontitis and decreased bone destruction when compared to the wild-type [118].
Several Treponema spp. appear consequent to P. gingivalis appearance and grow to high abundance in diseased periodontal pockets. Despite a rich diversity of phylotypes in healthy sites, only a small group of Treponema phylotypes show stronger association with diseased sites [119] fulfilling the characteristics of a pathobiont rather than a keystone pathogen.
Interestingly, as bacterial biomass and clinical periodontal inflammation increase, the ecological succession from health to disease is manifested as emergence of newly dominant community members rather than appearance of novel species [120]. Consequently, the emerging community may include members that can subvert or evade the immune response (e.g., keystone pathogens), thereby contributing to the stabilization of a disease-provoking biofilm dominated by pathobionts in individuals susceptible to periodontitis. Reflecting host genetic diversity and variability, susceptibility may be related to several immunoregulatory factors. For example, individuals of African descent show increased frequency and concurrent colonization with A. actinomycetemcomitans strain JP2 in association with aggressive and localized forms of periodontitis [121]. Interestingly, A. actinomycetemcomitans not only has a tropism and infects a certain host genetic background [122] but infection of the ecosystem generally follows a specific chronologic and site-specific pattern during the eruption of permanent incisors and first molars [123]. Hence, typically the incisor and molar teeth are selectively affected and A. actinomycetemcomitans is considered the causative agent of these aggressive forms of disease [124].
Challenges correlating shifts in the microbiota and disease progression
Despite very sophisticated efforts to analyze the periodontal microbiota at various stages of disease, knowing the microbial community that is predictive of disease progression is still not possible [125]. Indeed, the progression of periodontal disease is very difficult to measure accurately [126] unless the disease has destroyed at least 1 to 2 mm of connective tissue and bone support along the root length [127]. Despite the sophisticated methods to define the composition of the microbial community, we still measure periodontal disease crudely, with a metal pin known as the periodontal probe [127]. It will be extremely challenging, therefore, to predict the microbial community members that elicit tissue destruction, not because the specific species cannot be identified but because of the lack of sensitivity of the method to measure progression of periodontal disease.
Caries microbiome and immunity
Dental caries is the single most common disease in childhood with a prevalence rate five times higher than the next most prevalent disease, asthma. Over 50% of children in the United States ages 5–9 years have at least one cavity or filling. This number increases to 78% by the time children reach adulthood (17 years of age). The condition significantly contributes to the burden of pain, is associated with impaired development and marked decrease in quality of life [128,129]. For decades since the 1950s, Streptococcus mutans was held to be the etiological agent in dental caries. More recent attempts to define the specific etiological agent(s) of dental caries have proven to be elusive. Caries etiology is more complex and multi-faceted than previously recognized.
Caries active and caries-free individuals share approximately 50% of the supragingival microbiome [130]. Only 10 genera were expressed in high abundance including Streptococcus spp, Veillonella spp and Actinomyces spp. S. mitis (25.5%) and S. sanguinis, (9.1%) were predominant. Streptococcus mutans (1.2%) was a comparatively minor constituent. When caries-active and caries-free children were compared, significant differences in the microbiomes were attributed to low abundance phylotypes, Megasphera, Oribacterium, Moryella and Corynebacterium. The great majority of represented phenotypes were similar in both caries-free and caries-active children but decreased in caries-active samples. Phylotypes overrepresented in caries-active subjects included S. sanguinis, S. mutans, S. sobrinus, S. mitis, S. intermedius, S. gordonii, S. parasanguinis, S. constellatus, S. cristatus, S. oralis, S. equi, S. dentirousetti and S. peroris. Whereas S. mutans displayed greatest differential abundance of the observed phylotypes, the spectrum of other overrepresented bacteria suggests that a S. mutans etiology is ambiguous in dental caries.
Caries microbiota evolves with age and during disease progression
In predentate and dentate infants and adult mothers, the predominant bacterial phyla in saliva common to all are Firmicutes, Proteobacteria, Actinobacteria, and Fusobacteria [131]. The diversity of genera in the adult was greater than in the infant. Streptococcus spp. are the exception, predominating in infant saliva as 60% of clones while in the adult mother represent only 20%. The Veillonella, Neisseria, Rothia, Haemophilus, Gemella, Granulicatella, Leptotrichia, and Fusobacterium are also the also predominant genera in the infant, while Haemophilus, Neisseria, Veillonella, Fusobacterium, Oribacterium, Rothia, Treponema, and Actinomyces are predominant in adults [131]. In some Chinese populations, infants may show greater diversity in salivary phylotypes than adults by the age of 3 to 6 while maintaining proportions between different genera similar to other reports [132].
The buccal and approximal surfaces of 4-year-old children harbor caries-associated taxa Granulicatella elegans, Veillonella spp. as well as S. mutans and Bifidobacteriaceae spp. when analyzed with PCR. Alternatively, caries-free children harbor Capnocytophaga gingivalis, Abiotrophia defectiva, Lachnospiraceae spp, Streptococcus sanguinis and Streptococcus cristatus [133].
Environmental acidification is hypothesized to be the main driving force of the phenotypic and genotypic changes in the microbial community during the caries process. In dentin affected by severe caries common in children before the age of 16, acid-producing Lactobacillus spp. primarily L. gasseri-L. johnsonii and L casei-L. paracasei are dominant [134]. As caries progresses from initial demineralized lesions (white spots on the enamel) to deep cavitated lesions, levels of these species increase significantly. In recent studies S. mutans is often observed at high levels in the white spots but is also present in some healthy subjects in small numbers [134,135]. Surprisingly, S. mutans is only associated with caries initiation (white spots) but not with caries progression. S. mutans appears to have the characteristics of a keystone pathogen or of a pathobiont driven by a changing dietary environment. In some patients, Lactobacillus spp. and S. mutans are found at low levels or below detection suggesting that the initiation and progression of carious lesions cannot be attributed to S. mutans [134,135].
In white spots indicating early, demineralized enamel, other potential acid producers are observed at high levels including strains of Selenomonas, Neisseria, and S. mitis. Propionibacterium spp. are associated with caries progression but are not found at high levels. Attempts to discriminate the microbiome of caries-active and caries-free individuals at the specific ecological niche have proven technically difficult. The biomass available for analysis is limited at specific sites such as enamel pits and fissures or interproximal areas where the caries risk is the greatest (Fig. 8). Overall an initially diverse community in caries-free sites and in white spots appears to shift to progressive loss of families in caries-active sites. Species minimized in caries-active sites include Lachnospiraceae spp., the S. mitis-S. pneumoniae-S. infantis group, Corynebacterium matruchotii, S. gordonii, S. cristatus, Capnocytophaga gingivalis, Eubacterium IR009, and Campylobacter rectus [134].
