

Abstract
Synthetic methods have been developed for the lead Sortase A inhibitors identified from previous studies. Several derivatives of the lead inhibitor were synthesized to derive preliminary structure activity relationships (SAR). Different regions of the lead inhibitor that are critical for the enzyme activity have been determined by systematic SAR studies. The E stereochemistry of the lead compound was found to be critical for its activity. Replacement of the E double bond with Z double bond or a rigid triple bond reduced the enzyme inhibitory activity in most cases. Reduction of the double bond to a C-C single bond resulted in complete loss of activity. Amide carbonyl and NH groups were also found to be crucial for the activity of this class of inhibitors, as well. Morpholine ring oxygen atom was also found to be important factor for the activity of the lead inhibitor. Preliminary SAR studies led to the identification of compounds with improved enzyme inhibition. The most active compound was found to have an IC50 value of 58εM against the enzyme.
Keywords: Staphylococcus aureus, Sortase, Inhibitor, Antibacterial, Structure Activity Relationship, Synthesis
1. Introduction
S. aureus causes a variety of human infections, ranging from superficial abscesses to life threatening bacteremias. Staphylococcal infections within the hospital and in the community are increasing, and an ever-growing number of antibiotic-resistant strains make treatment options more difficult. MRSA strains were isolated from 2% of staphylococcal infections in 1974 and from 63% of staphylococcal infections in 2004. Many of the nosocomial MRSA strains are multi-drug resistant, and even methicillin-sensitive strains can be deadly. A recent report using population-based, active case finding revealed that 94,360 invasive MRSA infections occurred in the U.S. in 2005, and that the majority of these (58%) occurred outside of the hospital [1].
Notorious as a major source of nosocomial infections, S. aureus has recently taken on a new role in causing an escalating number of community-acquired infections in non-hospitalized persons without predisposing risk factors. A single S. aureus clone, designated as USA300, causes the majority of community acquired-MRSA infections in the U.S., and its dissemination has been observed globally [2–6]. Vancomycin is most commonly used for treatment of systemic infections caused by MRSA. However, S. aureus isolates with reduced susceptibility to vancomycin have been reported since 1997 [7]. These isolates are also methicillin resistant [8–10]. Because S. aureus cannot always be controlled by antibiotics and MRSA isolates are becoming increasingly prevalent in the community, additional control strategies and novel therapeutic approaches are sorely needed.
New approaches for the prevention and treatment of bacterial infections require greater understanding of the molecular structure and mechanisms of the chosen intervention targets and of the pathogenic role played by the target in the infection process. Bacterial infections are complex and involve the action of a large, sophisticated arsenal of virulence factors, many of which are surface-bound or secreted. Gram-positive bacteria such as S. aureus are endowed with a multitude of cell-wall anchored proteins that serve as an interface between the microbe and its host. Bacterial sortases are cysteine transpeptidases that participate in secretion and anchoring of many cell wall proteins by a mechanism conserved in almost the entire class of Gram-positive bacteria. Surface proteins can be attached to the bacterial surface in several fashions [11,12]. Proteins that are covalently attached to the cell wall share conserved regions known as the “sorting signal” or cell wall anchors [13,14]. The sorting signal includes a conserved amino acid motif, usually LPXTG. Precursor proteins are directed into a secretory pathway by their N-terminal signal peptides. They are translocated across the membrane and the signal peptide is cleaved [14,15]. Then, the C-terminal sorting signal retains the protein in the secretory pathway. The enzyme sortase acts at this point to cleave the protein between the threonine (T) and the glycine (G) of the LPXTG motif [11,16]. The carboxyl group of the Thr is then amide-linked to the amino group of a “cross-bridge” peptide in the lipid II precursor for cell wall synthesis [11,17]. Sortase-defective strains of various pathogens were shown to be faulty in the display of surface proteins and are less virulent [18,19]. In a number of studies, individual sortase genes have been deleted and the loss-of-sortase function has resulted in less virulence in several animal models of the disease [18, 20–24]. Hence, sortases are attractive pharmacotherapeutic targets [12].
Currently, there have only been a few reports of specific sortase inhibitors [25–28]. Recently, Oh et al [29] identified a small molecule reversible inhibitor of SrtA with a low micromolar IC50 value by structurally modifying a lead compound identified by random screening of a group of small molecules. We have recently identified an inhibitor (1) of S. aureus SrtA aided by in-silico virtual screening (Figure 1) [30]. We have conducted preliminary structure activity relationship (SAR) studies with the goal of improving the activity of inhibitor 1. This manuscript describes the synthesis of inhibitor 1 and several of its derivatives and the results of their in vitro enzymatic evaluation against SrtAΔ59, a fully active variant of SrtA with 59 N-terminus residues removed.
Figure 1.

Structure and IC50 value of inhibitor 1
2. Results and discussion
Inhibitor 1 was initially obtained from a commercial source in milligram quantities. For SAR studies we needed this compound in larger quantities. So, a general method for the synthesis of inhibitor 1 was developed in our laboratory. The synthetic procedures for all analogues were modelled around this synthesis. Chemistry employed for the synthesis of inhibitor 1 is outlined in Scheme 1.
Scheme 1.

Synthesis of inhibitor 1 and its furan analogue 6
Commercially available methyl 2-(N-morpholino)-5-nitrobenzoate (2) was reduced using hydrogen in the presence of Pd/C catalyst in anhydrous ethyl acetate to afford the corresponding amino compound 3. The compound 3 was coupled with commercially available trans-3-(thiophene-2-yl)acrylic acid (4a) in the presence of ethyl(N,N-dimethylaminopropyl) carbodiimide (EDAC) and N,N-dimethylaminopyridine (DMAP) in 1,2-dichloroethane to form the amide compound 5a [31]. Basic hydrolysis of the ester methyl group present in compound 5a afforded the inhibitor 1 as a white crystalline solid.
We have synthesized several derivatives of inhibitor 1 in order to derive preliminary structure activity relationship data. Their activities against SrtAΔ59 were determined using a fluorescence resonance energy transfer (FRET) assay which is a modification of a previously reported procedure. In this assay, the IC50 values were determined by monitoring the effect of the compounds on the steady state cleavage of a model substrate peptide, Dabcyl-QALPETGEE-EDANS [29,32,33].Structures and IC50 values of the newly synthesized derivatives of inhibitor 1 are given in Table 1.
Table 1.
S. aureus SrtAΔ59 inhibition of synthesized derivatives of inhibitor 1
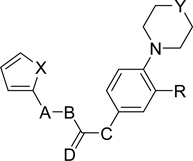 | ||||||||
|---|---|---|---|---|---|---|---|---|
| No | X | A-B | Stereochemistry | C | D | Y | R | IC50(µM)a |
| 5a | S | CH=CH | E | NH | O | O | COOCH3 | 71 ± 2.5 |
| 5b | O | CH=CH | E | NH | O | O | COOCH3 | 58 ± 4.9 |
| 6 | O | CH=CH | E | NH | O | O | COOH | 181 ± 14 |
| 7 | S | CH2-CH2 | -b | NH | O | O | COOH | >600 |
| 8 | S | CH2-CH2 | -b | NH | O | O | COOCH3 | >600 |
| 9 | O | CH2-CH2 | -b | NH | O | O | COOH | >600 |
| 10 | O | CH2-CH2 | -b | NH | O | O | COOCH3 | >600 |
| 12 | S | C≡C | -b | NH | O | O | COOCH3 | 165 ± 9 |
| 13 | S | C≡C | -b | NH | O | O | COOH | 183 ± 12 |
| 14 | S | CH=CH | Z | NH | O | O | COOCH3 | 61 ± 4 |
| 15 | S | CH=CH | Z | NH | O | O | COOH | 154 ± 5 |
| 16a | S | CH=CH | E | NMe | O | O | COOCH3 | 571 ± 43 |
| 16b | O | CH=CH | E | NMe | O | O | COOCH3 | 514 ± 39 |
| 17a | S | CH=CH | E | NMe | O | O | COOH | >600 |
| 17b | O | CH=CH | E | NMe | O | O | COOH | >600 |
| 19 | S | CH=CH | E | NH | HH | O | COOCH3 | 249 ± 9 |
| 20 | S | CH=CH | E | NH | HH | O | COOH | >600 |
| 24a | S | CH=CH | E | NH | O | CH2 | COOCH3 | 92 ± 5 |
| 24b | O | CH=CH | E | NH | O | CH2 | COOCH3 | 131 ± 14 |
| 25a | S | CH=CH | E | NH | O | CH2 | COOH | 181 ± 12 |
| 25b | O | CH=CH | E | NH | O | CH2 | COOH | 463 ± 19 |
| 26a | S | CH=CH | E | NH | O | O | CH2OH | 73 ± 2 |
| 26b | O | CH=CH | E | NH | O | O | CH2OH | 111 ± 5.7 |
| 27a | S | CH=CH | E | NH | O | O | CHO | 77 ± 6.5 |
| 27b | O | CH=CH | E | NH | O | O | CHO | 107 ± 8.5 |
| 28 | S | CH=CH | E | NH | O | O | CONH2 | 105 ± 3.7 |
IC50 values were determined by a Fluorescent Resonance Energy Transfer (FRET) assay. Reported values are an average of 3–5 measurements.
Not applicable
Compound 6 is a furan analogue of inhibitor 1. Compound 6 was synthesized by a similar procedure as described for the synthesis of inhibitor 1 (Scheme 1). We have used trans-3-(furan-2-yl)acrylic acid (4b) instead of trans-3-(thiophene-2-yl)acrylic acid (4a) in this procedure. Compound 3 was coupled with commercially available 4b in the presence of EDAC and DMAP in 1,2-dichloroethane to form the amide compound 5b. Basic hydrolysis of the ester present in compound 5b afforded compound 6. Compound 6 along with the synthetic intermediate methyl esters 5a and 5b were evaluated for their enzyme inhibitory activity. The results of the enzyme assays for these and all other compounds described in this report are summarized in Table 1. Compound 6 did not show an enhancement of activity. Instead, it was found to be less active compared to inhibitor 1 in the enzymatic assay (IC50 = 181 µM). But, the methyl ester intermediates (5a and 5b) showed improved inhibition as compared to the corresponding acid derivatives 1 and 6. Methyl ester derivative of the thiophene compound (5a) showed an IC50 value of 71 µM and methyl ester derivative of the furan compound (5b) showed a much improved inhibition at an IC50 value of 58 µM. Although this increase in activity of methyl ester analogues is counter intuitive of the FlexX model which showed a possible salt bridge interaction between the carboxylic acid and Arg197 side chain, the increased activity may be explained using the FlexX model of the methyl ester in the active site. This model shows a similar fit of the molecule in the active site with the ester methyl group resting in a small hydrophobic pocket alongside the central phenyl ring without completely destroying the electrostatic interaction between Arg197 side chain and ester group O atoms of the inhibitor. The increased activity could be a result of these additional hydrophobic interactions generated by the ester methyl group with hydrophobic residues in the active site.
