

Abstract
The chronic induction of inflammation underlies multiple pathological conditions, including metabolic, autoimmune disorders and cancer. The mitochondrial citrate carrier (CIC), encoded by the SLC25A1 gene, promotes the export of citrate from the mitochondria to the cytoplasm, a process that profoundly influences energy balance in the cells. We have previously shown that SLC25A1 is a target gene for lipopolysaccharide signaling and promotes the production of inflammatory mediators. We now demonstrate that SLC25A1 is induced at the transcriptional level by two key pro-inflammatory cytokines, tumor necrosis factor-α (TNFα) and interferon-γ (IFNγ), and such induction involves the activity of the nuclear factor kappa B and STAT1 transcription factors. By studying the down-stream events following SLC25A1 activation during signals that mimic inflammation, we demonstrate that CIC is required for regulating the levels of nitric oxide and of prostaglandins by TNFα or IFNγ. Importantly, we show that the citrate exported from mitochondria via CIC and its downstream metabolic intermediate, acetyl-coenzyme A, are necessary for TNFα or IFNγ to induce nitric oxide and prostaglandin production. These findings provide the first line of evidence that the citrate export pathway, via CIC, is central for cytokine-induced inflammatory signals and shed new light on the relationship between energy metabolism and inflammation.
Keywords: IFNγ, Mitochondrial citrate carrier, NFkB, Pro-inflammatory cytokine, STAT1, TNFα
1. Introduction
Tumor necrosis factor-α (TNFα) and interferon-γ (IFNγ) are endogenous pro-inflammatory cytokines produced by a variety of cells under various conditions of stress or cell damage. Abnormal or excessive cytokine activation is involved in various chronic inflammatory diseases such as Crohn's disease, rheumatoid arthritis, or asthma, as well as in cancer and in metabolic diseases [1,2]. Following tissue injury, the inflammatory response is primarily generated by activation of local macrophages and is subsequently amplified by migrating blood cells. Several key signal pathways are involved in this process, which induce dramatic changes in gene expression patterns in activated macrophages, in turn leading to the production of inflammatory molecules [3]. This process is energetically expensive, not only requiring the generation of a substantial amount of ATP but also involving a metabolic shift from oxidative phosphorylation to glycolysis [4]. As discussed in several recent reviews, this connection between metabolism and inflammation plays a role in various pathological conditions including autoimmune disorders, cancer, aging and cancer therapy [5].
Citrate is a key molecule for the generation of energy [6]. After its synthesis in the mitochondrion, citrate can have different fates supporting various pathways of energy production. In the mitochondrion citrate enters the Krebs cycle and promotes oxidative phosphorylation (OXPHOS), thus generating ATP. In the cytoplasm it is cleaved to acetyl-coenzyme A (acetyl-CoA), which is not only the precursor for fatty acid and sterol biosynthesis but also the universal donor for acetylation reactions [7]. A second metabolic product generated from the cleavage of citrate is oxaloacetate (OAA), which is reduced to malate and converted to pyruvate via malic enzyme in a reaction that generates cytosolic NADPH plus H+ (necessary for fatty acid and sterol synthesis) [6]. Thus, CIC plays a pivotal role in energy metabolism by regulating the amount of mitochondrial citrate and cytosolic citrate. We and others have also shown that CIC inhibition in the model organism zebrafish, or in children harboring germ line SLC25A1 mutations that inactivate the citrate export pathway, leads to severe mitochondrial dysfunction [8–10]. Therefore, CIC plays a key role in both the mitochondrial and cytoplasmic pathways of energy production.
The regulation of SLC25A1 expression by various signals is primarily achieved at the transcriptional level [11]. For example, fatty acids and insulin modulate hepatic SLC25A1 expression levels through the sterol response element-binding protein 1 (SREBP1) [12]. In addition, in the liver and pancreas, the SLC25A1 gene is also regulated by forkheadbox A1 (FOXA1) [13,14]. Interestingly, the human SLC25A1 proximal promoter encompasses multiple active Sp1 responsive elements, which can be targeted by epigenetic modifications [15]. Moreover, the repressor factor ZNF224 may play a role in controlling SLC25A1 gene expression during development [16]. This body of evidence clearly indicates the existence of a high extent of molecular plasticity and diversity in the signaling pathways that regulate SLC25A1 expression and activity.
There is currently little knowledge on the role played by the citrate export pathway in chronic inflammatory diseases and in the wider context of immune-metabolism. In this work we have investigated the role of CIC in inflammatory responses triggered by TNFα and IFNγ cytokines. We show that SLC25A1 gene expression is upregulated in TNFα- and IFNγ-induced macrophages and that CIC is required for important downstream events that follow cytokine stimulation. This role of CIC in pro-inflammatory cytokine signaling provides new insights on the relationship between energy metabolism and inflammation.
2. Materials and methods
2.1. Cell culture
Human monocytic/macrophage cells from histiocytoma, U937 cells (HTL 94002, Interlab Cell Line Collection, Genoa, Italy), were cultured in Roswell Park Memorial Institute 1640 (RPMI 1640) medium supplemented with 10% (v/v) fetal bovine serum, 2 mM l-glutamine, 100 U penicillin and 100 μg/ml streptomycin at 37 °C in 5% CO2 in a water-saturated atmosphere. U937 cells were differentiated with 10 ng/ml phorbol-12-myristate-13-acetate (PMA, Sigma-Aldrich, St. Louis, MO, USA).
2.2. Activating stimuli
U937/PMA (differentiated U937) cells were treated with 5 ng/ml TNFα (Sigma-Aldrich), 10 ng/ml IFNγ (ImmunoTools GmbH, Friesoythe, Germany) or combined IFNγ and TNFα in the presence or absence of 0.1 μg/ml cycloheximide. At various time periods after activation, U937/PMA cells were harvested and analyzed to detect CIC expression levels. Where indicated U937/PMA cells were treated with 20 μM IKK inhibitor VII (IKK VII, Millipore, Billerica, MA, USA), 10 μM nifuroxazide (NIFU, Sigma-Aldrich), 5 mM sodium acetate (Sigma-Aldrich) or 1 μM 4-chloro-3-{[(3-nitrophenyl)amino]sulfonyl}benzoic acid (CNFASB, Millipore), 1 h before stimulation with TNFα, IFNγ, or combined cytokines.