Directly associated with caries formation, organic acids derived from the hydrolysis of disaccharides like sucrose, are the final metabolic products [136]. Environmental acidification of dental plaque influences enzyme activities and also regulates transcription and translation (acid-induced adaptation through induction of proteins/enzymes). An acidified environment causes a shift of the microbial composition (acid-induced selection of acidogenic and aciduric microorganisms). This cycle will continue as long as the environmental acidification persists [137]. To fully explain the cariogenic potential of the microbial community at different stages of the caries process, the metabolic activities relevant to environmental acidification of the bacteria must be understood. Therefore studies of the metabolome may be more relevant for explaining caries activity than studies focusing exclusively on the microbiome.
Collectively these data suggest that like the microbiome in periodontal disease, the plaque microbiota at the initiation of the carious lesions is of greater complexity than in health to then fall to a lower diversity in the established carious lesion. For decades, S. mutans was viewed as a singular pathogen in caries. Recent studies of the caries microbiome suggest a complex microbial etiology with emergent pathobionts. A bacterial disease like dental caries, which appears to be caused by a complex microbiota rather than a single pathogen, may be associated with a microbiota of greater complexity only at the initiation of disease. Unlike periodontal disease, established caries shows decreases in microbiome complexity probably due to acid environment, which limits the microbiota to acidogenic and aciduric microorganisms.
Immunity against caries
Individuals with low or non-detectable levels of Mutans streptococci early in life remain caries-free into adulthood as reported in cross-sectional and longitudinal studies in Sweden [138]. Caries susceptibility is inversely related to the output of salivary IgA in children and young adults [139]. Indeed, salivary IgA antibody titers against S. mutans are inversely related to levels of early oral colonization and the colonized individual’s caries experience; the mechanistic immunogenic target of these relationships are the bacterial adhesins, glucosyltransferases (GTF), and glucan-binding proteins (GBP) [140,141]. These antibody specificities are not unexpected, given the life-long presence of mutans streptococci in the oral biofilm. IgG antibody to mutans streptococci can be detected in infant sera as a consequence of placental transfer and specific IgA is found in colostrum and breast milk, reflecting maternal experience with these microorganisms [142].
Early in childhood, children begin to synthesize serum IgG antibody to Mutans streptococcal antigens followed in time by production of IgA. Serum IgG antibody levels increase during childhood and remain detectable throughout life. In young adults, anti-S. mutans IgG titers are inversely related to disease levels [143]. In older adults, serum IgG antibody to cariogenic streptococci is directly related to cumulative dental caries experience, whereas IgA levels were inversely related [144]. This reciprocal relationship between IgA and IgG responses throughout life indicates that the initial adaptive immune responses to mutans streptococcal antigens may influence the time and rate at which these streptococci join the biofilms of the primary dentition.
The level and specificity of the immune response may also affect the ability of commensal Mutans streptococci to colonize newly erupting primary and permanent teeth. For example, salivary IgAs against Mutans streptococci in caries-active children react to different epitopes when compared to anti-S. mutans salivary IgA in caries-free individuals [145]. Secretory IgA antibody from parotid gland or serum IgG derived from the gingival crevicular fluid may influence the accumulation of a cariogenic microbiota at various stages of infection [146]. Caries and the associated microflora likely increase the antigenic load and stimulate the immune responses. Yet the antigenic load and history disease in adulthood may tell us little about the impact of the immune response on the establishment of cariogenic oral microorganisms and the clinical course of disease. In the presence of a seemingly protective immune response, environmental challenges like an increase in dietary sugar may cause an increase in production in bacterial acids and mark progression of caries (reviewed in [147]. After eruption of primary teeth, the formation of the initial oral biofilm may reflect the immune responses, but the caries microbiome and clinical caries involve relationships that remain to be characterized with complex environmental factors.
Caries Vaccine
Active and passive immunization strategies have been pursued to deal with infections leading to dental caries. Several acidogenic microorganisms have been associated with various stages of dental caries as we have seen, but S. mutans remains one of the species identified in white spots of initial enamel demineralization. Since S. mutans behaves as a keystone pathogen, it is a reasonable target for a caries vaccine. S. mutans, like other viridans streptococci, offer obvious cell wall and extracellular targets that are central to their attachment to the tooth and accumulation in the oral biofilm. Mutans streptococcal components that participate in adhesion (antigen I/II), glucan formation or binding, or cell wall synthesis, alone or in combination, have each been candidate immunogens of vaccine strategies to inhibit experimental dental caries formation in rats or mice [148]. The production of glucans and related dextrans stabilize the dental plaque microbial community. In animal models, therefore, glucosyltransferase (GTF), an enzyme produced by S. mutans or S. sobrinus that catalyzes the extracellular formation of α-1,3 and α-1,6-linked glucans from dietary sucrose, has been used to target adaptive immunity to explore the caries-protective effect of specific salivary IgA and IgG antibodies. Delivery of antigens with appropriate adjuvants via mucosal routes increases anti-GTF salivary (secretory) IgA and serum IgG levels and protects against experimental caries. Anti-caries protection in animals was obtained after immunization with intact GTF or glucan binding protein (GBP), their derived synthetic peptides, recombinant peptide-enzyme fusion proteins or, even DNA vaccines encoding one or more target antigens or their fragments [149,150]. Clearly, proof of principle exists for a dental caries vaccine.
Passive immune approaches have also been shown to be promising. Dietary supplements of polyclonal IgG or IgY antibody to GTF, GBP, or monoclonal or transgenic reagents that have specificity for other S. mutans cell surface antigens (Ag I/II), have each successfully reduced dental caries in experimental animals during infection with cariogenic streptococci [151]. Small-scale human clinical trials in which transgenic IgA/G antibody was administered in dental trays to adults over several weeks showed mixed success in preventing Mutans streptococcal recolonization of tooth surfaces after chlorhexidine treatment [152,153]. Passively administered antibody during the period of initial colonization with cariogenic streptococci in concert with adaptive immune approaches has been suggested to increase the effectiveness of protection. Effective methods of delivery of non-host-generated antibody need to be identified for human applications. Unfortunately, despite rising prevalence of caries in underserved children worldwide, the medical community is still reluctant to approve a vaccine for a normally non-life-threatening disease, and commercial entities are unwilling to invest because of the unfavorable risk-benefit ratio. The risk of mortality from the vaccine exceeds the reduction in morbidity represented by successful immunization of dental caries.