Compounds 7 – 10 were synthesized to evaluate the importance of the double bond present in compounds 1 and 6 for their activity. Compounds 7 and 9 are the saturated analogues of the carboxylic acid derivatives 1 and 6. Compounds 8 and 10 are the saturated analogues of the methyl ester derivatives 5a and 5b. Our assay showed that all four saturated compounds 7 – 10 were inactive up to a concentration of 600 µM showing that the double bond is crucial for the activity of these inhibitors (Table 1). Synthesis of compounds 7 – 10 is outlined in Scheme 2. Hydrogenation of the thiophene derivatives 1 and 5a using Pd/C as catalyst did not work, possibly due to the catalyst poisoning effect of the thiophene ring present in these molecules. Use of a stronger catalyst like Pd black in the presence of ammonium formate resulted in the hydrogenation of 1 and 5a to corresponding hydrogenated compounds 7 and 8. Hydrogenation of 6 and 5b were carried out using H2 in the presence of Pd/C catalyst to afford compounds 9 and 10 in good yields.
Scheme 2.

Synthesis of compounds 7–10
Our lead inhibitor 1 has a double bond with E stereochemistry. We already know that the double bond is critical for the activity of the inhibitor 1. Our next goal was to examine the effect of stereochemistry of the double bond on the activity. With this goal, we prepared the Z isomer 15. In order to examine the influence of a carbon-carbon triple bond on the activity, we prepared the compound 13. Synthesis of compounds 13 and 15 is outlined in Scheme 3.
Scheme 3.

Synthesis of acetylenic acid, 13 and the cis derivative, 15
The synthesis started from the known 3-(thiophen-2-yl)propiolic acid 11, which is prepared according to the literature procedure [34]. The acid 11 was coupled with the amino compound 3 in the presence of EDAC and DMAP in 1,2-dichloroethane afforded the alkyne ester 12. The ester group present in compound 12 was hydrolyzed using NaOH in MeOH to afford the acetylenic acid 13. The partial hydrogenation of the acetylenic ester 12 using Lindlar Pd catalyst did not work as expected possibly due to the catalytic poisoning effect of the thiophene ring already present in the molecule. However, reduction worked smoothly to a cis double bonded compound 14 when it was carried out with H2 in the presence of Pd/C in ethyl acetate. The ester group present in compound 14 was hydrolyzed using NaOH in MeOH to afford the cis derivative 15.
Compounds 13, 15 and the intermediate esters, 12 and 14 were all evaluated for their enzymatic activity (Table 1). The intermediate acetylenic compound 12 (165 µM) and the corresponding acid 13 (183 µM) showed decreased inhibition of the enzyme compared to the corresponding E alkene derivatives. The Z alkene ester derivative 14 showed more or less similar inhibition (61 µM) as compared to the E derivative, 5a. However, the Z alkene acid 15 showed a decreased inhibition (154 µM) of the enzyme as compared to E alkene acid, 1.
We have synthesized compounds 17a and 17b in order to examine the effect of interruption of possible H-bonding of NH group present in the inhibitors 1 and 6. Target compounds in this case were compounds 17a and 17b where the amide N was methylated. Synthesis of the target compounds are outlined in Scheme 4.
Scheme 4.

Synthesis of N-methyl compounds 17a and 17b
Compound 5a and 5b were methylated using methyl iodide in the presence of NaH in THF to afford the N-methylated esters 16a and 16b. Basic hydrolysis of 16a and 16b using 1N. NaOH in MeOH afforded the final acid products 17a and 17b, respectively.
The compounds 17a–b and intermediates 16a–b were evaluated for their enzymatic activity in our assay (Table 1). As expected, all four N-methyl derivatives showed decrease in inhibition as compared to corresponding NH analogues. Compounds 16a and 16b showed IC50 values 571 µM and 514 µM respectively and the final acids 17a–b were inactive up to a concentration of 600 µM. This demonstrates that the NH group of the inhibitors 1 and 6 is very important for their activity.
In order to examine the effect of amide carbonyl oxygen on the activity of the lead compound, we have prepared compound 20 in which carbonyl group is replaced with a CH2 group. Synthesis of compound 20 is outlined in Scheme 5.
Scheme 5.

Synthesis of compound 20
Reductive amination of the known aldehyde 18 with the amine 3 in the presence of NaCNBH3 and ZnCl2 in MeOH afforded the ester compound 19. Hydrolysis of the ester group present in compound 19 using NaOH in MeOH afforded the final acid product 20. The compound 20 and intermediate 19 were evaluated for their enzymatic activity (Table 1). Both the compounds showed decreased inhibition compared to corresponding amide analogues showing that amide carbonyl group is important for the activity. The acid 20 was inactive up to a concentration of 600 µM while the intermediate ester 19 had a reduced activity at 249 µM.
We wanted to inspect the importance of the oxygen atom in the morpholine ring of the inhibitors 1 and 6. With this objective, we prepared the compounds 25a and 25b where oxygen atom in the morpholine ring is replaced with carbon atom. Synthesis of the compounds 25a and 25b is outlined in Scheme 6.
Scheme 6.

Synthesis of compounds 25a–b
Commercially available methyl 2-fluoro-5-nitro benzoate (21) was treated with piperidine in THF to afford the compound 22, which upon hydrogenation using H2 in the presence of Pd/C gave the amine 23. Compound 23 was coupled with trans-3-(thiophene-2-yl)acrylic acid (4a) or trans-3-(furan-2-yl)acrylic acid (4b) in the presence of EDAC and DMAP in 1,2-dichloroethane to form the amides 24a or 24b. Basic hydrolysis of the ester methyl group present in compounds 24a or 24b afforded the acids 25a or 25b.
Compounds 25a–b along with the intermediate esters, 24a–b were all evaluated for their enzymatic activity (Table 1). Carboxylic acid derivatives 25a (181 µM) and 25b (463 µM) exhibited a decreased inhibition compared to corresponding morpholine analogues. The ester intermediates 24a (92 µM) and 24b (131 µM) also showed a similar decrease in activity compared to corresponding morpholine analogues.
We have also made a few derivatives of inhibitor 1 by incorporating different substituents such as a -CH2OH (26a), -CHO (27a) or -CONH2 (28) in the place of the carboxylic acid group of inhibitors 1. Substitution with -CH2OH and -CHO groups did not result in a major change in the activity (26a, IC50 = 73 µM and 27a, IC50 = 77 µM), while substitution with -CONH2 group resulted in a decrease in activity as compared to inhibitor 1 (28, IC50 = 105 µM) (Table 1). Derivatives of furan compound 6 incorporating substituents such as a -CH2OH (26b) and -CHO (27b) in the place of the carboxylic acid group were also made. These compounds showed improved inhibition as compared to the parent furan compound, 6. Compound 26b showed an IC50 value of 111 mM, and compound 27b showed an IC50 value of 107 µM. Synthesis of compounds 26a–b and 27a–b is outlined in Scheme 7.
Scheme 7.

Synthesis of compounds 26a,b and 27a,b
Compounds 5a–b were reduced using DIBAL in a mixture of anhydrous CH2Cl2 and THF to afford the alcohol derivatives 26a–b. Oxidation of alcohols 26a–b using PCC in anhydrous THF afforded the aldehydes, 27a–b. Synthesis of amide 28 is outlined in Scheme 8. Compound 28 was prepared from the inhibitor 1 by treatment with SOCl2, followed by the treatment of the acid chloride produced with ammonia.
Scheme 8.

Synthesis of compound 28
Strikingly, the IC50 values determined for all of the active compounds are well below the previously measured Km value of 5.5 mM for SrtA binding to the LPXTG peptide [32]. This suggests that these inhibitors bind ∼1–2 orders of magnitude tighter than the LPXTG peptide and thus should be effective at blocking the enzymes activity in vivo.
3. Conclusions
In conclusion, we have discovered a novel class of small-molecule inhibitors of Staphylococcus aureus Sortase A. Inhibitors were screened for their activity against the enzyme using a FRET assay. Micromolar inhibitors of the enzyme are identified. We have carried out preliminary structure activity relationship studies that have resulted in the identification of inhibitors with improved activity. Regions of the molecular structure of the lead inhibitor that are critical for its activity have been determined by systematic SAR studies. Further SAR studies to refine the activity of the lead using the information gained from these studies as well as attempts to obtain high resolution inhibitor / Sortase A complex co-crystal structures are currently in progress.
5. Experimental section
5.1. Fluorescence Resonance Energy Transfer (FRET) assay
To determine IC50 values for each potential inhibitor, we have implemented the previously published fluorescence resonance energy transfer (FRET) assay [32,33]. This FRET assay employs the use of the donor and quencher pair, EDANS and Dabcyl, respectively. We have purchased the peptide Dabcyl-QALPETGEE-EDANS from a commercial source. The EDANS fluorophore has an excitation wavelength, λex,d = 336 nm and an emission maximum, λem,d = 490 nm. The fluorescence emission spectra for EDANS overlaps very well with the absorption spectra of Dabcyl, where Dabcyl has an absorption maximum, λmax,a = 472 nm. As such, when the two molecules are spatially close, as they are in the intact peptide, the efficiency of transfer from EDANS to Dabcyl is high. Upon excitation of EDANS little emission from the donor is observed because most of the energy from fluorescence is transferred to the dabcyl acceptor and lost through non radiative decay pathways. In contrast, upon cleavage the newly formed fragments will diffuse apart and the two peptide fragments will become spatially separated and the FRET efficiency will diminish. Thus, the emission from the EDANS fluorophore will precipitously increase. Because of these properties, this FRET pair has been successfully employed to monitor SrtA catalyzed peptide cleavage.