2.3. Cell viability
Cell viability was evaluated by a modified MTT assay (CellTiter 96® Non-Radioactive Cell Proliferation Assay, Promega, Madison, WI, USA), as described previously [17]. In brief, U937/PMA cells growing in 96-well plates were treated for 48 h with CNFASB (1, 5, 10, 20, 50 μM) or vehicle. The level of formazan product was determined by measuring its absorbance at 570 nm using a 96-well plate reader (GloMax, Promega).
2.4. Transient transfection
To measure SLC25A1 gene promoter activity, U937/PMA cells were transiently transfected as described previously [18] using 0.5 μg of pGL3 basic-LUC vector containing the −1785/−20 bp region of the SLC25A1 gene promoter. Twenty-four hours after transfection, U937/PMA cells were treated with TNFα or IFNγ in the presence or absence of IKK VII and NIFU, respectively. The day after, cells were lysed in the luciferase cell culture lysis buffer provided with the Luciferase Assay Kit (Promega) and 15 μl of supernatant was analyzed for firefly luciferase activity [19]. Luminescence was measured as relative light units, taking the reading of luciferase assay substrate alone and then with lysate in GloMax plate reader (Promega). The extent of transfection was normalized by β-galactosidase activity [20]. RNA interference experiments were performed as described previously [21]. After 24 h, small interfering RNA-transfected U937/PMA cells were treated with TNFα and IFNγ alone or in combination. Reactive oxygen species (ROS) and nitric oxide (NO) were measured 24 h after the addition of inducers.
2.5. Real-time PCR, SDS-PAGE and Western blotting
Total RNA was extracted and reverse transcribed as reported [22]. Real-time PCR was performed by using human SLC25A1 (Hs00761590) and human β-actin (4326315E) TaqMan® assays (Life Technologies). For immunoblot analysis, U937 cells were rinsed with ice-cold PBS and lysed as described previously [23]. Thirty micrograms of total proteins were heated at 100 °C for 5 min, separated on 4–12% SDS polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were then blocked for 1 h in a PBS solution containing 2% bovine serum albumin and 0.1% Tween 20, and then treated at room temperature with anti-CIC (specific for the C-terminus of the human mitochondrial CIC protein) or anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies. The immunoreaction was detected by Immobilon Western Chemiluminescent HRP (horseradish peroxidase) Substrate (Millipore).
2.6. Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) experiments were performed as previously reported [24] with slight modification. The chromatin was immunoprecipitated for 2 h at 4 °C using protein G MicroBeads (MACS Miltenyi Biotec, Bergisch Gladbach, Germany) on a rocking platform with p65- (sc-109 X, Santa Cruz Biotechnology) or STAT1- (sc-346 X, Santa Cruz Biotechnology) specific antibodies. Mock samples were prepared by the immunoprecipitation procedure without antibody. Input (total chromatin extract) and mock samples were recovered and then used for PCR analysis. Magnetically labeled protein/DNA complexes were retained and washed in μ columns (MACS Miltenyi). After reverse cross-linking, DNA was purified and analyzed by PCR using a forward primer (5′-ACAACCGGGAAAGGTGTG GG-3′) and a reverse primer (5′-CTCCGAGCAGTGGGTCAAGGT-3′) that amplify a 289 bp region (at about −400 bp) or a forward primer (5′-AGGTTCTCTGGCTGACCCCACGC-3′) and a reverse primer (5′-TTAGTAACTCTGCAGGTAGGGTG-3′) that amplify a 324 bp region (at about − 1300 bp) on the CIC gene promoter. The enriched DNA fragments in ChIP were quantified by real-time PCR (qPCR) using Fast SYBR Green Master Mix (Life Technologies). The data from each immunoprecipitate were normalized to the corresponding inputs of chromatin and expressed as relative to the control.
2.7. Quantification of citrate
Cytosolic citrate levels were determined with the citrate assay kit (Biovision, Mountain View, CA, USA) following the manufacturer's protocol. Briefly, triplicate samples of treated and untreated U937/PMA cells were collected by centrifugation at 4 °C and washed twice in chilled water. Cell lysis was carried out in hypotonic buffer (20 mM Tris–HCl, pH 7.4, 10 mM NaCl, 0.5% Nonidet P40, 3 mM MgCl2). Then, cytoplasmatic fractions were isolated by centrifugation at 4 °C for 10 min at 600 ×g. Cytosolic extracts, deproteinized in Amicon Ultra column (10 kDa cutoff), were analyzed for citrate levels.
2.8. NO, ROS and PGE2 detection
Nitrite formation was detected by using 1H-naphthotriazole from 2,3-diaminonaphthalene (DAN, Life Technologies) [25]. For ROS analysis, U937/PMA activated cells were incubated with 10 μM DCFH2-DA (Life Technologies) for 30 min. The fluorescence was revealed by GloMax plate reader (Promega). Prostaglandin (PGE2) was detected by the PGE2 Enzyme Immunoassay Kit (Arbor Assays, Ann Arbor, MI, USA) according to the manufacturer's instructions.
2.9. Statistical analysis
Statistical significance of difference was determined using oneway ANOVA. Data are expressed as means ± standard error and differences were considered as significant (P < 0.05; *); very significant (P < 0.01; **) and highly significant (P < 0.001; ***).