Acknowledgments
Research in the investigators’ labs is supported by NIH/NIDCR 1 R01 DE021206 (MCH) and R21 DE022858 (MC)
References
- 1.Liljemark WF, Bloomquist C. Human oral microbial ecology and dental caries and periodontal diseases. Critical reviews in oral biology and medicine : an official publication of the American Association of Oral Biologists. 1996;7:180–198. doi: 10.1177/10454411960070020601. [DOI] [PubMed] [Google Scholar]
- 2.Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. Journal of clinical microbiology. 2005;43:5721–5732. doi: 10.1128/JCM.43.11.5721-5732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner ACR, Yu W-H, et al. The Human Oral Microbiome. Journal of bacteriology. 2010;192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang L, Chen T, Izard J, Tanner AC, Wade WG, Paster BJ, et al. The Human Oral Microbiome Database: Updates and New Features. Journal of dental research. 2014:93. [Google Scholar]
- 5.Palmer RJ., Jr Composition and development of oral bacterial communities. Periodontol 2000. 2014;64:20–39. doi: 10.1111/j.1600-0757.2012.00453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffen AL, Beall CJ, Firestone ND, Gross EL, DiFranco JM, Hardman JH, et al. CORE: A Phylogenetically-Curated 16S rDNA Database of the Core Oral Microbiome. Plos One. 2011:6. doi: 10.1371/journal.pone.0019051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Acinas SG, Klepac-Ceraj V, Hunt DE, Pharino C, Ceraj I, Distel DL, et al. Fine-scale phylogenetic architecture of a complex bacterial community. Nature. 2004;430:551–554. doi: 10.1038/nature02649. [DOI] [PubMed] [Google Scholar]
- 8.Wade WG. The oral microbiome in health and disease. Pharmacological research : the official journal of the Italian Pharmacological Society. 2013;69:137–143. doi: 10.1016/j.phrs.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Adler CJ, Dobney K, Weyrich LS, Kaidonis J, Walker AW, Haak W, et al. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the Neolithic and Industrial revolutions. Nature Genetics. 2013;45:450–455. doi: 10.1038/ng.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warinner C, Rodrigues JF, Vyas R, Trachsel C, Shved N, Grossmann J, et al. Pathogens and host immunity in the ancient human oral cavity. Nat Genet. 2014;46:336–344. doi: 10.1038/ng.2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Contreras M, Costello EK, Hidalgo G, Magris M, Knight R, Dominguez-Bello MG. The bacterial microbiota in the oral mucosa of rural Amerindians. Microbiology-Sgm. 2010;156:3282–3287. doi: 10.1099/mic.0.043174-0. [DOI] [PubMed] [Google Scholar]
- 12.Russell AL, Consolazio CF, White CL. Periodontal Disease and Nutrition in Eskimo Scouts of the Alaska National Guard. Journal of dental research. 1961;40:604–613. [Google Scholar]
- 13.Kristoffersen T, Bang G. Periodontal disease and oral hygiene in an Alaskan Eskimo population. Journal of dental research. 1973;52:791–796. doi: 10.1177/00220345730520042401. [DOI] [PubMed] [Google Scholar]
- 14.Loe H, Anerud A, Boysen H, Morrison E. Natural history of periodontal disease in man. Rapid, moderate and no loss of attachment in Sri Lankan laborers 14 to 46 years of age. Journal of clinical periodontology. 1986;13:431–445. doi: 10.1111/j.1600-051x.1986.tb01487.x. [DOI] [PubMed] [Google Scholar]
- 15.Preus HR, Ånerud Å, Boysen H, Dunford RG, Zambon JJ, Löe H. The natural history of periodontal disease. Journal of clinical periodontology. 1995;22:674–678. doi: 10.1111/j.1600-051x.1995.tb00825.x. [DOI] [PubMed] [Google Scholar]
- 16.Cadotte MW, Dinnage R, Tilman D. Phylogenetic diversity promotes ecosystem stability. Ecology. 2012;93:S223–S233. [Google Scholar]
- 17.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the healthy “core microbiome” of oral microbial communities. BMC microbiology. 2009;9:259. doi: 10.1186/1471-2180-9-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mager DL, Ximenez-Fyvie LA, Haffajee AD, Socransky SS. Distribution of selected bacterial species on intraoral surfaces. Journal of clinical periodontology. 2003;30:644–654. doi: 10.1034/j.1600-051x.2003.00376.x. [DOI] [PubMed] [Google Scholar]
- 20.Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, et al. Pyrosequencing analysis of the oral microflora of healthy adults. Journal of dental research. 2008;87:1016–1020. doi: 10.1177/154405910808701104. [DOI] [PubMed] [Google Scholar]
- 21.Carpenter GH. The secretion, components, and properties of saliva. Annual review of food science and technology. 2013;4:267–276. doi: 10.1146/annurev-food-030212-182700. [DOI] [PubMed] [Google Scholar]
- 22.Wickstrom C, Herzberg MC, Beighton D, Svensater G. Proteolytic degradation of human salivary MUC5B by dental biofilms. Microbiology. 2009;155:2866–2872. doi: 10.1099/mic.0.030536-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong K, Herzberg MC. Streptococcus sanguis expresses a 150-kilodalton two-domain adhesin: characterization of several independent adhesin epitopes. Infection and immunity. 1997;65:3815–3821. doi: 10.1128/iai.65.9.3815-3821.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong K, Mailloux L, Herzberg MC. Salivary film expresses a complex, macromolecular binding site for Streptococcus sanguis. The Journal of biological chemistry. 2000;275:8970–8974. doi: 10.1074/jbc.275.12.8970. [DOI] [PubMed] [Google Scholar]
- 25.Gong K, Ouyang T, Herzberg MC. A streptococcal adhesion system for salivary pellicle and platelets. Infection and immunity. 1998;66:5388–5392. doi: 10.1128/iai.66.11.5388-5392.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Lei Y, Nobbs A, Khammanivong A, Herzberg MC. Inactivation of Streptococcus gordonii SspAB alters expression of multiple adhesin genes. Infection and immunity. 2005;73:3351–3357. doi: 10.1128/IAI.73.6.3351-3357.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudiger SG, Carlen A, Meurman JH, Kari K, Olsson J. Dental biofilms at healthy and inflamed gingival margins. Journal of clinical periodontology. 2002;29:524–530. doi: 10.1034/j.1600-051x.2002.290609.x. [DOI] [PubMed] [Google Scholar]
- 28.Brandtzaeg P. Secretory immunity with special reference to the oral cavity. Journal of oral microbiology. 2013:5. doi: 10.3402/jom.v5i0.20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Offner GD, Troxler RF. Heterogeneity of high-molecular-weight human salivary mucins. Advances in dental research. 2000;14:69–75. doi: 10.1177/08959374000140011101. [DOI] [PubMed] [Google Scholar]