Typically, the EDANS fluorophore is excited at 350 nm and fluorescence from EDANS is observed at 495 nm over a period of time. However, to reduce the correction for the inner filter effect we have chosen to monitor emission at 590 nm, where the absorbance for either chromophore is insignificant. At the beginning of the observation, fluorescence is low due to a high FRET efficiency, but, as the peptide is cleaved the fluorescence increases linearly. The slope of this linear increase is taken to be the initial velocity, V0. Because of the large inner filter correction at high concentrations of substrate, we have chosen to monitor the effect of inhibitors on V0 at a select enzyme and substrate concentration, where the correction is negligible. These experiments are performed by mixing 5 µM SrtA with 20 µM Dabcyl-EDANS labelled peptide in a 0.5 cm cuvette. This mixture is excited at λex = 350 nm and fluorescence is observed at λem = 590 nm. From this experiment, a time course of fluorescence increase as a function of time is acquired. The slope of this line is considered the maximum velocity in the absence of inhibitor. The identical experiment is then performed in the presence of increasing concentrations of inhibitor and the slopes are compared until a 50 % decrease in the slope is observed. The concentration of inhibitor where 50 % of this maximum velocity is observed is taken to be the IC50. Each experiment was repeated at least three times to ensure the reproducibility and calculate standard deviation of the reported IC50 values.
5.2. General Methods for Synthesis
Solvent evaporations were carried out in vaccuo with a rotary evaporator. Analytical samples were prepared by drying the samples in vaccuo (0.2 mmHg) in an Abderhalden drying apparatus over P2O5 and ethyl acetate at reflux temperature. Thin layer chromatography (TLC) was performed on silica gel plates with fluorescent indicator (Whatmann, silica gel, UV254, 25 εm plates). Spots were visualized by UV light (254 and 365 nm). All analytical samples were single spots on TLC in at least two different solvent systems. Purification by column and flash chromatography was carried out using ‘BAKER’ silica gel (40 εm) in the solvent systems indicated. The amount (weight) of silica gel for column chromatography was in the range of 50–100 times the amount (weight) of the crude compounds being separated. Melting points were determined on a Mel-Temp II melting point apparatus and are uncorrected. Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were recorded on a Brucker DPX 300 spectrometer using TMS as internal standard. The values of chemical shifts (δ) are given in ppm and coupling constants (J) in Hz. The chemical shift values are reported as parts per million (ppm) relative to tetramethylsilane as internal standard. Mass spectra were recorded on a MicroMass Platform LCC instrument. Elemental analyses were performed by Atlantic Microlab, Norcross, Georgia and the results indicated by symbols for the elements were within ± 0.4% of theoretical values. Anhydrous solvents used for reactions were purchased in Sure-Seal™ bottles from Aldrich Chemical Company. Other reagents were purchased from Aldrich or Fisher chemical companies and used as received.
Methyl 5-amino-2-morpholinobenzoate (3)
To a solution of Methyl 5-nitro-2-morpholinobenzoate, 2 (1.0 g, 3.76 mmol) in EtOAc (35 mL) 10% Pd/C (100 mg) was added and stirred under an atmosphere of H2 from a balloon (∼1 atm) for 12 h. TLC analysis (1:1 EtOAc / CHCl3) revealed that the reaction is complete. The catalyst was removed by filtration through a celite bed and the filtrate was concentrated in vacuum to furnish 3 as a solid. (0.88 g, 99 %); Mp 121 – 122 °C; 1H NMR (CDCl3) δ 2.91 – 2.97 (m, 4H), 3.60 (bs, 2H), 3.79 – 3.85 (m, 4H), 3.87 (s, 3H), 6.78 (dd, 1H, J1 = 2.7 Hz, J2 = 8.7 Hz), 6.95 (d, 1H, J = 8.7 Hz) and 7.05 (d, 1H, J = 2.7 Hz); 13C NMR (CDCl3) δ 51.7, 53.3, 67.1, 116.7, 118.8, 120.8, 127.0, 141.9, 143.7 and 168.0; MS (ES) m/z 237 (M + H); Anal Calcd for C12H16N2O3: C, 61.00; H, 6.83; N, 11.86. Found: C, 60.87; H, 6.81; N, 11.75.
Methyl 5-((E)-3-(thiophen-2-yl)acrylamido)-2-morpholinobenzoate (5a)
To a solution of 3 (0.430 g, 1.82 mmol) in 1,2-dichloroethane (20 mL) 3-(2-thienyl)acrylic acid 4a (0.337 g, 2.18 mmol), EDAC (1.12 g, 5.82 mmol), and DMAP (0.022 g, 0.18 mmol) were added and stirred for 16 h at room temperature. TLC analysis (1:1 EtOAc / CHCl3) revealed that the reaction is complete. The reaction mixture was diluted with CH2Cl2 (50 mL) and the organic layer was washed with saturated NaHCO3 solution (2 × 25 mL), water (1 × 25 mL), brine (1 × 25 mL) and dried over anhydrous Na2SO4. The drying agent was filtered off and the solvent was concentrated in vacuo to afford the crude product which was crystallized form a mixture of CHCl3 and hexanes to furnish 5a as a yellow solid. (0.62 g, 91%); Mp 196 – 197 °C; 1H NMR (CDCl3) δ 3.00 – 3.05 (m, 4H), 3.83 – 3.88 (m, 4H), 3.89 (s, 3H), 6.31 (d, 1H, J = 15.3 Hz), 7.03 – 7.08 (m, 2H), 7.24 – 7.28 (m, 2H), 7.33 – 7.38 (m,1H), 7.80 (d, 1H, J = 8.7 Hz), 7.83 – 7.90 (m, 1H) and 7.92 (d, 1H, J = 2.3 Hz); 13C NMR (CDCl3) δ 52.2, 53.1, 67.2, 119.3, 119.8, 123.2, 124.6, 124.9, 127.9, 128.2, 130.9, 132.5, 135.1, 139.8, 149.0, 163.7 and 167.5; MS (ES) m/z 373 (M + H); Anal Calcd for C19H20N2O4S: C, 61.27; H, 5.41; N, 7.52. Found: C, 60.98; H, 5.39; N, 7.45.
Methyl 5-((E)-3-(furan-2-yl)acrylamido)-2-morpholinobenzoate (5b)
Compound 5b was synthesized, following a similar procedure as the one used for preparation of 5a starting from compound 3 (0.426 g, 1.8 mmol), 3-(2-furyl)acrylic acid 4b (0.298 g, 2.16 mmol), EDAC (1.10 g, 5.76 mmol), and DMAP (0.022 g, 0.182 mmol) to obtain the pure product. (0.639 g, 99 %); Mp 172 – 173 °C; 1H NMR (CDCl3) δ 2.98–3.07 (m, 4H), 3.82–3.88 (m, 4H), 3.89 (s, 3H), 6.42 (d, 1H, J = 15.0 Hz), 6.48 (dd, 1H, J1 = 1.8 Hz, J2 = 3.3 Hz), 6.60 (d, 1H, J = 3.3 Hz), 7.04 (d, 1H, J = 9.0 Hz), 7.40 (bs, 1H), 7.46 (d, 1H, J = 1.5 Hz), 7.52 (d, 1H, J = 15.0 Hz), 7.82 (d, 1H, J = 7.2 Hz) and 7.91 (s, 1H); 13C NMR (CDCl3) δ 52.1, 53.0, 67.1, 112.3, 114.4, 118.3, 119.6, 123.2, 124.6, 128.8, 132.7, 144.2, 148.8, 151.1 (2C), 164.1 and 167.6; MS (ES) m/z 357 (M + H); Anal Calcd for C19H20N2O5: C, 64.04; H, 5.66; N, 7.86. Found: C, 64.33; H, 5.63; N, 7.64.
5-((E)-3-(Thiophen-2-yl)acrylamido)-2-morpholinobenzoic acid (1)
To a solution of compound 5a (0.3g, 0.81 mmol) in a mixture of THF (1.5 mL) and MeOH (1.5 mL), 3N. NaOH solution (2.5 mL) was added at room temperature and the resulting mixture was refluxed gently for 45 min. TLC analysis (1:1 EtOAc / CHCl3) revealed that the reaction is complete. The solvent was completely removed in vaccuo and the residue obtained was taken up in water (3 mL). The resulting mixture was cooled to 0 °C and acidified to pH ∼2 by carefully adding 3N HCl (∼2.5 mL). The resulting white solid was filtered and dried under vacuum to furnish pure compound 1. (0.283 g, 98%); Mp 278–279 °C; 1H NMR (DMSO-d6) δ 3.00–3.09 (m, 4H), 3.74–3.85 (m, 4H), 6.55 (d, 1H, J = 15.3 Hz), 7.14 (dd, 1H, J1 = 3.6 Hz, J2 = 5.1 Hz), 7.47 (d, 1H, J = 3.3 Hz), 7.67 (s, 1H), 7.68 (d, 1H, J = 3.0 Hz), 7.77 (d, 1H, J = 15.3 Hz), 7.99 (dd, 1H, J1 = 2.7 Hz, J2 = 8.7 Hz), 8.29 (d, 1H, J = 2.7 Hz) and 10.4 (bs, 1H); 13C NMR (DMSO-d6) δ 52.7, 66.3, 120.3, 120.9, 123.8, 124.0, 125.4, 128.5, 128.7, 131.6, 133.7, 137.8, 139.6, 145.2, 163.4 and 166.4; MS (ES) m/z 357 (M– H); Anal Calcd for C18H18N2O4S: C, 60.32; H, 5.06; N, 7.82. Found: C, 60.37; H, 5.14; N, 7.59.
5-((E)-3-(Furan-2-yl)acrylamido)-2-morpholinobenzoic acid (6)
Compound 6 was synthesized following a similar procedure as the one used for preparation of compound 1 starting from 5b (0.200 g, 0.57 mmol) and 3N. NaOH solution to afford the pure product as a white solid. (0.192 g, 99%); Mp 281–282 °C; 1H NMR (DMSO-d6) δ 3.01–3.11 (m, 4H), 3.75–3.86 (m, 4H), 6.59 (d, 1H, J = 15.3 Hz), 6.60–6.69 (m, 1H), 6.87 (d, 1H, J = 3.6 Hz), 7.41 (d, 1H, J = 15.6 Hz), 7.68 (d, 1H, J = 9.0 Hz), 7.84 (d, 1H, J = 1.5 Hz), 8.00 (dd, 1H, J1 = 2.7 Hz, J2 = 8.7 Hz), 8.29 (d, 1H, J = 2.7 Hz) and 10.4 (bs, 1H); 13C NMR (DMSO-d6) δ 52.8, 66.4, 112.5, 115.0, 118.9, 120.9, 123.8, 124.0, 125.4, 127.8, 137.8, 145.2, 145.4, 150.8, 163.6 and 166.4; MS (ES) m/z 341 (M – H); Anal. (C18H18N2O5) C, H, N. Anal Calcd for C18H18N2O5: C, 63.15; H, 5.30; N, 8.18. Found: C, 63.06; H, 5.53; N, 7.89.