3. Results
3.1. The pro-inflammatory cytokines TNFα and IFNγ induce SLC25A1 gene expression levels
To shed light on the relationship between the mitochondrial citrate carrier (CIC), which is the only protein responsible for the efflux of citrate from the mitochondrial matrix to the cytosol [6], and inflammation we first investigated whether pro-inflammatory cytokines can modulate SLC25A1 gene expression. To this end, we employed the human differentiated U937 (U937/PMA) cells which provide an ideal model being representative of a large repertoire of macrophage functions [26,27]. Moreover, when appropriately stimulated with cytokines, U937 cells also mimic the inflammatory response of activated macrophages [28]. Thus, we induced an inflammatory response by treating U937 cells with TNFα and IFNγ either alone or in combination, and we subsequently performed time course experiments to analyze the SLC25A1 mRNA levels. In U937/PMA cells treated with TNFα alone, the levels of the SLC25A1 mRNA were unchanged for the first hours of treatment, began to slightly increase at nine hours, and reached a maximum peak at 16 h after treatment (Fig. 1A). At this time point a consistent 4-fold activation was seen followed by a decline of the SLC25A1 mRNA levels after 24 h of continuous TNFα treatment (Fig. 1A). When IFNγ was used as stimulus to trigger inflammation, an increase of SLC25A1 mRNA levels was also seen at nine hours after treatment (Fig. 1B). However and interestingly, the combination of TNFα and IFNγ changed the kinetics of SLC25A1 gene expression. Indeed, in this case we observed a first increase of the SLC25A1 mRNA earlier compared to treatment with either agents alone, specifically at one hour after stimulation, followed by a delayed but robust peak 16 h later (Fig. 1C). These experiments suggested that TNFα and IFNγ can induce SLC25A1 gene expression through distinct cellular signals.
Fig. 1.

Time course of CIC mRNA levels in activated macrophages. U937/PMA cells were treated with 5 ng/ml TNFα (A), 10 ng/ml IFNγ (B), a combination of 10 ng/ml IFNγ and 5 ng/ml TNFα (C) or vehicle for 1, 3, 9, 16, and 24 h in the presence (light gray bars) or absence (dark gray bars) of cycloheximide (CHX). Then, cells were lysed and used to quantify CIC mRNA by real-time PCR. CIC mRNA levels of induced cells at each time point were normalized against vehicle-treated controls. Real-time PCR values represent mean ± s.e.m. (N = 5). *** P < 0.001 versus time 0.
To better understand the mechanisms underlying CIC activation in cytokine-triggered inflammation, we next determined the time-dependent expression profile of SLC25A1 under the same conditions described above in the presence of cycloheximide, a potent inhibitor of protein synthesis [29]. As illustrated in Fig. 1A–B (light gray bars), the ability of either TNFα or IFNγ alone to upregulate SLC25A1 was abolished when U937/PMA cells were treated with cycloheximide. Of note, when macrophages were induced with a combination of TNFα and IFNγ, cycloheximide did not impair the early response of SLC25A1 gene expression (Fig. 1C, light gray bars). On the contrary, de novo protein synthesis was required for the late SLC25A1 activation during a costimulation with TNFα plus IFNγ (Fig. 1C, light gray bars).
These results show that the TNFα and IFNγ pro-inflammatory cytokines activate SLC25A1 gene expression in macrophages. They also indicate that TNFα and IFNγ, when co-produced during the inflammatory response, generate an early and late activation of SLC25A1. Furthermore, our findings show a role of the de novo protein synthesis in CIC gene upregulation during inflammation.
3.2. TNFα induces SLC25A1 expression via the transcription factor NFkB
It is well established that TNFα signaling acts through the nuclear factor kappa B (NFkB) [30]. We previously showed that the human SLC25A1 promoter contains two NFkB responsive elements at positions −414/−405 bp and −1314/−1305 bp [21], but it is still unknown whether these elements actually play a role in the regulation of SLC25A1 expression by cytokines. Therefore, we tested the hypothesis that TNFα induces activation of SLC25A1 via NFkB. Several approaches were employed to test this possibility. First, we used a well-known inhibitor of the IκB kinase, whose activity is required for NFkB function, specifically IKK inhibitor VII [31]. We found that the addition of inhibitor VII to U937/PMA cells co-treated with TNFα led to a reduction of both the SLC25A1 mRNA and protein levels compared to treatment with TNFα alone (Fig. 2A).
To verify that such differences were due to alterations in the transcription rate of the SLC25A1 promoter, we conducted luciferase assays by employing the previously described SLC25A1-promoter encompassing the −1785/−20 bp region of the SLC25A1 gene, cloned upstream of the luciferase reporter gene (SLC25A1pGL3) [12]. Fig. 2B shows that TNFα induces an increase of SLC25A1 promoter activity of more than 50%, which is completely abolished by co-treatment with IKK inhibitor VII. Finally, we performed ChIP assays to directly test the idea that NFkB binds to its responsive elements in the SLC25A1 gene promoter. We detected a strong binding activity of NFkB p65 subunit to the SLC25A1 gene promoter when macrophages were activated with TNFα (Figs. 2C and S1). This interaction, however, disappeared in the presence of IKK inhibitor VII (Figs. 2C and S1). The binding of p65 increased about 4-fold relative to input in TNFα-induced cells (Fig. 2D). IKK inhibitor VII abolished this effect when added to TNFα-induced cells (Fig. 2D). Similar results were obtained when a p50-specific antibody was used (data not shown). Thus, our experiments clearly demonstrate that TNFα induces SLC25A1 upregulation through NFkB.
Fig. 2.
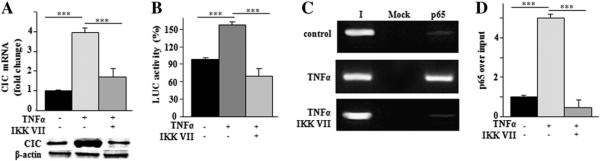
Molecular mechanism of TNFα-triggered SLC25A1 upregulation. (A) CIC mRNA (upper panel) and protein (lower panel) levels from U937/PMA untreated (−) or treated (+) with TNFα and incubated with (+) or without (−) IKK VII inhibitor. Real-time PCR values represent mean ± s.e.m. (N = 6). Western blot data are representative of three independent experiments with similar results. (B) U937/PMA cells transfected with the pGL3 basic-LUC vector containing the −1785/−20 bp region of the CIC gene promoter and incubated with (+) or without (−) IKK VII inhibitor were treated with TNFα and assayed for luciferase activity. Luciferase values represent mean ± s.e.m. (N = 5). (C) ChIP analysis was done with anti-p65 in TNFα-treated U937/PMA cells incubated as above. Lane Mock, PCR of immunoprecipitates without antibody; lane I, PCR of input DNA. (D) qPCR-ChIP was quantified by real-time PCR. Data are expressed as fold change versus control. qPCR values represent mean ± s.e.m. (N = 3).