- 30.Leito JT, Ligtenberg AJ, Nazmi K, de Blieck-Hogervorst JM, Veerman EC, Nieuw Amerongen AV. A common binding motif for various bacteria of the bacteria-binding peptide SRCRP2 of DMBT1/gp-340/salivary agglutinin. Biological chemistry. 2008;389:1193–1200. doi: 10.1515/BC.2008.135. [DOI] [PubMed] [Google Scholar]
- 31.Murakami J, Terao Y, Morisaki I, Hamada S, Kawabata S. Group A streptococcus adheres to pharyngeal epithelial cells with salivary proline-rich proteins via GrpE chaperone protein. The Journal of biological chemistry. 2012;287:22266–22275. doi: 10.1074/jbc.M112.350082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Esberg A, Lofgren-Burstrom A, Ohman U, Stromberg N. Host and bacterial phenotype variation in adhesion of Streptococcus mutans to matched human hosts. Infection and immunity. 2012;80:3869–3879. doi: 10.1128/IAI.00435-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siqueira WL, Custodio W, McDonald EE. New insights into the composition and functions of the acquired enamel pellicle. Journal of dental research. 2012;91:1110–1118. doi: 10.1177/0022034512462578. [DOI] [PubMed] [Google Scholar]
- 34.Iavarone F, Melis M, Platania G, Cabras T, Manconi B, Petruzzelli R, et al. Characterization of salivary proteins of schizophrenic and bipolar disorder patients by top-down proteomics. Journal of proteomics. 2014;103C:15–22. doi: 10.1016/j.jprot.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 35.Tsai H, Bobek LA. Studies of the mechanism of human salivary histatin-5 candidacidal activity with histatin-5 variants and azole-sensitive and -resistant Candida species. Antimicrobial agents and chemotherapy. 1997;41:2224–2228. doi: 10.1128/aac.41.10.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruissen AL, Groenink J, Helmerhorst EJ, Walgreen-Weterings E, Van’t Hof W, Veerman EC, et al. Effects of histatin 5 and derived peptides on Candida albicans. The Biochemical journal. 2001;356:361–368. doi: 10.1042/0264-6021:3560361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suomalainen K, Saxen L, Vilja P, Tenovuo J. Peroxidases, lactoferrin and lysozyme in peripheral blood neutrophils, gingival crevicular fluid and whole saliva of patients with localized juvenile periodontitis. Oral diseases. 1996;2:129–134. doi: 10.1111/j.1601-0825.1996.tb00213.x. [DOI] [PubMed] [Google Scholar]
- 38.Salazar MG, Jehmlich N, Murr A, Dhople VM, Holtfreter B, Hammer E, et al. Identification of periodontitis associated changes in the proteome of whole human saliva by mass spectrometric analysis. Journal of clinical periodontology. 2013;40:825–832. doi: 10.1111/jcpe.12130. [DOI] [PubMed] [Google Scholar]
- 39.Lis M, Bhatt S, Schoenly NE, Lee AY, Nislow C, Bobek LA. Chemical genomic screening of a Saccharomyces cerevisiae genomewide mutant collection reveals genes required for defense against four antimicrobial peptides derived from proteins found in human saliva. Antimicrobial agents and chemotherapy. 2013;57:840–847. doi: 10.1128/AAC.01439-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou M, Meng HX, Zhao YB, Chen ZB. Changes of four proinflammatory proteins in whole saliva during experimental gingivitis. The Chinese journal of dental research : the official journal of the Scientific Section of the Chinese Stomatological Association. 2012;15:121–127. [PubMed] [Google Scholar]
- 41.Kolenbrander PE, Palmer RJ, Periasamy S, Jakubovics NS. Oral multispecies biofilm development and the key role of cell-cell distance. Nat Rev Microbiol. 2010;8:471–480. doi: 10.1038/nrmicro2381. [DOI] [PubMed] [Google Scholar]
- 42.Ding AM, Palmer RJ, Jr, Cisar JO, Kolenbrander PE. Shear-enhanced oral microbial adhesion. Applied and environmental microbiology. 2010;76:1294–1297. doi: 10.1128/AEM.02083-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valm AM, Welch JLM, Rieken CW, Hasegawa Y, Sogin ML, Oldenbourg R, et al. Systems-level analysis of microbial community organization through combinatorial labeling and spectral imaging. Proceedings of the National Academy of Sciences. 2011;108:4152–4157. doi: 10.1073/pnas.1101134108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar PS, Brooker MR, Dowd SE, Camerlengo T. Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. Plos One. 2011;6:e20956. doi: 10.1371/journal.pone.0020956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simon-Soro A, Tomas I, Cabrera-Rubio R, Catalan MD, Nyvad B, Mira A. Microbial Geography of the Oral Cavity. Journal of dental research. 2013;92:616–621. doi: 10.1177/0022034513488119. [DOI] [PubMed] [Google Scholar]
- 46.Bosshardt DD, Lang NP. The Junctional Epithelium: from Health to Disease. Journal of dental research. 2005;84:9–20. doi: 10.1177/154405910508400102. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi Y, Matsunaga T, Yamamoto G, Nishii K, Usui M, Yamamoto M, et al. Comprehensive analysis of gene expression in the junctional epithelium by laser microdissection and microarray analysis. Journal of Periodontal Research. 2010;45:618–625. doi: 10.1111/j.1600-0765.2010.01276.x. [DOI] [PubMed] [Google Scholar]
- 48.Tonetti MS, Imboden MA, Gerber L, Lang NP, Laissue J, Mueller C. Localized expression of mRNA for phagocyte-specific chemotactic cytokines in human periodontal infections. Infection and immunity. 1994;62:4005–4014. doi: 10.1128/iai.62.9.4005-4014.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ertugrul AS, Sahin H, Dikilitas A, Alpaslan N, Bozoglan A. Comparison of CCL28, interleukin-8, interleukin-1beta and tumor necrosis factor-alpha in subjects with gingivitis, chronic periodontitis and generalized aggressive periodontitis. J Periodontal Res. 2013;48:44–51. doi: 10.1111/j.1600-0765.2012.01500.x. [DOI] [PubMed] [Google Scholar]
- 50.Gamonal J, Acevedo A, Bascones A, Jorge O, Silva A. Levels of interleukin-1 beta, -8, and -10 and RANTES in gingival crevicular fluid and cell populations in adult periodontitis patients and the effect of periodontal treatment. Journal of periodontology. 2000;71:1535–1545. doi: 10.1902/jop.2000.71.10.1535. [DOI] [PubMed] [Google Scholar]
- 51.Rindom Schiött C, Löe H. The origin and variation in number of leukocytes in the human saliva. Journal of Periodontal Research. 1970;5:36–41. doi: 10.1111/j.1600-0765.1970.tb01835.x. [DOI] [PubMed] [Google Scholar]
- 52.Dixon DR, Reife RA, Cebra JJ, Darveau RP. Commensal Bacteria Influence Innate Status Within Gingival Tissues: A Pilot Study. Journal of periodontology. 2004;75:1486–1492. doi: 10.1902/jop.2004.75.11.1486. [DOI] [PubMed] [Google Scholar]