5-(3-(Thiophen-2-yl)propanamido)-2-morpholinobenzoic acid (7)
To a solution of compound 1 (0.071 g, 0.20 mmol) in a mixture of MeOH (4 mL) and EtOAc (3 mL), HCOONH4 (0.188 g, 2.97 mmol) and Pd black (40 mg) were added and refluxed for 6 h. TLC analysis (1:1 EtOAc / CHCl3) and mass spectrum revealed that the reaction was complete. The catalyst was removed by filtration through a celite bed, washed with a mixture of MeOH and CHCl3, and the filtrate was concentrated in vaccuo to afford the crude product. The crude product was purified by washing thoroughly with water to remove the inorganic salts, filtered and dried under vacuum to furnish pure product 7. (0.049 g, 69%); Mp 226 – 227 °C; 1H NMR (DMSO-d6) δ 2.68 (t, 2H, J = 7.5 Hz), 2.99 – 3.08 (m, 4H), 3.13 (t, 2H, J = 7.5 Hz), 3.74 – 3.86 (m, 4H), 6.86 – 6.97 (m, 2H), 7.30 (dd, 1H, J1 = 1.0 Hz, J2 = 4.9 Hz), 7.65 (d, 1H, J = 8.4 Hz), 7.90 (dd, 1H, J1 = 2.7 Hz, J2 = 8.7 Hz), 8.21 (d, 1H, J = 2.4 Hz) and 10.2 (bs, 1H); 13C NMR (DMSO-d6) δ 24.9, 38.0, 52.7, 66.3, 120.8, 123.7, 123.8, 123.9, 124.7, 125.2, 126.9, 137.7, 143.4, 145.0, 166.3 and 170.1; MS (ES) m/z 361 (M + H); Anal Calcd for C18H20N2O4S: C, 59.98; H, 5.59; N, 7.77. Found: C, 60.15; H, 5.75; N, 7.64.
Methyl 5-(3-(thiophen-2-yl)propanamido)-2-morpholinobenzoate (8)
To a solution of compound 5a (0.100 g, 0.27 mmol) in MeOH (5 mL), HCOONH4 (0.255 g, 4.05 mmol) and Pd black (0.05 g) were added and refluxed for 6 h. TLC analysis (1:1 EtOAc / CHCl3) and mass spectrum revealed that the reaction is complete. The catalyst was removed by filtration through a celite bed, washed with CHCl3, and the filtrate was concentrated in vacuo. The residue obtained was dissolved in CH2Cl2 (20 mL) and water (15 mL) was added. The organic layer was separated and washed with saturated NaHCO3 solution (2 × 15 mL), water (2 × 15 mL), brine (1 × 15 mL) and dried (Na2SO4). The drying agent was filtered off and the solvent was removed in vaccuo to obtain the crude product. This crude product was purified by column chromatography over Si gel (4 × 2 cm) using 1:5 EtOAc / CHCl3 as eluent to afford the pure compound 8. (0.082 g, 82 %); Mp 115 – 116 °C; 1H NMR (CDCl3) δ 2.70 (t, 2H, J = 7.5 Hz), 2.97 – 3.05 (m, 4H), 3.27 (t, 2H, J = 7.2 Hz), 3.81 – 3.87 (m, 4H), 3.88 (s, 3H), 6.86 (dd, 1H, J1 = 0.9 Hz, J2 = 3.3 Hz), 6.90 – 6.96 (m, 1H), 7.01 (d, 1H, J = 9.0 Hz), 7.11 (bs, 1H), 7.14 (dd, 1H, J1 = 1.2 Hz, J2 = 5.1 Hz), 7.64 (dd, 1H, J1 = 2.7 Hz, J2 = 8.7 Hz) and 7.76 (d, 1H, J = 2.7 Hz); 13C NMR (CDCl3) δ 25.5, 38.9, 52.0, 52.9, 67.0, 119.5, 123.2, 123.4, 124.6, 124.7 (2C), 126.8, 132.3, 143.0, 148.6, 167.4 and 170.1. MS (ES) m/z 375 (M + H). Anal Calcd for C19H22N2O4S. 0.25H2O: C, 60.26; H, 5.99; N, 7.40. Found: C, 60.27; H, 6.13; N, 7.22.
5-(3-(Furan-2-yl)propanamido)-2-morpholinobenzoic acid (9)
To a solution of compound 6 (0.075 g, 0.22 mmol) in EtOAc (15 mL) 10% Pd/C (0.02 g) was added and stirred under a hydrogen atmosphere from balloon (∼1 atm) for 25 min. TLC analysis (1:1 EtOAc: CHCl3) and 1H NMR of aliquot revealed that the reaction is complete. (Note: Prolonged reaction times lead to hydrogenation of furan ring). The catalyst was removed by filtration through a celite bed and the filtrate was concentrated in vaccuo to afford the crude product which was purified by column chromatography over Si gel (4 × 2 cm) using EtOAc as eluent to afford pure compound 9 as a white solid. (0.066 g, 88 %); Mp 230 – 231 °C; 1H NMR (DMSO-d6) δ 2.65 (t, 2H, J = 7.5 Hz), 2.93 (t, 2H, J = 7.3 Hz), 3.00–3.07 (m, 4H), 3.73–3.83 (m, 4H), 6.11 (dd, 1H, J1 = 1.2 Hz, J2 = 2.1 Hz), 6.34 (dd, 1H, J1 = 1.8 Hz, J2 = 3.0 Hz), 7.51 – 7.52 (m, 1H), 7.65 (d, 1H, J = 8.7 Hz), 7.89 (dd, 1H, J1 = 2.7 Hz, J2 = 8.7 Hz), 8.21 (d, 1H, J = 2.7 Hz) and 10.2 (bs, 1H); 13C NMR (DMSO-d6) δ 23.2, 34.5, 52.8, 66.4, 105.2, 110.4, 120.8, 123.8, 123.9, 125.3, 137.8, 141.5, 145.0, 154.4, 166.4 and 170.2; MS (ES) m/z 343 (M – H); Anal Calcd for C18H20N2O5: C, 62.78; H, 5.85; N, 8.13. Found: C, 62.50; H, 5.89; N, 8.01.
Methyl 5-(3-(furan-2-yl)propanamido)-2-morpholinobenzoate (10)
Compound 10 was synthesized, following procedure used for the preparation of compound 9 starting from compound 5b (0.136 g, 0.38 mmol) in EtOAc (15 mL) and 10% Pd/C (0.02 g) under a hydrogen atmosphere. (0.106 g, 77 %); Mp 119 – 120 °C; 1H NMR (CDCl3) δ 2.69 (t, 2H, J = 7.5 Hz), 2.97–3.03 (m, 4H), 3.07 (t, 2H, J = 7.3 Hz), 3.81–3.87 (m, 4H), 3.88 (s, 3H), 6.07 (d, 1H, J = 3.3 Hz), 6.27–6.32 (m, 1H), 7.01 (d, 1H, J = 9.0 Hz), 7.16 (bs, 1H), 7.32 (d, 1H, J = 1.2 Hz), 7.65 (dd, 1H, J1 = 2.7 Hz, J2 = 8.7 Hz) and 7.78 (d, 1H, J = 2.4 Hz); 13C NMR (CDCl3) δ 23.7, 35.4, 52.0, 52.9, 67.1, 105.5, 110.2, 119.5, 123.2, 124.6, 124.7, 132.3, 141.1, 148.7, 154.0, 167.5 and 170.3; MS (ES) m/z 357 (M – H). Anal Calcd for C19H22N2O5.0.25H2O: C, 62.92; H, 6.25; N, 7.72. Found: C, 62.60; H, 6.13; N, 7.59.
Methyl 5-(3-(thiophen-2-yl)propiolamido)-2-morpholinobenzoate (12)
Compound 12 was prepared similar to compound 5a by the treatment of methyl 4-amino-2-(4-morpholino)-benzoate 3 (0.542 g, 2.3 mmol) and 3-(thiophen-2-yl)propiolic acid, 11 (0.428 g, 2.8 mmol) in the presence of EDAC (1.42 g, 7.4 mmol) and a DMAP (0.028 g, 0.23 mmol). The crude product was purified by flash chromatography over Si gel using 10% EtOAc in CHCl3 as eluent to afford compound 12 as oil. (0.407 g, 37%); 1HNMR (CDCl3) δ 3.03 (t, 4H, J = 4.5 Hz), 3.84 – 3.89 (m, 7H), 7.03 – 7.07 (m, 2H), 7.44 – 7.46 (m, 2H), 7.65 (bs, 1H), 7.77 (dd, 1H, J1 = 2.7 Hz, J2 = 9 Hz), 7.86 (d, 1H, J = 2.7 Hz); 13CNMR (CDCl3) δ 52.3, 52.9, 67.1, 79.9, 87.4, 119.7, 123.3, 124.6, 124.8, 127.5 (2C), 130.5, 131.9, 135.7, 149.2, 151.1, 167.5; MS (ES+) m/z 371 (M+H); Anal Calcd for C19H18N2O4S: C, 61.61; H, 4.90; N, 7.56. Found: C, 61.47; H, 4.88; N, 7.30.
5-(3-(Thiophen-2-yl)propiolamido)-2-morpholinobenzoic acid (13)
Compound 13 was prepared similar to compound 1 by hydrolysis of 12 (0.049 g, 0.13 mmol) using 1N. NaOH in a MeOH / THF (4 mL, 1:1) medium to afford (0.03 g; 64%) as a white solid. Mp. 232 °C (decomp.); 1HNMR (DMSO-d6) δ 3.04 (t, 4H, J = 4.5 Hz), 3.79 (t, 4H, J = 4.5 Hz), 7.21 (dd, 1H, J1 = 3.6 Hz, J2 = 5.1 Hz), 7.64 – 7.67 (m, 2H), 7.85 – 7.89 (m, 2H), 8.28 (d, 1H, J = 2.7 Hz) and 11.08 (s, 1H); 13CNMR (DMSO-d6) δ 52.6, 66.4, 78.9, 87.8, 118.7, 121.4, 123.7, 124.5, 125.5, 128.3, 132.2, 136.3, 136.7, 146.0, 150.3, 166.4; MS (ES+) m/z 357 (M+H); Anal Calcd for C18H16N2O4S.0.25H2O: C, 59.90; H, 4.61; N, 7.76. Found: C, 59.89; H, 4.56; N, 7.62.