3.3. IFNγ induces SLC25A1 transcriptional activation via STAT1
In response to type II interferon (IFNγ), the transcription factor STAT1 is phosphorylated and forms a homodimer. In this dimeric form STAT1 migrates into the nucleus and binds to the IFNγ-activated sequence (GAS) to drive the expression of target genes, thereby inducing a cellular inflammatory state [32]. Consistent with our findings that IFNγ activates SLC25A1 expression (Fig. 1), an in silico analysis of the human SLC25A1 gene promoter revealed indeed the presence of one GAS element at position −440/−450 bp. Therefore, we verified its involvement in IFNγ-mediated activation of SLC25A1. To this end, we treated U937/PMA cells with IFNγ in the presence or absence of nifuroxazide, a specific inhibitor of STAT signaling, and evaluated SLC25A1 promoter activity and SLC25A1 mRNA expression. Real-time PCR and Western-blot experiments showed a strong reduction of both CIC mRNA and protein levels in cells co-treated with nifuroxazide and IFNγ compared to cells treated with IFNγ alone (Fig. 3A). Moreover, we again performed luciferase reporter assays by employing the SLC25A1pGL3 vector in the presence or absence of nifuroxazide. As seen in Fig. 3B, SLC25A1 promoter activity increased in IFNγ-activated U937/PMA cells and such enhancement was reduced upon treatment with nifuroxazide. We also detected STAT1 binding to the GAS sequences of the SLC25A1 promoter in U937/PMA cells treated with IFNγ (Fig. 3C). Importantly, nifuroxazide abrogated STAT1 binding activity to the promoter, thus demonstrating the specificity of this interaction (Fig. 3C). STAT1 immunoprecipitation increased of about 1,2-fold relative to input in IFNγ-induced cells (Fig. 3D). This effect disappeared when nifuroxazide was added to IFNγ-induced cells (Fig. 3D). Collectively, these results demonstrate that IFNγ activates SLC25A1 gene expression in macrophages via STAT signaling.
Fig. 3.
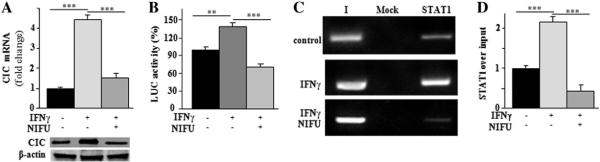
Molecular mechanism of IFNγ-triggered SLC25A1 upregulation. (A) CIC mRNA (upper panel) and protein (lower panel) levels from U937/PMA untreated (−) or treated (+) with IFNγ and incubated with (+) or without (−) nifuroxazide. Real-time PCR values represent mean ± s.e.m. (N = 6). Western blot data are representative of three independent experiments with similar results. (B) U937/PMA cells transfected with the pGL3 basic-LUC vector containing the −1785/−20 bp region of the CIC gene promoter and incubated with (+) or without (−) nifuroxazide were treated with IFNγ and assayed for luciferase activity. Luciferase values represent mean ± s.e.m. (N = 5). (C) ChIP analysis was done with anti-STAT1 in IFNγ-treated U937/PMA cells incubated as above. Lane Mock, PCR of immunoprecipitates without antibody; lane I, PCR of input DNA.(D) qPCR-ChIP was quantified by real-time PCR. Data are expressed as fold change versus control. qPCR values represent mean ± s.e.m. (N = 3).
3.4. Cytosolic citrate levels increase in U937/PMA cells stimulated by cytokines
To assess the significance of the CIC-dependent citrate export pathway, in the next set of experiments we monitored the cytosolic levels of citrate in U937/PMA cells treated with TNFα, IFNγ or both in the presence or absence of a CIC inhibitor, CNFASB [33]. In these experiments 1 μM CNFASB inhibitor was used, because this concentration had no influence on cell viability but was still able to inhibit CIC activity (Fig. S2). Interestingly, the amounts of citrate determined in the cytosol at different time periods parallel the time course of SLC25A1 mRNA presented in Fig. 1A–C. In fact, a significant increase in cytosolic citrate in TNFα- and IFNγ-exposed cells at 16 and 9 h, respectively, was found as compared to controls (Fig. 4A–B). Furthermore, when inflammation was triggered by both cytokines, two peaks of cytosolic citrate levels (one hour and 16 h after cytokine addition) were observed (Fig. 4C). In addition, the increase in cytosolic citrate levels induced by TNFα and/or IFNγ was prevented by the addition of CNFASB (Fig. 4A–C, light gray bars). These data prove that cytosolic citrate levels in cytokine triggered-inflammation are dependent on SLC25A1 upregulation.
Fig. 4.

Time course of cytosolic citrate levels in cytokine-induced U937/PMA cells. U937/PMA cells were treated with 5 ng/ml TNFα (A), 10 ng/ml IFNγ (B), a combination of 10 ng/ml IFNγ and 5 ng/ml TNFα (C) or vehicle for 0, 2, 6, 10, 16, and 24 h in the presence (light gray bars) or absence (dark gray bars) of 1 μM CNFASB. The amount of cytosolic citrate in each sample was normalized to cell number. Cytosolic citrate levels of induced cells at each time point were normalized against vehicle-treated controls. Values represent mean ± s.e.m. (N = 3). *** P < 0.001 versus time 0.
3.5. Effect of CIC inhibition on PGE2 production
In keeping with the above findings, in the next set of experiments we assessed the physiological significance of SLC25A1 upregulation in the inflammatory response induced by cytokines as well as the effect of the CIC inhibitor CNFASB on important downstream signaling events that follow macrophage activation. The inflammatory response in U937/PMA cells was triggered by treatment with TNFα and IFNγ employed alone or in combination. First we measured the secretion of PGE2, which is the principal mediator of inflammatory responses, after 24 h of cytokine treatment [34,35]. As shown in Fig. 5A–C, a marked reduction in PGE2 production was seen in cells treated with CNFASB compared to untreated cells.
Fig 5.