- 53.Nishii K, Usui M, Yamamoto G, Yajima S, Tsukamoto Y, Tanaka J, et al. The distribution and expression of S100A8 and S100A9 in gingival epithelium of mice. J Periodontal Res. 2013;48:235–242. doi: 10.1111/jre.12000. [DOI] [PubMed] [Google Scholar]
- 54.Greer A, Zenobia C, Darveau RP. Defensins and LL-37: a review of function in the gingival epithelium. Periodontol 2000. 2013;63:67–79. doi: 10.1111/prd.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Champagne CME, Buchanan W, Reddy MS, Preisser JS, Beck JD, Offenbacher S. Potential for gingival crevice fluid measures as predictors of risk for periodontal diseases. Periodontology 2000. 2003;31:167–180. doi: 10.1034/j.1600-0757.2003.03110.x. [DOI] [PubMed] [Google Scholar]
- 56.Ross KF, Herzberg MC. Calprotectin expression by gingival epithelial cells. Infection and immunity. 2001;69:3248–3254. doi: 10.1128/IAI.69.5.3248-3254.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zou X, Sorenson BS, Ross KF, Herzberg MC. Augmentation of epithelial resistance to invading bacteria by using mRNA transfections. Infection and immunity. 2013;81:3975–3983. doi: 10.1128/IAI.00539-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science. 2008;319:962–965. doi: 10.1126/science.1152449. [DOI] [PubMed] [Google Scholar]
- 59.Palmer LJ, Cooper PR, Ling MR, Wright HJ, Huissoon A, Chapple IL. Hypochlorous acid regulates neutrophil extracellular trap release in humans. Clinical and experimental immunology. 2012;167:261–268. doi: 10.1111/j.1365-2249.2011.04518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Champaiboon C, Sappington KJ, Guenther BD, Ross KF, Herzberg MC. Calprotectin S100A9 calcium-binding loops I and II are essential for keratinocyte resistance to bacterial invasion. The Journal of biological chemistry. 2009;284:7078–7090. doi: 10.1074/jbc.M806605200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sorenson BS, Khammanivong A, Guenther BD, Ross KF, Herzberg MC. IL-1 receptor regulates S100A8/A9-dependent keratinocyte resistance to bacterial invasion. Mucosal immunology. 2012;5:66–75. doi: 10.1038/mi.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith QT, Harriman L, Au GS, Stoltenberg JL, Osborn JB, Aeppli DM, et al. Neutrophil elastase in crevicular fluid: comparison of a middle-aged general population with healthy and periodontitis groups. Journal of clinical periodontology. 1995;22:935–941. doi: 10.1111/j.1600-051x.1995.tb01798.x. [DOI] [PubMed] [Google Scholar]
- 63.Golub LM, Lee HM, Greenwald RA, Ryan ME, Sorsa T, Salo T, et al. A matrix metalloproteinase inhibitor reduces bone-type collagen degradation fragments and specific collagenases in gingival crevicular fluid during adult periodontitis. Inflammation research : official journal of the European Histamine Research Society [et al] 1997;46:310–319. doi: 10.1007/s000110050193. [DOI] [PubMed] [Google Scholar]
- 64.Hernandez M, Gamonal J, Tervahartiala T, Mantyla P, Rivera O, Dezerega A, et al. Associations between matrix metalloproteinase-8 and -14 and myeloperoxidase in gingival crevicular fluid from subjects with progressive chronic periodontitis: a longitudinal study. Journal of periodontology. 2010;81:1644–1652. doi: 10.1902/jop.2010.100196. [DOI] [PubMed] [Google Scholar]
- 65.Eskan MA, Jotwani R, Abe T, Chmelar J, Lim JH, Liang S, et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nature immunology. 2012;13:465–473. doi: 10.1038/ni.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eggert FM, Drewell L, Bigelow JA, Speck JE, Goldner M. The pH of gingival crevices and periodontal pockets in children, teenagers and adults. Archives of Oral Biology. 1991;36:233–238. doi: 10.1016/0003-9969(91)90091-8. [DOI] [PubMed] [Google Scholar]
- 67.Armitage GC. Clinical evaluation of periodontal diseases. Periodontol 2000. 1995;7:39–53. doi: 10.1111/j.1600-0757.1995.tb00035.x. [DOI] [PubMed] [Google Scholar]
- 68.Armitage GC. Periodontal diagnoses and classification of periodontal diseases. Periodontol 2000. 2004;34:9–21. doi: 10.1046/j.0906-6713.2002.003421.x. [DOI] [PubMed] [Google Scholar]
- 69.Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. The ISME journal. 2012;6:1176–1185. doi: 10.1038/ismej.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamanaka W, Takeshita T, Shibata Y, Matsuo K, Eshima N, Yokoyama T, et al. Compositional stability of a salivary bacterial population against supragingival microbiota shift following periodontal therapy. Plos One. 2012;7:e42806. doi: 10.1371/journal.pone.0042806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reinhardt RA, McDonald TL, Bolton RW, DuBois LM, Kaldahl WB. IgG subclasses in gingival crevicular fluid from active versus stable periodontal sites. Journal of periodontology. 1989;60:44–50. doi: 10.1902/jop.1989.60.1.44. [DOI] [PubMed] [Google Scholar]
- 72.Johnson V, Johnson BD, Sims TJ, Whitney CW, Moncla BJ, Engel LD, et al. Effects of treatment on antibody titer to Porphyromonas gingivalis in gingival crevicular fluid of patients with rapidly progressive periodontitis. Journal of periodontology. 1993;64:559–565. doi: 10.1902/jop.1993.64.6.559. [DOI] [PubMed] [Google Scholar]
- 73.Lamster IB, Celenti R, Ebersole JL. The relationship of serum IgG antibody titers to periodontal pathogens to indicators of the host response in crevicular fluid. Journal of clinical periodontology. 1990;17:419–425. doi: 10.1111/j.1600-051x.1990.tb02340.x. [DOI] [PubMed] [Google Scholar]
- 74.Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends in Immunology. 2014;35:3–11. doi: 10.1016/j.it.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang J, Qi J, Zhao H, He S, Zhang Y, Wei S, et al. Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease. Scientific Reports. 2013:3. doi: 10.1038/srep01843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL. Microbial complexes in subgingival plaque. Journal of clinical periodontology. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 77.Hinrichs JE, Wolff LF, Pihlstrom BL, Schaffer EM, Liljemark WF, Bandt CL. Effects of scaling and root planing on subgingival microbial proportions standardized in terms of their naturally occurring distribution. Journal of periodontology. 1985;56:187–194. doi: 10.1902/jop.1985.56.4.187. [DOI] [PubMed] [Google Scholar]
- 78.Kumar PS, Griffen AL, Barton JA, Paster BJ, Moeschberger ML, Leys EJ. New Bacterial Species Associated with Chronic Periodontitis. Journal of dental research. 2003;82:338–344. doi: 10.1177/154405910308200503. [DOI] [PubMed] [Google Scholar]