(Z)-Methyl 5-(-3-(thiophen-2-yl)acrylamido)-2-morpholinobenzoate (14)
To a solution of 12 (0.042 g, 0.11 mmol) in EtOAc (6 mL) 10% Pd/C (0.02 g) was added and stirred under H2 atmosphere for 30 min. TLC examination (1:9 EtOAc / CHCl3) showed that the reaction is complete. Catalyst was filtered off through a bed of celite. The filtrate was concentrated in vaccuo to obtain the crude product which was purified by flash chromatography over Si gel using 5% EtOAc in CHCl3) to yield compound 14 as a yellow gum. (0.035 g, 82%); 1HNMR (CDCl3) δ 2.99 – 3.02 (m, 4H), 3.83 – 3.88 (m, 7H), 5.78 (d, 1H, J = 12.6 Hz), 6.99 – 7.06 (m, 3H), 7.38 – 7.39 (m, 1H), 7.46 – 7.48 (m, 2H), 7.83 (m, 2H). 13CNMR (CDCl3) δ 52.2, 53.0, 67.1, 117.1, 199.7, 123.3, 124.8, 124.9, 126.6, 131.7, 132.4, 133.9, 134.8, 137.6, 148.9, 164.4, 167.6. MS (ES+) m/z 373 (M+H). Anal Calcd for C19H20N2O4S: C, 61.27; H, 5.41; N, 7.52. Found: C, 61.28; H, 5.50; N, 7.50.
(Z)-5-(3-(thiophen-2-yl)acrylamido)-2-morpholinobenzoic acid (15)
Compound 15 was prepared similar to compound 1 by hydrolysis of compound 14 (0.029 g, 0.08 mmol) using 1N. NaOH in a MeOH / THF (2 mL, 1:1) to afford as a white solid. (0.018 g, 65% yield); Mp 190 °C (decomp.); 1H NMR (DMSO-d6) δ 3.06 (bs, 4H), 3.80 (bs, 4H), 6.02 (d, 1H, J = 12.3 Hz), 7.08 – 7.11 (m, 1H), 7.19 (d, 1H, J = 12.6 Hz), 7.52 (d, 1H, J = 3.0 Hz), 7.68 – 7.71 (m, 2H), 7.98 (dd, 1H, J1 = 2.1 Hz, J2 = 8.7 Hz), 8.38 (d, 1H, J = 2.1 Hz), 10.59 (s, 1H); 13CNMR (DMSO-d6) δ 53.6, 67.2, 117.9, 121.7, 124.6, 124.9, 126.1, 127.5, 133.5, 134.7, 136.6, 138.5, 138.7, 145.9, 165.2, 167.3; MS (ES+) m/z 359 (M+H); HRMS Calcd for C18H18N2O4S: 358.0987. Found: 358.0980.
(E)-Methyl 5-(N-methyl-3-(thiophen-2-yl)acrylamido)-2-morpholinobenzoate (16a)
To a solution of compound 5a (0.04 g, 0.11 mmol) in anhydrous THF (10 mL), NaH (0.013 g, 60 %; 0.33 mmol) was added and stirred at 0 °C under a N2 atm for 30 min. Iodomethane (0.03 mL, 0.4 mmol) was added to the reaction mixture and the solution was stirred for 20 min. at rt. MS analysis confirmed product formation (m/z = 387). The reaction was quenched with aqueous NaHCO3 (1M, 20 mL). It was extracted with EtOAc (3 × 25 mL), washed with water (2 × 25 mL) and brine (25 mL), and dried over Na2SO4. The drying agent was filtered off and the filtrate was concentrated in vaccuo to afford the crude product which was purified by flash chromatography over Si gel (10 × 2 cm) using EtOAc / hexanes (1:1) as eluent to afford compound 16a as a colorless oil. (0.043 g, 100%); 1HNMR (CDCl3) δ 3.09 – 3.12 (m, 4H), 3.36 (s, 3H), 3.87 – 3.91 (m, 7H), 6.16 (d, 1H, J = 15.3 Hz), 6.98 (dd, 1H, J1 = 3.6 Hz, J2 = 5.1 Hz), 7.07 (d, 1H, J = 8.4 Hz), 7.15 (d, 1H, J = 3.6 Hz), 7.25 – 7.29 (m, 2H), 7.66 (d, 1H, J = 2.7 Hz), 7.76 – 7.81 (d, 1H, J = 15 Hz); 13CNMR (CDCl3) δ 37.6, 52.3, 52.7, 66.9, 117.2, 119.7, 124.4, 127.4, 127.9, 130.1, 130.5, 131.8, 134.7, 136.9, 140.3, 151.4, 165.9, 167.1; MS (ES+) m/z 387 (M+H); Anal Calcd for C20H22N2O4S.0.5C6H14: C, 64.31; H, 6.80; N, 6.52 Found: C, 64.19; H, 6.56; N, 6.53.
(E)-Methyl 5-(3-(furan-2-yl)-N-methylacrylamido)-2-morpholinobenzoate (16b)
Compound 16b was prepared in a similar fashion to compound 16a using compound 5b (0.048 g, 0.13 mmol), NaH (0.016 g, 60 %; 0.4 mmol) and iodomethane (0.03 mL, 0.48 mmol) to afford (0.044 g, 88% yield) as a colorless oil; 1H NMR (CDCl3) δ 3.10 – 3.12 (m, 4H), 3.36 (s, 3H), 3.88 – 3.91 (m, 7H), 6.23 (d, 1H, J = 15.1 Hz), 6.39 – 6.41 (m, 1H), 6.51 (d, 1H, J = 3.4 Hz), 7.07 (d, 1H, 8.7 Hz), 7.28 (dd, 1H, J1 = 2.7 Hz, J2 = 8.7 Hz), 7.35 (d, 1H, J = 1 Hz), 7.45 (d, 1H, J = 15.2 Hz), 7.66 (d, 1H, J = 2.7 Hz); 13C NMR (CDCl3) δ 38.0, 52.7, 53.1, 67.4, 112.5, 114.4, 116.3, 120.1, 124.8, 129.2, 130.99, 132.3, 137.4, 144.4, 151.8, 151.9, 166.6, 167.5; MS (ES+) m/z 371 (M+H); Anal Calcd for C20H22N2O5: C, 64.85; H, 5.99; N, 7.56. Found: C, 64.27; H, 6.04; N, 7.39.
(E)-5-(N-methyl-3-(thiophen-2-yl)acrylamido)-2-morpholinobenzoic acid (17a)
Compound 17a was prepared similar to compound 1 by hydrolysis of 16a (0.035 g, 0.09 mmol) using 1N. NaOH in a MeOH / THF (2 mL, 1:1) to yield (0.026 g, 78%) as a white solid; Mp 220 °C (decomp.); 1HNMR (CDCl3) δ 3.14 (bs, 4H), 3.42 (s, 3H), 3.99 (bs, 4H), 6.14 (d, 1H, J = 15.3 Hz), 6.99 (dd, 1H, J1 = 3.6 Hz, J2 = 5 Hz), 7.18 (d, 1H, J = 3.1 Hz), 7.26 – 7.29 (m, 1H), 7.48 – 7.54 (m, 2H), 7.82 (d, 1H, J = 15.1 Hz), 8.20 (d, 1H, J = 2.6 Hz); 13CNMR (CDCl3) δ 37.9, 54.0, 67.2, 117.2, 124.2, 127.1, 128.1, 128.5, 130.8, 130.9, 133.3, 135.9, 140.4, 143.5, 149.1, 166.1, 166.2; MS (ES+) m/z 373 (M+H); Anal Calcd for C19H20N2O4S: C, 61.27; H, 5.41; N, 7.52. Found: C, 61.34; H, 5.54; N, 7.34.
(E)-5-(3-(furan-2-yl)-N-methylacrylamido)-2-morpholinobenzoic acid (17b)
Compound 17b was prepared similar to compound 1 by hydrolysis of 16b (0.035 g, 0.095 mmol) using 1N. NaOH in a MeOH / THF (2 mL, 1:1) to afford (0.033 g; 97%) as oil; 1HNMR (CDCl3) δ 3.05–3.15 (m, 4H), 3.42 (s, 3H), 3.90–4.02 (m, 4H), 6.24 (d, 1H, J = 14.7 Hz), 6.42 (dd, 1H, J1 = 1.8 Hz, J2 = 3.4 Hz), 6.55 (d, 1H, J = 3.4 Hz), 7.35 (s, 1H), 7.47 – 7.54 (m, 3H), 8.20 (d, 1H, J = 2.9 Hz); 13CNMR (CDCl3) δ 38.0, 54.0, 67.2, 112.7, 115.1, 115.7, 124.3, 127.1, 130.1, 130.9, 133.4, 143.5, 144.6, 149.1, 151.7, 166.2, 166.4; MS (ES+) m/z 357 (M+H); Anal Calcd for C19H20N2O5: C, 64.04; H, 5.66; N, 7.86. Found: C, 63.84; H, 6.06; N, 7.56.
(E)-Methyl 5-(3-(thiophen-2-yl)allylamino)-2-morpholinobenzoate (19)
To a solution of (E)-3-(thiophen-2-yl)acrylaldehyde, 18 (0.74 g, 5.32 mmol) in MeOH (30 mL). Methyl 5-amino-2-(4-morpholino)-benzoate 3 (1.25 g, 5.32 mmol) was added and the mixture stirred at rt for 30 min. A solution of ZnCl2 (0.362 g, 2.66 mmol) and NaCNBH3 (0.367 g, 5.85 mmol) in MeOH (20 mL) was added to the reaction mixture and stirred at rt for 24h. The reaction was then quenched with water (20 mL) and solvents were completely evaporated in vaccuo. The crude residue obtained was dissolved in CH2Cl2 (60 mL), washed with water (3 × 50 mL) and brine (1 × 50 mL), and dried over anhydrous Na2SO4. Drying agent was filtered off and the filtrate was concentrated to afford the clean product 19 (1.68 g, 88% yield); Mp 131 °C; 1H NMR (CDCl3) δ 2.94 (t, 4H, J = 4.5 Hz), 3.82 (t, 4H, J = 4.5 Hz), 3.87 – 3.89 (m, 5H), 6.06 – 6.19 (m, 1H), 6.70 – 6.76 (m, 2H), 6.93 – 7.01 (m, 4H), 7.14 (d, 1H, J = 5.1 Hz); 13C NMR (CDCl3) δ 46.2, 52.0, 53.7, 67.5, 114.9, 116.8, 121.3, 124.2, 124.8, 125.6, 126.4, 127.4, 127.6, 141.9, 143.3, 143.7, 168.5; MS (ES+) m/z 359 (M+H); Anal Calcd for C19H22N2O3S.0.25H2O: C, 62.87; H, 6.25; N, 7.72. Found: C, 62.74; H, 6.13; N, 7.52.