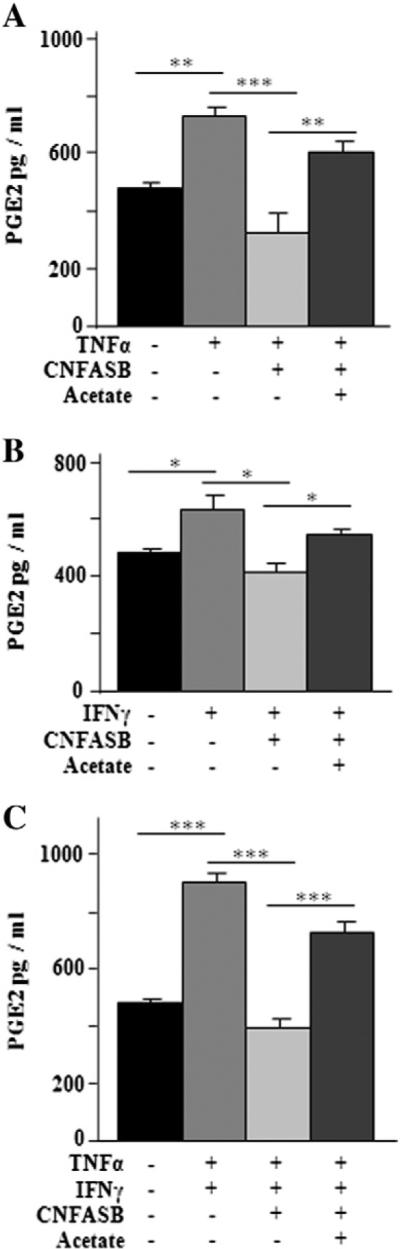
Effect of SLC25A1 inhibition on PGE2 production. U937/PMA cells untreated (black bar) or treated with TNFα (A), IFNγ (B), a combination of TNFα and IFNγ (C) in the presence (+) or absence (−) of 1 μM CNFASB or 5 mM sodium acetate were assayed for PGE2 secretion. Values represent mean ± s.e.m. (N = 5).
It is known that acetate is permeable through the plasma membrane and excess acetate can promote the synthesis of acetyl-CoA through the action of acetyl-CoA synthase [36,37]. Therefore, to more specifically probe the role of the citrate export pathway in PGE2 synthesis, we performed an additional set of experiments where cells treated with the CIC inhibitor CNFASB were exposed to supra-physiological levels of acetate. As shown in Fig. 5A–C, the addition of exogenous acetate nearly entirely prevented the inhibition of PGE2 synthesis by CNFASB. These results provide a strong mechanistic link between the CIC-dependent citrate export pathway, and consequent citrate-derived acetyl-CoA production, and PGE2 synthesis. They also imply that the citrate export activity of CIC is required for the production of key inflammatory molecules.
3.6. Effects of CIC inhibition on TNFα- and IFNγ-induced ROS production
To further explore CIC function in cytokine-activated macrophages we monitored the levels of inflammatory mediators closely associated with cellular energy state after 24 h of cytokine treatment. NO is synthesized from l-arginine in a reaction catalyzed by inducible nitric oxide synthase (iNOS). The conversion of l-arginine to NO and l-citrulline requires NADPH and O2 as substrates. Inhibition of CIC activity by CNFASB caused a marked reduction in NO levels in immune cells induced with both cytokines alone or together (Fig. 6A–C).
Fig. 6.

Analysis of NO and ROS production in CIC-inhibited macrophages. U937/PMA cells untreated (black bar) or treated with TNFα (A), IFNγ (B), a combination of TNFα and IFNγ (C) in the presence (+) or absence (−) of 1 μM CNFASB, were assayed for NO production. U937/PMA cells untreated (black bar) or treated with TNFα (D), IFNγ (E), a combination of TNFα and IFNγ(F) in the presence (+) or absence (−) of 1 μM CNFASB were assayed for ROS production. Values represent mean ± s.e.m. (N = 5).
Next, the effect of CNFASB on ROS production, which also requires NADPH, was tested. ROS are generated by the NADPH oxidase complex in the presence of molecular oxygen and NADPH [38]. While this reaction initiates a key step in immune defense response, overproduction of ROS, however, frequently due to excessive stimulation of NADPH oxidase by pro-inflammatory stimuli, results in oxidative stress [39,40].
As shown for NO, when U937/PMA cells were treated as above in the presence of both cytokines, the CNFASB inhibitor was able to reduce ROS to control levels (Fig. 6D–F). These observations imply that citrate transported by CIC is required for ROS production in cytokine-activated macrophages.
3.7. SLC25A1 knocking down and inflammatory mediators' production
In order to deepen the role of CIC in cytokine-triggered inflammation, we performed SLC25A1 gene silencing experiments by using the RNA interference approach. A specific small interfering RNA (siRNA) against SLC25A1 or a non targeting siRNA negative control was used in U937/PMA cells. The efficacy of siRNA in inducing SLC25A1 knock-down was confirmed by the significant decrease of CIC mRNA and protein levels (data not shown). In SLC25A1-silenced cells induced with TNFα and IFNγ, alone or in combination, there was a prominent decrease in PGE2, NO and ROS levels (Fig. 7A–C). These findings strengthen the effects of the CIC inhibitor CNFASB described above and highlight CIC ability to control the production of main inflammatory mediators.
Fig. 7.

Effect of SLC25A1 silencing on inflammatory response. U937/PMA cells untreated (black bar) or treated with TNFα, IFNγ or a combination of TNFα and IFNγ, and transfected with the siRNA targeting human CIC (+) or control siRNA (−) were assayed for PGE2 (A), NO (B) and ROS (C) production. Values represent mean ± s.e.m. (N = 5).
4. Discussion
In this study we demonstrate the importance of CIC, encoded by the SLC25A1 gene, and of the CIC-mediated citrate export pathway in cytokine-triggered inflammatory signals. Our findings suggest that the TNFα and IFNγ pro-inflammatory cytokines, alone or together, induce SLC25A1 gene expression and activity in macrophages through different molecular pathways. The prototypic pro-inflammatory cytokine TNFα exerts its effect on SLC25A1 gene expression through NFkB signaling. In support of this conclusion we have shown that NFkB binds to two consensus elements located in the SLC25A1 gene promoter in TNFα-activated macrophages. Moreover, TNFα induces a stark increase in SLC25A1 promoter activity, which is eliminated when NFkB is inhibited.