- 79.Kumar PS, Griffen AL, Moeschberger ML, Leys EJ. Identification of Candidate Periodontal Pathogens and Beneficial Species by Quantitative 16S Clonal Analysis. Journal of clinical microbiology. 2005;43:3944–3955. doi: 10.1128/JCM.43.8.3944-3955.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kumar PS, Leys EJ, Bryk JM, Martinez FJ, Moeschberger ML, Griffen AL. Changes in Periodontal Health Status Are Associated with Bacterial Community Shifts as Assessed by Quantitative 16S Cloning and Sequencing. Journal of clinical microbiology. 2006;44:3665–3673. doi: 10.1128/JCM.00317-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ge X, Rodriguez R, My T, Gunsolley J, Xu P. Oral Microbiome of Deep and Shallow Dental Pockets In Chronic Periodontitis. Plos One. 2013:8. doi: 10.1371/journal.pone.0065520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vartoukian SR, Palmer RM, Wade WG. Diversity and morphology of members of the phylum “synergistetes” in periodontal health and disease. Applied and environmental microbiology. 2009;75:3777–3786. doi: 10.1128/AEM.02763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hutter G, Schlagenhauf U, Valenza G, Horn M, Burgemeister S, Claus H, et al. Molecular analysis of bacteria in periodontitis: evaluation of clone libraries, novel phylotypes and putative pathogens. Microbiology. 2003;149:67–75. doi: 10.1099/mic.0.25791-0. [DOI] [PubMed] [Google Scholar]
- 84.Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001;183:3770–3783. doi: 10.1128/JB.183.12.3770-3783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feres M, Haffajee AD, Allard K, Som S, Goodson JM, Socransky SS. Antibiotic resistance of subgingival species during and after antibiotic therapy. Journal of clinical periodontology. 2002;29:724–735. doi: 10.1034/j.1600-051x.2002.290809.x. [DOI] [PubMed] [Google Scholar]
- 86.Nguyen-Hieu T, Khelaifia S, Aboudharam G, Drancourt M. Methanogenic archaea in subgingival sites: a review. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 2013;121:467–477. doi: 10.1111/apm.12015. [DOI] [PubMed] [Google Scholar]
- 87.Lepp PW, Brinig MM, Ouverney CC, Palm K, Armitage GC, Relman DA. Methanogenic Archaea and human periodontal disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6176–6181. doi: 10.1073/pnas.0308766101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mikx FH. Environmental effects on the growth and proteolysis of Treponema denticola ATCC 33520. Oral microbiology and immunology. 1997;12:249–253. doi: 10.1111/j.1399-302x.1997.tb00387.x. [DOI] [PubMed] [Google Scholar]
- 89.Vianna ME, Holtgraewe S, Seyfarth I, Conrads G, Horz HP. Quantitative Analysis of Three Hydrogenotrophic Microbial Groups, Methanogenic Archaea, Sulfate-Reducing Bacteria, and Acetogenic Bacteria, within Plaque Biofilms Associated with Human Periodontal Disease. Journal of bacteriology. 2008;190:3779–3785. doi: 10.1128/JB.01861-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Slots J. Human viruses in periodontitis. Periodontol 2000. 2010;53:89–110. doi: 10.1111/j.1600-0757.2009.00325.x. [DOI] [PubMed] [Google Scholar]
- 91.Zijnge V, van Leeuwen MB, Degener JE, Abbas F, Thurnheer T, Gmur R, et al. Oral biofilm architecture on natural teeth. Plos One. 2010;5:e9321. doi: 10.1371/journal.pone.0009321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nguyen-Hieu T. Microbial sampling process can change results of microbiological analysis in periodontitis diagnosis. A minireview. Journal of investigative and clinical dentistry. 2013;4:151–159. doi: 10.1111/jicd.12010. [DOI] [PubMed] [Google Scholar]
- 93.Dennison DK, Van Dyke TE. The acute inflammatory response and the role of phagocytic cells in periodontal health and disease. Periodontol 2000. 1997;14:54–78. doi: 10.1111/j.1600-0757.1997.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 94.Yamazaki K, Yoshie H, Seymour GJ. T cell regulation of the immune response to infection in periodontal diseases. Histol Histopathol. 2003;18:889–896. doi: 10.14670/HH-18.889. [DOI] [PubMed] [Google Scholar]
- 95.Berglundh T, Donati M. Aspects of adaptive host response in periodontitis. Journal of clinical periodontology. 2005;32 (Suppl 6):87–107. doi: 10.1111/j.1600-051X.2005.00820.x. [DOI] [PubMed] [Google Scholar]
- 96.Teng YT. The role of acquired immunity and periodontal disease progression. Critical reviews in oral biology and medicine : an official publication of the American Association of Oral Biologists. 2003;14:237–252. doi: 10.1177/154411130301400402. [DOI] [PubMed] [Google Scholar]
- 97.Baker PJ, Dixon M, Evans RT, Dufour L, Johnson E, Roopenian DC. CD4(+) T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infection and immunity. 1999;67:2804–2809. doi: 10.1128/iai.67.6.2804-2809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Baker PJ, Garneau J, Howe L, Roopenian DC. T-cell contributions to alveolar bone loss in response to oral infection with Porphyromonas gingivalis. Acta Odontol Scand. 2001;59:222–225. doi: 10.1080/00016350152509247. [DOI] [PubMed] [Google Scholar]
- 99.Takahashi K, Azuma T, Motohira H, Kinane DF, Kitetsu S. The potential role of interleukin-17 in the immunopathology of periodontal disease. Journal of clinical periodontology. 2005;32:369–374. doi: 10.1111/j.1600-051X.2005.00676.x. [DOI] [PubMed] [Google Scholar]
- 100.Bittner-Eddy PD, Fischer LA, Costalonga M. Identification of gingipain-specific I-A(b) -restricted CD4+ T cells following mucosal colonization with Porphyromonas gingivalis in C57BL/6 mice. Mol Oral Microbiol. 2013;28:452–466. doi: 10.1111/omi.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Costalonga M, Bittner-Eddy PD, Fischer L. Gingipain-specific Th17 do not Sustain Anti-Porphyromonas gingivalis IgGs. Journal of dental research. 2013;92:Abstract #2169. [Google Scholar]
- 102.Bittner-Eddy PD, Fischer L, Thieu K, Costalonga M. Langerhans cells drive gingipain-specific Th17 differentiation in murine periodontitis. Journal of dental research. 2014;93:Abstract #43. [Google Scholar]
- 103.Rovin S, Costich ER, Gordon HA. The influence of bacteria and irritation in the initiation of periodontal disease in germfree and conventional rats. Journal of Periodontal Research. 1966;1:193–203. doi: 10.1111/j.1600-0765.1966.tb01860.x. [DOI] [PubMed] [Google Scholar]