(E)-5-(3-(thiophen-2-yl)allylamino)-2-morpholinobenzoic acid (20)
Compound 20 was prepared similar to compound 1 by hydrolysis of 19 (1.0 g, 2.79 mmol) using 1N. NaOH in a MeOH / THF (14 mL, 1:1) to yield (0.745 g, 78% yield) as a white solid; Mp 208 °C; 1H NMR (CDCl3) δ 2.92–3.17 (m, 4H), 3.84–4.0 (m, 6H), 6.12 (dt, 1H, J1 = 15.9 Hz, J2 = 5.7 Hz), 6.73 (d, 1H, J = 16.5 Hz), 6.82 (dd, 1H, J1 = 8.7 Hz, J2 = 3.0 Hz), 6.92–6.96 (m, 2H), 7.15 (d, 1H, J = 4.8 Hz), 7.23 (d, 1H, J = 8.7 Hz), 7.52 (d, 1H, J = 3.0 Hz); 13C NMR (CDCl3) δ 45.6, 53.7, 67.0, 115.0, 117.5, 123.3, 124.3, 125.1, 125.6, 125.75, 125.8, 127.4, 139.5, 141.7, 147.1, 167.5; MS (ES) m/z 345 (M+H); Anal. Calc for C18H20N2O3S: C, 62.77; H, 5.85; N, 8.13. Found: C, 62.31; H, 5.83; N, 8.02.
Methyl 5-nitro-2-(piperidin-1-yl)benzoate (22)
To a solution of Methyl 2-fluoro-5-nitrobenzoate, 21 (0.082 g, 0.41 mmol) and triethylamine (0.11 mL, 0.82 mmol) in THF, piperidine (0.054 g, 0.64 mmol) was added and the reaction mixture was stirred at rt for 30 min. TLC (25% EtOAc in CHCl3) examination revealed the completion of the reaction. The solvent was completely removed and the residue was dissolved in CHCl3 (20 mL), washed with water (2 × 15 mL) and brine (15 mL), and dried over anhydrous Na2SO4. Drying agent was filtered off and the filtrate was concentrated to afford clean compound 22 (0.115 g, 100%) as a yellow gum. 1HNMR (CDCl3) δ 1.69 – 1.72 (m, 6H), 3.23 – 3.26 (m, 4H), 3.93 (s, 3H), 6.95 (d, 1H, J = 9.3 Hz), 8.15 (dd, 1H, J1 = 2.7 Hz, J2 = 9.3 Hz), 8.55 (d, 1H, J = 2.7 Hz); 13CNMR (CDCl3) δ 23.8, 25.6, 52.4, 52.6, 117.2, 119.5, 127.7, 128.8, 138.2, 156.3, 167.0; MS (ES+) m/z 265 (M+H); Anal. Calcd. for C13H16N2O4: C, 59.08; H, 6.10; N, 10.60. Found: C, 58.90; H, 6.29; N, 10.25.
Methyl 5-amino-2-(piperidin-1-yl)benzoate (23)
To a solution of compound 22 (0.102 g, 0.38 mmol) in EtOAc (5 mL) was added 10% Pd/C (37 mg) and stirred under a hydrogen atmosphere (∼1 atm) for 2 h. TLC analysis (25% EtOAc / CHCl3) revealed that the reaction is complete. The catalyst was filtered off on a celite bed and the filtrate was concentrated on vacuum to furnish compound 23 (0.083 g, 93%) as a yellow oil. 1H NMR (CDCl3) δ 1.51 – 1.53 (m, 2H), 1.64 – 1.69 (m, 4H), 2.84 – 2.87 (m, 4H), 3.58 (bs, 2H), 3.87 (s, 3H), 6.74 (dd, 1H, J1 = 3 Hz, J2 = 8.7 Hz), 6.92 (d, 1H, J = 8.4 Hz), 7.02 (d, 1H, J = 3 Hz); 13C NMR (CDCl3) δ 24.2, 26.5, 51.9, 54.7, 117.3, 119.2, 120.9, 126.7, 141.0, 145.7, 168.8; MS (ES+) m/z 235 (M+H); Anal. Calcd. for C13H18N2O2: C, 66.64; H, 7.74; N, 11.96; Found C, 67.01; H, 7.59; N, 11.13.
(E)-Methyl 5-(3-(thiophen-2-yl)acrylamido)-2-(piperidin-1-yl)benzoate (24a)
Compound 24a was prepared similar to compound 5a by the treatment of 23 (0.066 g, 0.28 mmol) and 3-(thiophen-2-yl)acrylic acid, 4a (0.052 g, 0.34 mmol) in the presence of EDAC (0.162 g, 0.84 mmol) and a DMAP (0.005 g, 0.041 mmol) to yield (0.064 g, 62%) as a yellow gum. 1H NMR (CDCl3) δ 1.49 – 1.55 (m, 2H), 1.64 – 1.71 (m, 4H), 2.91 (t, 4H, J = 5.1 Hz), 3.85 (s, 3H), 6.43 (d, 1H, J = 15.3 Hz), 6.93 – 7.01 (m, 2H), 7.15 (d, 1H, J = 3.3 Hz), 7.28 (d, 1H, J = 6 Hz), 7.79 – 7.87 (m, 3H), 8.28 (bs, 1H); 13C NMR (CDCl3) δ 24.2, 26.2, 52.1, 54.1, 119.6, 119.9, 123.2, 124.2, 124.7, 127.6, 128.0, 130.5, 131.7, 134.5, 139.9, 150.1, 164.0, 168.4; MS (ES+) m/z 371 (M+H); HRMS Calcd for C20H22N2O3S: 370.1351. Found: 370.1341.
(E)-Methyl 5-(3-(furan-2-yl)acrylamido)-2-(piperidin-1-yl)benzoate (24b)
Compound 24b was prepared similar to compound 5a by the treatment of 23 (0.068 g, 0.29 mmol) and 3-(2-furyl)acrylic acid, 4b (0.047 g, 0.34 mmol) in the presence of EDAC (0.14 g, 0.73 mmol) and a DMAP (0.005 g, 0.041 mmol) to yield (0.081 g, 78% yield) as a yellow gum. 1H NMR (CDCl3) δ 1.54 – 1.58 (m, 2H), 1.66 – 1.73 (m, 4H), 2.92 – 2.96 (m, 4H), 3.88 (s, 3H), 6.44 – 6.49 (m, 2H), 6.56 (d, 1H, J = 3.3 Hz), 6.98 (d, 1H, J = 9.3 Hz), 7.43 (d, 1H, J = 1.8 Hz), 7.50 (d, 1H, J = 15.3 Hz), 7.76 – 7.8 (m, 3H); 13C NMR (CDCl3) δ 24.6, 26.7, 52.7, 54.5, 112.6, 114.6, 119.2, 119.9, 123.6, 124.5, 125.1, 128.9, 132.2, 144.6, 150.4, 151.7, 164.6, 168.9; MS (ES+) m/z 355 (M+H); Anal Calcd for C20H22N2O4: C, 67.78; H, 6.26; N, 7.90. Found: C, 67.90; H, 6.50; N, 7.70.
(E)-5-(3-(thiophen-2-yl)acrylamido)-2-(piperidin-1-yl)benzoic acid (25a)
Compound 25a was prepared similar to compound 1 by hydrolysis of 24a (0.056 g, 0.15 mmol) using 1N. NaOH in a MeOH / THF (2 mL, 1:1) to afford (0.048 g; 89%) as a white solid. Mp 239 °C (decomp.); 1H NMR (DMSO-d6) δ 1.61–1.63 (m, 2H), 1.75 (bs, 4H), 3.02 – 3.04 (m, 4H), 6.56 (d, 1H, J = 15.6 Hz), 7.15 (t, 1H, J = 3.6 Hz), 7.48 (d, 1H, J = 3.3 Hz), 7.67 – 7.79 (m, 3H), 8.01 (dd, 1H, J1 = 2.4 Hz, J2 = 8.7 Hz), 8.29 (d, 1H, J = 2.4 Hz), 10.44 (s, 1H); 13C NMR (DMSO-d6) δ 22.6, 26.0, 54.2, 120.8, 121.1, 124.1, 124.2, 125.9, 128.9, 129.2, 132.0, 134.1, 138.8, 140.0, 145.5, 163.9, 167.0; MS (ES+) m/z 357 (M+H); Anal Calcd for C19H20N2O3S: C, 64.02; H, 5.66; N, 7.86. Found: C, 63.69; H, 5.67; N, 7.62.
(E)-5-(3-(furan-2-yl)acrylamido)-2-(piperidin-1-yl)benzoic acid (25b)
Compound 25b was prepared similar to compound 1 by hydrolysis of 24b (0.08 g, 0.23 mmol) using 1N. NaOH in MeOH / THF (2 mL, 1:1) to afford (0.078 g; 100%) as oil; 1H NMR (CD3OD) δ 1.82 – 2.07 (m, 6H), 3.66 – 3.73 (m, 4H), 6.55 – 6.75 (m, 3H), 7.47 (d, 1H, J = 14.4 Hz), 7.63 (s, 1H), 7.89 – 8.16 (m, 2H), 8.59 (bs, 1H); 13C NMR (CD3OD) δ 22.9, 26.8, 58.6, 114.0, 116.7, 119.4, 124.2, 124.5, 126.9, 130.9, 140.6, 142.4, 146.9 (2C), 152.9, 167.3 (2C); MS (ES+) m/z 341 (M+H); HRMS Calcd for C19H20N2O4: 340.1423. Found: 340.1413.