The STAT1 pathway is responsible for IFNγ-induced SLC25A1 upregulation as demonstrated by its binding to the GAS site of the SLC25A1 gene promoter in ChIP analysis when IFNγ triggers inflammation. We also report the specific involvement of STAT1 in SLC25A1 promoter activation given that when STAT1 signaling is blocked IFNγ is not able to exert its effect.
The early and late activations of SLC25A1 expression observed upon cell treatment with both TNFα and IFNγ cytokines warrant some comments. It can be hypothesized that the SLC25A1 rapid upregulation involves nuclear translocation of both STAT1 and NFkB transcription factors induced by the presence of both TNFα and IFNγ cytokines. This is consistent with the well-known synergic effect of TNFα and IFNγ cytokines by different mechanisms such as increase in NFkB nuclear translocation and STAT1-NFkB p65 subunit interaction [41,42].
Regarding the late de novo protein synthesis-dependent SLC25A1 upregulation, this finding might be accounted for by an increase in STAT1 and IFNγ expression levels upon IFNγ treatment as well as by an increase in TNFα expression via NFkB [41,43]. Secretion of newly synthesized IFNγ and TNFα would lead to an amplification of the proinflammatory stimuli and to the second phase of the cellular immune response. However, other factors such as turnover of IFNγ and TNFα receptors cannot be excluded in the de novo protein synthesis-dependent late SLC25A1 activation.
Based on the available data, CIC plays a key role in multiple aspects of metabolism. Thus, its upregulation in cytokine-triggered inflammation may be necessary to provide energy as well as the intermediate metabolites necessary for the metabolic switches needed for macrophage activation and/or for the inflammatory response. Consistently, both CIC inhibition and SLC25A1 gene silencing strongly reduce NO, PGE2, and ROS levels in TNFα- and IFNγ-induced macrophages. This result strongly implies that NADPH produced by oxaloacetate, one of the two products of cytosolic citrate cleavage, is an important cofactor in the generation of ROS (by NADPH-oxidase) and NO (by iNOS), whereas acetyl-CoA can be directly used for fatty acid biosynthesis, providing a precursor for PGE2 synthesis (depicted in Fig. 8). Taken together, these findings, provide the first line of evidence that the citrate export pathway, via CIC, is central for cytokine-induced inflammation and shed light on the relationship between energy metabolism and inflammation.
Fig. 8.

Proposed mechanism for mitochondria-derived citrate in cytokine-induced inflammation. Tumor necrosis factor-α (TNFα) induces CIC gene activation through NFkB signaling. Interferon-γ (IFNγ) acts through STAT transcription factors. Then, CIC activity supplies acetyl-CoA for prostaglandin (PGE2) synthesis and NADPH for reactive oxygen species (ROS) and nitric oxide (NO) production.
It is noteworthy that the TNFα pro-inflammatory cytokine is not usually detectable in healthy individuals, but elevated serum and tissue levels are found in inflammatory infectious conditions (lung diseases, rheumatoid arthritis, Crohn's disease) as well as in cancer [44]. While the biological effects of TNFα have been extensively explored in the context of inflammatory signal pathways, very little is known about the metabolic effects of this important molecule [44]. Our work, although performed only in culture cells, clearly demonstrates that CIC is essential for inflammatory responses in TNFα-induced macrophages, leading us to propose a central role for mitochondria-derived citrate in many inflammatory conditions. Once inflammation is initiated, IFNγ is produced and subsequently acts through various molecules and pathways of the immune system to amplify the inflammatory cascade [45]. Overwhelming evidence has implicated IFNγ to be crucially involved in various chronic inflammatory diseases, such as rheumatoid arthritis [46]. Herein we show that CIC activity is also engaged in IFNγ-triggered inflammation.
5. Conclusion
In conclusion, this work shows that 1) TNFα and IFNγ, alone or together, induce SLC25A1 gene expression and activity in U937 cells through different molecular pathways and different timing patterns; 2) the early activation of SLC25A1 expression (at 1 h) by TNFα and IFNγ together is de novo protein synthesis-independent, whereas the late activation (at 16 h) is de novo protein synthesis-dependent; 3) the biphasic cytosolic citrate level increase parallels the biphasic SLC25A1 upregulation; 4) silencing of SLC25A1 gene or inhibition of citrate efflux from mitochondria abolish NO, ROS and PGE2 production and 5) acetate, a citrate by-product, restores the increase of PGE2 when the efflux of citrate is inhibited. All together these data provide clear evidence for the existence of a “citrate pathway” required in pro-inflammatory signals and in inflammatory mediator production (Fig. 8). This study also highlights the important role of the intermediary metabolism in inflammation. In keeping with the deleterious role played by inflammation in disorders of the immune system and in cancer, CIC inhibition may offer new therapeutic strategies for the treatment of chronic inflammatory diseases and, as we previously showed [9], of cancer.
Supplementary Material
Acknowledgements
This work was supported by grants from the Ministero dell'Università e della Ricerca (MIUR) No. 20109Z2XRJ 009, the Comitato Telethon Fondazione Onlus No. GGP11139, the Center of Excellence in Genomics (CEGBA) and the Italian Human Proteome Net No. RBRN07BMCT_009.
Abbreviations
- acetyl-CoA
acetyl-coenzyme A
- ChIP
chromatin immunoprecipitation
- CHX
cycloheximide
- CIC
mitochondrial citrate carrier
- CNFASB
4-chloro-3-{[(3-nitrophenyl)amino]sulfonyl}benzoic acid
- DAN
2,3-diaminonaphthalene
- FOXA1
forkhead-box A1
- GAS
IFNγ-activated sequence
- IFNγ
interferon-γ
- NFkB
nuclear factor kappa B
- LUC
luciferase
- OAA
oxaloacetate
- OXPHOS
oxidative phosphorylation
- PMA
phorbol-12-myristate-13-acetate
- SREBP1
sterol response element-binding protein 1
- STAT1
signal transducer and activator of transcription 1
- TNFα
tumor necrosis factor-α
Footnotes
Author contribution
Vittoria Infantino, Vito Iacobazzi, Maria Laura Avantaggiati and Ferdinando Palmieri designed the research. Vittoria Infantino, Vito Iacobazzi and Alessio Menga performed the experiments. Vittoria Infantino, Vito Iacobazzi and Alessio Menga analyzed the data. Vittoria Infantino, Vito Iacobazzi, Maria Laura Avantaggiati and Ferdinando Palmieri wrote the paper.