- 104.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-Abundance Biofilm Species Orchestrates Inflammatory Periodontal Disease through the Commensal Microbiota and Complement. Cell Host & Microbe. 2011;10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Colombo APV, Bennet S, Cotton SL, Goodson JM, Kent R, Haffajee AD, et al. Impact of Periodontal Therapy on the Subgingival Microbiota of Severe Periodontitis: Comparison Between Good Responders and Individuals With Refractory Periodontitis Using the Human Oral Microbe Identification Microarray. Journal of periodontology. 2012;83:1279–1287. doi: 10.1902/jop.2012.110566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Darveau RP, Hajishengallis G, Curtis MA. Porphyromonas gingivalis as a Potential Community Activist for Disease. Journal of dental research. 2012;91:816–820. doi: 10.1177/0022034512453589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hajishengallis G, Lambris JD. Microbial manipulation of receptor crosstalk in innate immunity. Nature reviews Immunology. 2011;11:187–200. doi: 10.1038/nri2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol. 2012;27:409–419. doi: 10.1111/j.2041-1014.2012.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, et al. The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol. 2011;186:869–877. doi: 10.4049/jimmunol.1003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Popadiak K, Potempa J, Riesbeck K, Blom AM. Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J Immunol. 2007;178:7242–7250. doi: 10.4049/jimmunol.178.11.7242. [DOI] [PubMed] [Google Scholar]
- 111.Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P, et al. Microbial hijacking of complement-toll-like receptor crosstalk. Science signaling. 2010;3:ra11. doi: 10.1126/scisignal.2000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hajishengallis G, Wang M, Liang S, Triantafilou M, Triantafilou K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13532–13537. doi: 10.1073/pnas.0803852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Reife RA, Coats SR, Al-Qutub M, Dixon DM, Braham PA, Billharz RJ, et al. Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: differential activities of tetra- and penta-acylated lipid A structures on E-selectin expression and TLR4 recognition. Cellular microbiology. 2006;8:857–868. doi: 10.1111/j.1462-5822.2005.00672.x. [DOI] [PubMed] [Google Scholar]
- 114.Coats SR, Reife RA, Bainbridge BW, Pham TT, Darveau RP. Porphyromonas gingivalis lipopolysaccharide antagonizes Escherichia coli lipopolysaccharide at toll-like receptor 4 in human endothelial cells. Infection and immunity. 2003;71:6799–6807. doi: 10.1128/IAI.71.12.6799-6807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Harokopakis E, Hajishengallis G. Integrin activation by bacterial fimbriae through a pathway involving CD14, Toll-like receptor 2, and phosphatidylinositol-3-kinase. European journal of immunology. 2005;35:1201–1210. doi: 10.1002/eji.200425883. [DOI] [PubMed] [Google Scholar]
- 116.Sendide K, Reiner NE, Lee JS, Bourgoin S, Talal A, Hmama Z. Cross-talk between CD14 and complement receptor 3 promotes phagocytosis of mycobacteria: regulation by phosphatidylinositol 3-kinase and cytohesin-1. J Immunol. 2005;174:4210–4219. doi: 10.4049/jimmunol.174.7.4210. [DOI] [PubMed] [Google Scholar]
- 117.Hajishengallis G, Shakhatreh MA, Wang M, Liang S. Complement receptor 3 blockade promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J Immunol. 2007;179:2359–2367. doi: 10.4049/jimmunol.179.4.2359. [DOI] [PubMed] [Google Scholar]
- 118.Jiao Y, Darzi Y, Tawaratsumida K, Marchesan JT, Hasegawa M, Moon H, et al. Induction of bone loss by pathobiont-mediated Nod1 signaling in the oral cavity. Cell Host Microbe. 2013;13:595–601. doi: 10.1016/j.chom.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.You M, Mo S, Leung WK, Watt RM. Comparative analysis of oral treponemes associated with periodontal health and disease. Bmc Infectious Diseases. 2013:13. doi: 10.1186/1471-2334-13-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, et al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. Isme Journal. 2013;7:1016–1025. doi: 10.1038/ismej.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Haubek D. The highly leukotoxic JP2 clone of Aggregatibacter actinomycetemcomitans: evolutionary aspects, epidemiology and etiological role in aggressive periodontitis. APMIS Supplementum. 2010:1–53. doi: 10.1111/j.1600-0463.2010.02665.x. [DOI] [PubMed] [Google Scholar]
- 122.Meng H, Xu L, Li Q, Han J, Zhao Y. Determinants of host susceptibility in aggressive periodontitis. Periodontol 2000. 2007;43:133–159. doi: 10.1111/j.1600-0757.2006.00204.x. [DOI] [PubMed] [Google Scholar]
- 123.Haubek D, Ennibi OK, Poulsen K, Vaeth M, Poulsen S, Kilian M. Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans in Morocco: a prospective longitudinal cohort study. Lancet. 2008;371:237–242. doi: 10.1016/S0140-6736(08)60135-X. [DOI] [PubMed] [Google Scholar]
- 124.Fine DH, Markowitz K, Furgang D, Fairlie K, Ferrandiz J, Nasri C, et al. Aggregatibacter actinomycetemcomitans and Its Relationship to Initiation of Localized Aggressive Periodontitis: Longitudinal Cohort Study of Initially Healthy Adolescents. Journal of clinical microbiology. 2007;45:3859–3869. doi: 10.1128/JCM.00653-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Teles R, Teles F, Frias-Lopez J, Paster B, Haffajee A. Lessons learned and unlearned in periodontal microbiology. Periodontology 2000. 2013;62:95–162. doi: 10.1111/prd.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Michalowicz BS, Hodges JS, Pihlstrom BL. Is change in probing depth a reliable predictor of change in clinical attachment loss? J Am Dent Assoc. 2013;144:171–178. doi: 10.14219/jada.archive.2013.0096. [DOI] [PubMed] [Google Scholar]
- 127.Pihlstrom BL. Measurement of attachment level in clinical trials: probing methods. Journal of periodontology. 1992;63:1072–1077. doi: 10.1902/jop.1992.63.12s.1072. [DOI] [PubMed] [Google Scholar]
- 128.Acs G, Shulman R, Ng MW, Chussid S. The effect of dental rehabilitation on the body weight of children with early childhood caries. Pediatric dentistry. 1999;21:109–113. [PubMed] [Google Scholar]
- 129.Low W, Tan S, Schwartz S. The effect of severe caries on the quality of life in young children. Pediatric dentistry. 1999;21:325–326. [PubMed] [Google Scholar]