(E)-N-(3-(Hydroxymethyl)-4-morpholinophenyl)-3-(thiophen-2-yl)acrylamide (26a)
To a solution of compound 5a (0.500 g, 1.34 mmol) in a mixture of anhydrous CH2Cl2 (35 mL) and THF (5 mL), DIBAL-H (6.7 mL, 1M in CH2Cl2, excess) was added and stirred at 0 °C for 1 h. Then it was allowed to attain room temperature at which it was stirred for an additional 2 h. TLC analysis (1:1 EtOAc / CHCl3) revealed that the reduction is complete. The reaction mixture was cooled to 0 °C and was quenched by careful addition of MeOH (3 mL) and 1N. HCl (0.5 mL). The solvent was removed under reduced pressure and the residue obtained was dissolved in CHCl3 (300 mL) and washed with water (2 × 75 mL), brine (1 × 150 mL) and dried (Na2SO4). The drying agent was filtered off and the solvent was removed in vaccuo to afford the crude product which was purified by flash column chromatography on silica gel (20 × 3 cm) using EtOAc / hexanes (3 : 1) as eluent to furnish compound 26a as a yellow solid. (0.354 g, 77 %); Mp 184 – 185 °C; 1H NMR (DMSO-d6) δ 2.79 – 2.83 (m, 4H), 3.65 – 3.75 (m, 4H), 4.54 (d, 2H, J = 5.1 Hz), 5.12 (t, 1H, J = 5.4 Hz), 6.58 (d, 1H, J = 15.0 Hz), 7.02 (d, 1H, J = 8.7 Hz), 7.13 (dd, 1H, J1 = 3.6 Hz, J2 = 5.1 Hz), 7.42 (d, 1H, J = 3.3 Hz), 7.60–7.74 (m, 4H) and 10.1 (bs, 1H); 13C NMR (DMSO-d6) δ 52.7, 58.5, 66.6, 118.1, 119.0, 119.1, 121.1, 128.3, 128.4, 131.1, 132.7, 135.0, 137.1, 139.8, 145.6 and 162.9; MS (ES) m/z 343 (M – H); Anal Calcd for C18H20N2O3S: C, 62.77; H, 5.85; N, 8.13. Found: C, 62.78; H, 5.95; N, 8.08.
(E)-3-(Furan-2-yl)-N-(3-(hydroxymethyl)-4-morpholinophenyl)acrylamide (26b)
Compound 26b was synthesized, following the procedure used for preparation of 26a, by the reaction of 5b (0.500 g, 1.40 mmol) and DIBAL-H (7.02 mL, 1M in CH2Cl2) in CH2Cl2 (35 mL) and THF (5 mL) for 2 h at room temperature to furnish pure product. (0.372 g, 81 %); Mp 182 – 183 °C; 1H NMR (DMSO – d6) δ 2.74–2.83 (m, 4H), 3.65–3.74 (m, 4H), 4.54 (d, 2H, J = 5.1 Hz), 5.12 (t, 1H, J = 5.4 Hz), 6.58–6.67 (m, 2H), 6.82 (d, 1H, J = 3.3 Hz), 7.02 (d, 1H, J = 8.7 Hz), 7.35 (d, 1H, J = 15.3 Hz), 7.61–7.71 (m, 2H), 7.81 (d, 1H, J = 1.5 Hz) and 10.1 (bs, 1H); 13C NMR (DMSO – d6) δ 52.6, 58.5, 66.6, 112.5, 114.3, 118.2, 119.0, 119.1, 119.6, 126.8, 134.9, 137.0, 145.0, 145.5, 150.9 and 163.0; MS (ES) m/z 327 (M – H); Anal Calcd for C18H20N2O4: C, 65.84; H, 6.14; N, 8.53. Found: C, 65.55; H, 6.27; N, 8.37.
(E)-N-(3-Formyl-4-morpholinophenyl)-3-(thiophen-2-yl)acrylamide (27a)
To a solution of compound 26a (0.500 g, 0.44 mmol) in anhydrous THF at 0 °C, pyridinium chlorochromate (0.188 g, 0.87 mmol) was added and stirred for 30 min and then at room temperature for 1 h. TLC analysis (1:1 EtOAc / CHCl3) revealed that the reaction was complete. The solvent was removed under vacuum and the crude product obtained was purified by column chromatography over Si gel using EtOAc/CHCl3 (1:4) as the eluent to afford pure 27a. (0.107 g, 72 %); Mp 218 – 219 °C; 1H NMR (DMSO) δ 2.94–3.03 (m, 4H), 3.72–3.82 (m, 4H), 6.53 (d, 1H, J = 15.3 Hz), 7.11–7.17 (m, 1H), 7.25 (d, 1H, J = 8.7 Hz), 7.45 (d, 1H, J = 3.3 Hz), 7.66 (d, 1H, J = 5.1 Hz), 7.74 (d, 1H, J = 15.6 Hz), 7.88 (dd, 1H, J1 = 2.4 Hz, J2 = 8.7 Hz), 8.06 (d, 1H, J = 2.7 Hz), 10.2 (s, 1H) and 10.3 (bs, 1H); 13C NMR (CD3COCD3) δ 55.3, 67.4, 120.1, 121.0, 121.4, 126.8, 128.7, 129.1, 130.1, 131.7, 134.6, 136.1, 141.0, 152.5, 164.2 and 191.0; MS (ES) m/z 341 (M – H); Anal Calcd for C18H18N2O3S: C, 63.14; H, 5.30; N, 8.18. Found: C, 63.39; H, 5.24; N, 8.00.
(E)-N-(3-Formyl-4-morpholinophenyl)-3-(furan-2-yl)acrylamide (27b)
27b was synthesized, following the procedure used for preparation of 27a, by the reaction of 26b (0.188 g, 0.57 mmol) and pyridinium chlorochromate (0.247 g, 1.15 mmol) in anhydrous THF (10 mL) to furnish the pure product. (0.115 g, 61 %); Mp 231 – 232 °C; 1H NMR (DMSO-d6) δ 2.94–3.03 (m, 4H), 3.73–3.82 (m, 4H), 6.53–6.65 (m, 2H), 6.86 (d, 1H, J = 3.6 Hz), 7.24 (d, 1H, J = 9.0 Hz), 7.38 (d, 1H, J = 15.6 Hz), 7.82 (d, 1H, J = 1.5 Hz), 7.89 (dd, 1H, J1 = 2.5 Hz, J2 = 8.8 Hz), 8.06 (d, 1H, J = 2.7 Hz), 10.2 (s, 1H) and 10.3 (bs, 1H); 13C NMR (DMSO-d6) δ 54.1, 66.2, 112.6, 114.8, 119.0, 119.1, 120.2, 126.0, 127.4, 128.2, 134.6, 145.3, 150.9, 151.2, 163.4 and 190.7; MS (ES) m/z 325 (M – H); Anal Calcd for C18H18N2O4. 0.3H2O: C, 65.20; H, 5.65; N, 8.45. Found: C, 64.90; H, 5.35; N, 8.33.
5-((E)-3-(Thiophen-2-yl)acrylamido)-2-morpholinobenzamide (28)
To a solution of compound 1 (0.050 g, 0.14 mmol) in anhydrous CH2Cl2 (4 mL) at room temperature a mixture of thionyl chloride (0.2 mL) and anhydrous DMF (0.2 mL) was added and the resulting mixture was stirred for 4 h at room temperature. The solvent was removed under reduced pressure. The resulting gum was dissolved in anhydrous CH2Cl2 (3 mL) and excess 50% aqueous ammonium hydroxide (3 mL) was added to achieve basic pH (∼12) and stirred at room temperature for 2 h. The solvent was removed under reduced pressure and the crude compound was purified by flash column chromatography (20 × 1 cm) on silica gel using EtOAc / CHCl3 (1:1) as eluent to furnish the pure amide 28. (0.013 g, 26 %); 1H NMR (DMSO-d6) δ 2.86–2.94 (m, 4H), 3.71–3.78 (m, 4H), 6.55 (d, 1H, J = 15.3 Hz), 7.14 (dd, 1H, J1 = 3.6 Hz, J2 = 5.1 Hz), 7.20 (d, 1H, J = 8.7 Hz), 7.45 (d, 1H, J = 3.0 Hz), 7.57 (bs, 1H), 7.66 (d, 1H, J = 5.1 Hz), 7.73 (d, 1H, J = 15.6 Hz), 7.84 (dd, 1H, J1 = 2.7 Hz, J2 = 8.7 Hz), 7.99 (d, 1H, J = 2.7 Hz), 8.64 (bs, 1H) and 10.2 (bs, 1H); 13C NMR (DMSO-d6) δ 52.9, 66.4, 120.4, 120.8, 121.1, 122.0, 128.5 (2C), 129.3, 131.3, 133.1, 135.1, 139.7, 146.2, 163.1 and 167.7; MS (ES) m/z 356 (M – H).
Acknowledgements
Authors wish to acknowledge the generous financial support from the Department of Chemistry, University of Alabama at Birmingham (UAB). Beginning Grant-in-Aid (AHA0865323E) and Grant-in-Aid (AHA0855076E) from American Heart Association Greater Southeast Affiliate are acknowledged. Breast Spore Pilot grant from the UAB Comprehensive Cancer Center and a translational research grant from UAB Center for Clinical and Translational Science are also acknowledged.