Supplementary data to this article can be found online at http://dx. doi.org/10.1016/j.bbagrm.2014.07.013.
References
- 1.Roberts-Thomson IC, Fon J, Uylaki W, Cummins AG, Barry S. Cells, cytokines and inflammatory bowel disease: a clinical perspective, Expert Rev. Gastroenterol. Hepatol. 2011;5:703–716. doi: 10.1586/egh.11.74. [DOI] [PubMed] [Google Scholar]
- 2.Romas E, Gillespie MT, Martin TJ. Involvement of receptor activator of NFkappaB ligand and tumor necrosis factor-alpha in bone destruction in rheumatoid arthritis. Bone. 2002;30:340–346. doi: 10.1016/s8756-3282(01)00682-2. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 4.O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 5.Tornatore L, Thotakura AK, Bennett J, Moretti M, Franzoso G. The nuclear factor kappa B signaling pathway: integrating metabolism with inflammation. Trends Cell Biol. 2012;22:557–566. doi: 10.1016/j.tcb.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Iacobazzi V, Infantino V. Citrate — new functions for an old metabolite. Biol. Chem. 2014;395:387–399. doi: 10.1515/hsz-2013-0271. [DOI] [PubMed] [Google Scholar]
- 7.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edvardson S, Porcelli V, Jalas C, Soiferman D, Kellner Y, Shaag A, Korman SH, Pierri CL, Scarcia P, Fraenkel ND, Segel R, Schechter A, Frumkin A, Pines O, Saada A, Palmieri L, Elpeleg O. Agenesis of corpus callosum and optic nerve hypoplasia due to mutations in SLC25A1 encoding the mitochondrial citrate transporter. J. Med. Genet. 2013;50:240–245. doi: 10.1136/jmedgenet-2012-101485. [DOI] [PubMed] [Google Scholar]
- 9.Catalina-Rodriguez O, Kolukula VK, Tomita Y, Preet A, Palmieri F, Wellstein A, Byers S, Giaccia AJ, Glasgow E, Albanese C, Avantaggiati ML. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget. 2012;3:1220–1235. doi: 10.18632/oncotarget.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nota B, Struys EA, Pop A, Jansen EE, Fernandez Ojeda MR, Kanhai WA, Kranendijk M, van Dooren SJ, Bevova MR, Sistermans EA, Nieuwint AW, Barth M, Ben-Omran T, Hoffmann GF, de Lonlay P, McDonald MT, Meberg A, Muntau AC, Nuoffer JM, Parini R, Read MH, Renneberg A, Santer R, Strahleck T, van Schaftingen E, van der Knaap MS, Jakobs C, Salomons GS. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2013;92:627–631. doi: 10.1016/j.ajhg.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iacobazzi V, Infantino V, Palmieri F. Transcriptional regulation of the mitochondrial citrate and carnitine/acylcarnitine transporters: two genes involved in fatty acid biosynthesis and beta-oxidation. Biology. 2013;2:284–303. doi: 10.3390/biology2010284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Infantino V, Iacobazzi V, De Santis F, Mastrapasqua M, Palmieri F. Transcription of the mitochondrial citrate carrier gene: role of SREBP-1, upregulation by insulin and downregulation by PUFA. Biochem. Biophys. Res. Commun. 2007;356:249–254. doi: 10.1016/j.bbrc.2007.02.114. [DOI] [PubMed] [Google Scholar]
- 13.Iacobazzi V, Infantino V, Bisaccia F, Castegna A, Palmieri F. Role of FOXA in mitochondrial citrate carrier gene expression and insulin secretion. Biochem. Biophys. Res. Commun. 2009;385:220–224. doi: 10.1016/j.bbrc.2009.05.030. [DOI] [PubMed] [Google Scholar]
- 14.Menga A, Infantino V, Iacobazzi F, Convertini P, Palmieri F, Iacobazzi V. Insight into mechanism of in vitro insulin secretion increase induced by antipsychotic clozapine: role of FOXA1 and mitochondrial citrate carrier, Eur. Neuropsychopharmacol. J. Eur. Coll. Eur. Neuropsychopharmacol. 2013;23:978–987. doi: 10.1016/j.euroneuro.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 15.Iacobazzi V, Infantino V, Palmieri F. Epigenetic mechanisms and Sp1 regulate mitochondrial citrate carrier gene expression. Biochem. Biophys. Res. Commun. 2008;376:15–20. doi: 10.1016/j.bbrc.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 16.Iacobazzi V, Infantino V, Convertini P, Vozza A, Agrimi G, Palmieri F. Transcription of the mitochondrial citrate carrier gene: identification of a silencer and its binding protein ZNF224. Biochem. Biophys. Res. Commun. 2009;386:186–191. doi: 10.1016/j.bbrc.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Infantino V, Iacobazzi V, Palmieri F, Menga A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Biophys. Res. Commun. 2013;440:105–111. doi: 10.1016/j.bbrc.2013.09.037. [DOI] [PubMed] [Google Scholar]
- 18.Infantino V, Convertini P, Menga A, Iacobazzi V. MEF2C exon alpha: role in gene activation and differentiation. Gene. 2013;531:355–362. doi: 10.1016/j.gene.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 19.Laghezza A, Pochetti G, Lavecchia A, Fracchiolla G, Faliti S, Piemontese L, Di Giovanni C, Iacobazzi V, Infantino V, Montanari R, Capelli D, Tortorella P, Loiodice F. New 2-(aryloxy)-3-phenylpropanoic acids as peroxisome proliferator-activated receptor alpha/gamma dual agonists able to upregulate mitochondrial carnitine shuttle system gene expression. J. Med. Chem. 2013;56:60–72. doi: 10.1021/jm301018z. [DOI] [PubMed] [Google Scholar]
- 20.Iacobazzi V, Infantino V, Costanzo P, Izzo P, Palmieri F. Functional analysis of the promoter of the mitochondrial phosphate carrier human gene: identification of activator and repressor elements and their transcription factors. Biochem. J. 2005;391:613–621. doi: 10.1042/BJ20050776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, Palmieri F, Iacobazzi V. The mitochondrial citrate carrier: a new player in inflammation. Biochem. J. 2011;438:433–436. doi: 10.1042/BJ20111275. [DOI] [PubMed] [Google Scholar]