- 130.Peterson SN, Snesrud E, Liu J, Ong AC, Kilian M, Schork NJ, et al. The Dental Plaque Microbiome in Health and Disease. Plos One. 2013;8:e58487. doi: 10.1371/journal.pone.0058487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cephas KD, Kim J, Mathai RA, Barry KA, Dowd SE, Meline BS, et al. Comparative Analysis of Salivary Bacterial Microbiome Diversity in Edentulous Infants and Their Mothers or Primary Care Givers Using Pyrosequencing. Plos One. 2011:6. doi: 10.1371/journal.pone.0023503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ling Z, Liu X, Wang Y, Li L, Xiang C. Pyrosequencing Analysis of the Salivary Microbiota of Healthy Chinese Children and Adults. Microbial Ecology. 2013;65:487–495. doi: 10.1007/s00248-012-0123-x. [DOI] [PubMed] [Google Scholar]
- 133.Kanasi E, Dewhirst FE, Chalmers NI, Kent R, Jr, Moore A, Hughes CV, et al. Clonal Analysis of the Microbiota of Severe Early Childhood Caries. Caries Research. 2010;44:485–497. doi: 10.1159/000320158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gross EL, Leys EJ, Gasparovich SR, Firestone ND, Schwartzbaum JA, Janies DA, et al. Bacterial 16S Sequence Analysis of Severe Caries in Young Permanent Teeth. Journal of clinical microbiology. 2010;48:4121–4128. doi: 10.1128/JCM.01232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Belda-Ferre P, Alcaraz LD, Cabrera-Rubio R, Romero H, Simon-Soro A, Pignatelli M, et al. The oral metagenome in health and disease. Isme Journal. 2012;6:46–56. doi: 10.1038/ismej.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Takahashi N, Washio J, Mayanagi G. Metabolomic approach to oral biofilm characterization—A future direction of biofilm research. Journal of Oral Biosciences. 2012;54:138–143. [Google Scholar]
- 137.Takahashi N, Nyvad B. The role of bacteria in the caries process: ecological perspectives. Journal of dental research. 2011;90:294–303. doi: 10.1177/0022034510379602. [DOI] [PubMed] [Google Scholar]
- 138.Axelsson P. The effect of a needs-related caries preventive program in children and young adults - results after 20 years. BMC oral health. 2006;6 (Suppl 1):S7. doi: 10.1186/1472-6831-6-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Taubman MA, Smith DJ. Significance of salivary antibody in dental disease. Annals of the New York Academy of Sciences. 1993;694:202–215. doi: 10.1111/j.1749-6632.1993.tb18354.x. [DOI] [PubMed] [Google Scholar]
- 140.Nogueira RD, Alves AC, Napimoga MH, Smith DJ, Mattos-Graner RO. Characterization of salivary immunoglobulin A responses in children heavily exposed to the oral bacterium Streptococcus mutans: influence of specific antigen recognition in infection. Infection and immunity. 2005;73:5675–5684. doi: 10.1128/IAI.73.9.5675-5684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nogueira RD, Talarico Sesso ML, Loureiro Borges MC, Mattos-Graner RO, Smith DJ, Leme Ferriani VP. Salivary IgA antibody responses to Streptococcus mitis and Streptococcus mutans in preterm and fullterm newborn children. Archives of Oral Biology. 2012;57:647–653. doi: 10.1016/j.archoralbio.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 142.Luo Z, Smith DJ, Taubman MA, King WF. Cross-sectional analysis of serum antibody to oral streptococcal antigens in children. Journal of dental research. 1988;67:554–560. doi: 10.1177/00220345880670030601. [DOI] [PubMed] [Google Scholar]
- 143.Gregory RL, Filler SJ, Michalek SM, McGhee JR. Salivary immunoglobulin A and serum antibodies to Streptococcus mutans ribosomal preparations in dental caries-free and caries-susceptible human subjects. Infection and immunity. 1986;51:348–351. doi: 10.1128/iai.51.1.348-351.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kent R, Smith DJ, Joshipura K, Soparkar P, Taubman MA. Humoral IgG antibodies to oral microbiota in a population at risk for root-surface caries. Journal of dental research. 1992;71:1399–1407. doi: 10.1177/00220345920710070801. [DOI] [PubMed] [Google Scholar]
- 145.Bratthall D, Serinirach R, Hamberg K, Widerstrom L. Immunoglobulin A reaction to oral streptococci in saliva of subjects with different combinations of caries and levels of mutans streptococci. Oral microbiology and immunology. 1997;12:212–218. doi: 10.1111/j.1399-302x.1997.tb00381.x. [DOI] [PubMed] [Google Scholar]
- 146.Smith DJ, van Houte J, Kent R, Taubman MA. Effect of antibody in gingival crevicular fluid on early colonization of exposed root surfaces by mutans streptococci. Oral microbiology and immunology. 1994;9:65–69. doi: 10.1111/j.1399-302x.1994.tb00036.x. [DOI] [PubMed] [Google Scholar]
- 147.Smith DJ, Mattos-Graner RO. Secretory immunity following mutans streptococcal infection or immunization. Curr Top Microbiol Immunol. 2008;319:131–156. doi: 10.1007/978-3-540-73900-5_6. [DOI] [PubMed] [Google Scholar]
- 148.Dinis M, Tavares D, Veiga-Malta I, Fonseca AJMM, Andrade EB, Trigo G, et al. Oral Therapeutic Vaccination with Streptococcus sobrinus Recombinant Enolase Confers Protection against Dental Caries in Rats. Journal of Infectious Diseases. 2009;199:116–123. doi: 10.1086/594372. [DOI] [PubMed] [Google Scholar]
- 149.Smith DJ, King WF, Rivero J, Taubman MA. Immunological and protective effects of diepitopic subunit dental caries vaccines. Infection and immunity. 2005;73:2797–2804. doi: 10.1128/IAI.73.5.2797-2804.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Culshaw S, LaRosa K, Tolani H, Han X, Eastcott JW, Smith DJ, et al. Immunogenic and protective potential of mutans streptococcal glucosyltransferase peptide constructs selected by major histocompatibility complex class II allele binding. Infection and immunity. 2007;75:915–923. doi: 10.1128/IAI.01582-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Robinette RA, Oli MW, McArthur WP, Brady LJ. A therapeutic anti-Streptococcus mutans monoclonal antibody used in human passive protection trials influences the adaptive immune response. Vaccine. 2011;29:6292–6300. doi: 10.1016/j.vaccine.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ma JK, Hikmat BY, Wycoff K, Vine ND, Chargelegue D, Yu L, et al. Characterization of a recombinant plant monoclonal secretory antibody and preventive immunotherapy in humans. Nature medicine. 1998;4:601–606. doi: 10.1038/nm0598-601. [DOI] [PubMed] [Google Scholar]
- 153.Weintraub JA, Hilton JF, White JM, Hoover CI, Wycoff KL, Yu L, et al. Clinical trial of a plant-derived antibody on recolonization of mutans streptococci. Caries Res. 2005;39:241–250. doi: 10.1159/000084805. [DOI] [PubMed] [Google Scholar]
- 154.Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Systematic biology. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- 155.Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]