Abbreviations
- Dabsyl
4-(4-Dimethylaminophenylazo)-benzenesulfonyl
- DIBAL
Diisobutylaluminum hydride
- DMAP
N, N-dimethylaminopyridine
- DMF
N,N-Dimethylformamide
- EDAC
Ethyl (N, N-dimethylaminopropyl) carbodiimide
- EDANS
5-[(2-aminoethyl)amino]naphthalene-1-sulfonyl
- FRET
Fluorescence Resonance Energy Transfer
- IC50
Half maximal inhibitory concentration of inhibitor
- MRSA
Methicillin resistant Staphylococcus aureus
- MS
Mass spectrometry
- PCC
Pyridinium chlorochromate
- Pd black
Palladium black
- Pd/C
Palladium on activated carbon
- ppm
Parts per million
- SAR
Structure activity relationship
- S. aureus
Staphylococcus aureus
- SrtA
Sortase A
- THF
Tetrahydrofuran
- Thr
Threonine
- TLC
Thin layer chromatography
- TMS
Tetramethylsilane
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. J. Am. Med. Assoc. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 2.Kazakova SV, Hageman JC, Matava M, Srinivasan A, Phelan L, Garfinkel B, Boo T, McAllister S, Anderson J, Jensen B, Dodson D, Lonsway D, McDougal LK, Arduino M, Fraser VJ, Killgore G, Tenover FC, Cody S, Jernigan DB. A clone of methicillin-resistant Staphylococcus aureus among professional football players. N. Engl. J. Med. 2005;352:468–475. doi: 10.1056/NEJMoa042859. [DOI] [PubMed] [Google Scholar]
- 3.Gosbell IB, Barbagiannakos T, Neville SA, Mercer JL, Vickery AM, O’Brien FG, Coombs GW, Malkowski MJ, Pearson JC. Non-multiresistant methicillin-resistant Staphylococcus aureus bacteraemia in Sydney, Australia: emergence of EMRSA-15 Oceania, Queensland and Western Australian MRSA strains. Pathology. 2006;38:239–244. doi: 10.1080/00313020600699227. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert M, MacDonald J, Gregson D, Siushansian J, Zhang K, Elsayed S, Laupland K, Louie T, Hope K, Mulvey M, Gillespie J, Nielsen D, Wheeler V, Louie M, Honish A, Keays G, Conly J. Outbreak in Alberta of community-acquired (USA300) methicillin-resistant Staphylococcus aureus in people with a history of drug use, homelessness or incarceration. Canad. Med. Assoc. J. 2006;175:149–154. doi: 10.1503/cmaj.051565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.David MD, Kearns AM, Gossain S, Ganner M, Holmes A. Community-associated meticillin-resistant Staphylococcus aureus: nosocomial transmission in a neonatal unit. J. Hosp. Infect. 2006;64:244–250. doi: 10.1016/j.jhin.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 6.Baggett HC, Hennessy TW, Rudolph K, Bruden D, Reasonover A, Parkinson A, Sparks R, Donlan RM, Martinez P, Mongkolrattanothai K, Butler JC. Community-onset methicillin-resistant Staphylococcus aureus associated with antibiotic use and the cytotoxin Panton-Valentine leukocidin during a furunculosis outbreak in rural Alaska. J. Infect. Dis. 2004;189:1565–1573. doi: 10.1086/383247. [DOI] [PubMed] [Google Scholar]
- 7.Zhu W, Clark NC, McDougal LK, Hageman J, McDonald LC, Patel JB. Vancomycin-resistant Staphylococcus aureus isolates associated with Inc18-like vanA plasmids in Michigan. Antimicrob. Agents chemother. 2008;52:452–457. doi: 10.1128/AAC.00908-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang S, Sievert DM, Hageman JC, Boulton ML, Tenover FC, Downes FP, Shah S, Rudrik JT, Pupp GR, Brown WJ, Cardo D, Fridkin SK. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N. Engl. J. Med. 2003;348:1342–1347. doi: 10.1056/NEJMoa025025. [DOI] [PubMed] [Google Scholar]
- 9.Anonymous, Staphylococcus aureus resistant to vancomycin-United States, MMWR. 2002;51:565–566. [PubMed] [Google Scholar]
- 10.Anonymous, Vancomycin-resistant Staphylococcus aureus--New York, MMWR. 2004;53:322. [PubMed] [Google Scholar]
- 11.Navarre WW, Schneewind O. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 1999;63:174–229. doi: 10.1128/mmbr.63.1.174-229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cossart P, Jonquieres R. Sortase, a universal target for therapeutic agents against gram-positive bacteria? Proc. Natl. Acad. Sci. U.S.A. 2000;97:5013–5015. doi: 10.1073/pnas.97.10.5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischetti VA, Pancholi V, Schneewind O. Conservation of a hexapeptide sequence in the anchor region of surface proteins from gram-positive cocci. Mol. Microbiol. 1990;4:1603–1605. doi: 10.1111/j.1365-2958.1990.tb02072.x. [DOI] [PubMed] [Google Scholar]
- 14.Schneewind O, Model P, Fischetti V. Sorting of protein A to the staphylococcal cell wall. Cell. 1992;70:267–281. doi: 10.1016/0092-8674(92)90101-h. [DOI] [PubMed] [Google Scholar]
- 15.Schneewind O, Mihaylova-Petkov D, Model P. Cell wall sorting signals in surface proteins of gram-positive bacteria. Embo. J. 1993;12:4803–4811. doi: 10.1002/j.1460-2075.1993.tb06169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Navarre WW, Schneewind O. Proteolytic cleavage and cell wall anchoring at the LPXTG motif of surface proteins in gram-positive bacteria. Mol. Microbiol. 1994;14:115–121. doi: 10.1111/j.1365-2958.1994.tb01271.x. [DOI] [PubMed] [Google Scholar]
- 17.Perry AM, Ton-That H, Mazmanian SK, Schneewind O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. III. Lipid II is an in vivo peptidoglycan substrate for sortase-catalyzed surface protein anchoring. J. Biol. Chem. 2002;277:16241–16248. doi: 10.1074/jbc.M109194200. [DOI] [PubMed] [Google Scholar]
- 18.Jonsson IM, Mazmanian SK, Schneewind O, Bremell T, Tarkowski A. The role of Staphylococcus aureus sortase A and sortase B in murine arthritis. Microb. Infect. 2003;5:775–780. doi: 10.1016/s1286-4579(03)00143-6. [DOI] [PubMed] [Google Scholar]
- 19.Mazmanian SK, Liu G, Jensen ER, Lenoy E, Schneewind O. Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5510–5515. doi: 10.1073/pnas.080520697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnett TC, Scott JR. Differential recognition of surface proteins in Streptococcus pyogenes by two sortase gene homologs. J. bacteriol. 2002;184:2181–2191. doi: 10.1128/JB.184.8.2181-2191.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bierne H, Mazmanian SK, Trost M, Pucciarelli MG, Liu G, Dehoux P, Jansch L, Garcia-del Portillo F, Schneewind O, Cossart P. Inactivation of the srtA gene in Listeria monocytogenes inhibits anchoring of surface proteins and affects virulence. Mol. Microbiol. 2002;43:869–881. doi: 10.1046/j.1365-2958.2002.02798.x. [DOI] [PubMed] [Google Scholar]
- 22.Bolken TC, Franke CA, Jones KF, Zeller GO, Jones CH, Dutton EK, Hruby DE. Inactivation of the srtA gene in Streptococcus gordonii inhibits cell wall anchoring of surface proteins and decreases in vitro and in vivo adhesion. Infect. Immun. 2001;69:75–80. doi: 10.1128/IAI.69.1.75-80.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garandeau C, Reglier-Poupet H, Dubail I, Beretti JL, Berche P, Charbit A. The sortase SrtA of Listeria monocytogenes is involved in processing of internalin and in virulence. Infect. Immun. 2002;70:1382–1390. doi: 10.1128/IAI.70.3.1382-1390.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jonsson IM, Mazmanian SK, Schneewind O, Verdrengh M, Bremell T, Tarkowski A. On the role of Staphylococcus aureus sortase and sortase-catalyzed surface protein anchoring in murine septic arthritis. J. Infect. Dis. 2002;185:1417–1424. doi: 10.1086/340503. [DOI] [PubMed] [Google Scholar]
- 25.Kang SS, Kim JG, Lee TH, Oh KB. Flavonols inhibit sortases and sortase-mediated Staphylococcus aureus clumping to fibrinogen. Biol. Pharm. Bull. 2006;29:1751–1755. doi: 10.1248/bpb.29.1751. [DOI] [PubMed] [Google Scholar]
- 26.Kim SH, Shin DS, Oh MN, Chung SC, Lee JS, Chang IM, Oh KB. Inhibition of sortase a bacterial surface protein anchoring transpeptidase, by beta-sitosterol-3-O-glucopyranoside from Fritillaria verticillata. Biosci. Biotechnol. Biochem. 2003;67:2477–2479. doi: 10.1271/bbb.67.2477. [DOI] [PubMed] [Google Scholar]
- 27.Kim SW, Chang IM, Oh KB. Inhibition of the bacterial surface protein anchoring transpeptidase sortase by medicinal plants. Biosci. Biotechnol. Biochem. 2002;66:2751–2754. doi: 10.1271/bbb.66.2751. [DOI] [PubMed] [Google Scholar]
- 28.Scott CJ, McDowell A, Martin SL, Lynas JF, Vandenbroeck K, Walker B. Irreversible inhibition of the bacterial cysteine protease-transpeptidase sortase (SrtA) by substrate-derived affinity labels. Biochem. J. 2002;366:953–958. doi: 10.1042/BJ20020602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oh KB, Kim SH, Lee J, Cho WJ, Lee T, Kim S. Discovery of diarylacrylonitriles as a novel series of small molecule sortase A inhibitors. J. med. chem. 2004;47:2418–2421. doi: 10.1021/jm0498708. [DOI] [PubMed] [Google Scholar]
- 30.Chenna BC, Shinkre BA, King JR, Lucius AL, Narayana SV, Velu SE. Identification of novel inhibitors of bacterial surface enzyme Staphylococcus aureus Sortase A. Bioorg. Med. Chem. Lett. 2008;18:380–385. doi: 10.1016/j.bmcl.2007.10.051. [DOI] [PubMed] [Google Scholar]
- 31.Skotnicki JS, Kearney RM, Smith AL. Synthesis of secorapamycin esters and amides Tetrahedron Lett. 1994;35:197–200. [Google Scholar]
- 32.Kruger RG, Dostal P, McCafferty DG. Development of a high-performance liquid chromatography assay and revision of kinetic parameters for the Staphylococcus aureus sortase transpeptidase SrtA. Anal. Biochem. 2004;326:42–48. doi: 10.1016/j.ab.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 33.Ton-That H, Mazmanian SK, Faull KF, Schneewind O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. Sortase catalyzed in vitro transpeptidation reaction using LPXTG peptide and NH(2)-Gly(3) substrates. J. Biol. Chem. 2000;275:9876–9881. doi: 10.1074/jbc.275.13.9876. [DOI] [PubMed] [Google Scholar]
- 34.Wadsworth DH, Geer SM, Detty MR. Preparation of arylpropiolate esters from trichlorocyclopropenium cation and elaboration of the esters to unsymmetrical 1,4-pentadiyn-3-ones and unsymmetrical tellurapyranones. J. Org. Chem. 1987;52:3662–3668. [Google Scholar]