- 22.Infantino V, Castegna A, Iacobazzi F, Spera I, Scala I, Andria G, Iacobazzi V. Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down's syndrome. Mol. Genet. Metab. 2011;102:378–382. doi: 10.1016/j.ymgme.2010.11.166. [DOI] [PubMed] [Google Scholar]
- 23.Infantino V, Convertini P, Iacobazzi F, Pisano I, Scarcia P, Iacobazzi V. Identification of a novel Sp1 splice variant as a strong transcriptional activator. Biochem. Biophys. Res. Commun. 2011;412:86–91. doi: 10.1016/j.bbrc.2011.07.047. [DOI] [PubMed] [Google Scholar]
- 24.Convertini P, Infantino V, Bisaccia F, Palmieri F, Iacobazzi V. Role of FOXA and Sp1 in mitochondrial acylcarnitine carrier gene expression in different cell lines. Biochem. Biophys. Res. Commun. 2011;404:376–381. doi: 10.1016/j.bbrc.2010.11.126. [DOI] [PubMed] [Google Scholar]
- 25.Sala R, Rotoli BM, Colla E, Visigalli R, Parolari A, Bussolati O, Gazzola GC, Dall'Asta V. Two-way arginine transport in human endothelial cells: TNF-alpha stimulation is restricted to system y(+), American journal of physiology. Am. J. Physiol. Cell Physiol. 2002;282:C134–C143. doi: 10.1152/ajpcell.2002.282.1.C134. [DOI] [PubMed] [Google Scholar]
- 26.Sheth B, Dransfield I, Partridge LJ, Barker MD, Burton DR. Dibutyryl cyclic AMP stimulation of a monocyte-like cell line, U937: a model for monocyte chemotaxis and Fc receptor-related functions. Immunology. 1988;63:483–490. [PMC free article] [PubMed] [Google Scholar]
- 27.Cathcart MK, Chisolm GM, 3rd, McNally AK, Morel DW. Oxidative modification of low density lipoprotein (LDL) by activated human monocytes and the cell lines U937 and HL60, In vitro cellular & developmental biology. J. Tissue Cult. Assoc. 1988;24:1001–1008. doi: 10.1007/BF02620873. [DOI] [PubMed] [Google Scholar]
- 28.Sirois P, Cadieux A, Rola-Pleszcynski M, Begin R. Perifused alveolar macrophages. A technique to study the effects of toxicants on prostaglandin release. Experientia. 1982;38:1125–1127. doi: 10.1007/BF01955404. [DOI] [PubMed] [Google Scholar]
- 29.Obrig TG, Culp WJ, McKeehan WL, Hardesty B. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J. Biol. Chem. 1971;246:174–181. [PubMed] [Google Scholar]
- 30.Beg AA, Finco TS, Nantermet PV, Baldwin AS., Jr Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: a mechanism for NF-kappa B activation. Mol. Cell. Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin G, Field JJ, Yu JC, Ken R, Neuberg D, Nathan DG, Linden J. NF-kappaB is activated in CD4(+) iNKT cells by sickle cell disease and mediates rapid induction of adenosine A2A receptors. PLoS One. 2013;8:e74664. doi: 10.1371/journal.pone.0074664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 33.Aluvila S, Sun J, Harrison DH, Walters DE, Kaplan RS. Inhibitors of the mitochondrial citrate transport protein: validation of the role of substrate binding residues and discovery of the first purely competitive inhibitor. Mol. Pharmacol. 2010;77:26–34. doi: 10.1124/mol.109.058750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCoy JM, Wicks JR, Audoly LP. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J. Clin. Invest. 2002;110:651–658. doi: 10.1172/JCI15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe J-M, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson P-J, Carty TJ, Perez JR, Audoly LP. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scheppach W, Pomare EW, Elia M, Cummings JH. The contribution of the large intestine to blood acetate in man. Clin. Sci. (Lond.) 1991;80:177–182. doi: 10.1042/cs0800177. [DOI] [PubMed] [Google Scholar]
- 37.Starai VJ, Escalante-Semerena JC. Acetyl-coenzyme A synthetase (AMP forming) Cell Mol. Life Sci. CMLS. 2004;61:2020–2030. doi: 10.1007/s00018-004-3448-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Babior BM. NADPH oxidase. Curr. Opin. Immunol. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 39.Octavia Y, Brunner-La Rocca HP, Moens AL. NADPH oxidase-dependent oxidative stress in the failing heart: From pathogenic roles to therapeutic approach. Free Radic. Biol. Med. 2012;52:291–297. doi: 10.1016/j.freeradbiomed.2011.10.482. [DOI] [PubMed] [Google Scholar]
- 40.Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011;63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 41.Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol. Rev. 2008;226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheshire JL, Baldwin AS., Jr Synergistic activation of NF-kappaB by tumor necrosis factor alpha and gamma interferon via enhanced I kappaB alpha degradation and de novo I kappaBbeta degradation. Mol. Cell. Biol. 1997;17:6746–6754. doi: 10.1128/mcb.17.11.6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pekalski J, Zuk PJ, Kochanczyk M, Junkin M, Kellogg R, Tay S, Lipniacki T. Spontaneous NF-kappaB activation by autocrine TNFalpha signaling: a computational analysis. PLoS One. 2013;8:e78887. doi: 10.1371/journal.pone.0078887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bradley JR. TNF-mediated inflammatory disease. J. Pathol. 2008;214:149–160. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J. Yin and yang interplay of IFN-gamma in inflammation and autoimmune disease. J. Clin. Invest. 2007;117:871–873. doi: 10.1172/JCI31860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skurkovich B, Skurkovich S. Anti-interferon-gamma antibodies in the treatment of autoimmune diseases. Curr. Opin. Mol. Ther. 2003;5:52–57. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.