

Abstract
Following the discovery that nearly half of all cutaneous melanomas harbour a mutation in the BRAF gene, molecular targeted kinase inhibitors have been developed for the treatment of metastatic melanoma and have dramatically improved outcomes for those patients with BRAF mutant disease, achieving high levels of objective response and prolonging survival. Since 2011, the specific BRAF targeted agents, vemurafenib and dabrafenib, and the MEK inhibitor, trametinib, have been licensed for the treatment of patients with unresectable or metastatic BRAF mutant melanoma. As with other biological targeted agents, these drugs are associated with predictable patterns of adverse events. Proactive toxicity management is important to ensure maximum treatment benefit and avoid unnecessary treatment discontinuation. We review the most common and serious adverse events associated with BRAF targeted agents and suggest management algorithms to guide practitioners in using these drugs effectively in the clinic.
Keywords: BRAF inhibitor, dabrafenib, management, MEK inhibitor, melanoma, toxicity, trametinib, vemurafenib
Introduction
Melanoma is the most lethal skin cancer and its incidence continues to increase worldwide. Until recently, the only effective treatment was surgery, since neither chemotherapy nor radiotherapy has been shown to impact on overall survival of patients with advanced, unresectable disease. Better understanding of the molecular biology of melanoma has radically changed how this disease is now managed. Dysregulation of mitogen-activated protein kinase (MAPK) signalling is a hallmark of melanoma and approximately half of all patients with metastatic cutaneous melanoma have a mutation in the BRAF gene [Chapman et al. 2011]. In particular, BRAFV600-mutated melanoma cells are dependent on RAF/MEK/ERK signalling [Ribas and Flaherty, 2011]. Based on improved overall survival confirmed in international multicentre randomized phase III trials, two small molecule inhibitors of BRAF, vemurafenib and dabrafenib, and one inhibitor of MEK, trametinib, have been licensed for the treatment of metastatic BRAF mutant melanoma. The combination of dabrafenib plus trametinib is now also being registered for approved use in various countries around the world.
Several other mechanistically similar targeted agents are in development as single agents or in combination regimens. As selective rather than specific kinase inhibitors, these oral agents administered chronically to patients are associated with characteristic patterns of drug-related toxicities and their optimal use depends on prompt and active side effect management in order to maintain patients on therapy. Certain toxicities appear to be common to all agents, while others are more specific to individual drugs. The purpose of this paper is to summarize the most commonly reported and serious side effects of BRAF inhibitors (BRAFi) and MEK inhibitors (MEKi) approved for clinical use and offer recommendations for their management. Since data regarding management of specific side effects are currently limited, it should be noted that some recommendations are based on the authors’ experience from using these agents in clinical trials as well as routine clinical practice.
Commonly occurring toxicities associated with BRAFi and MEKi
Vemurafenib (Zelboraf, Roche Products Ltd, Hertfordshire, UK) was licensed in the USA in August 2011 and in Europe in March 2012 and is the best studied of all BRAFi. The standard dose of vemurafenib is 960 mg taken orally twice a day, continuously. In the BRIM-3 registration trial comparing vemurafenib with dacarbazine chemotherapy, the most common adverse events (AEs) graded as moderate or severe (grade 2 or more) using international common toxicity criteria (CTC) associated with vemurafenib treatment were cutaneous [including rash, keratoacanthoma, cutaneous squamous cell carcinoma (SCC) and photosensitivity], arthralgia, diarrhoea and fatigue [Chapman et al. 2011]. In BRIM-3, vemurafenib AEs led to dose modification or treatment interruption in 38% of treated patients (Table 1) compared with 16% of patients receiving dacarbazine. The higher event frequency associated with vemurafenib is in part explained by the fact that patients remained on oral therapy for longer than with dacarbazine. Of note, there were no vemurafenib-related deaths reported. The incidence of SCC in 19% of patients, although easily managed with local treatment, was cause for concern. Therefore, an extensive international single arm safety study was undertaken, which included 3226 patients with advanced melanoma treated with vemurafenib [Larkin et al. 2014]. This confirmed all that was already known regarding vemurafenib toxicity, although in some cases the incidence rates were lower in the larger population. The most common AEs of all severity grades were rash (49%), arthralgia (39%), fatigue (34%), photosensitivity (31%), alopecia (26%) and nausea (19%). Forty-six percent of patients experienced grade 3 or 4 AEs, which were most commonly cutaneous SCC (12%), rash (5%), liver function abnormalities (5%), arthralgia (3%) and fatigue (3%). The pattern of onset of vemurafenib side effects is also predictable: photosensitivity and rash occur usually within days of starting the drug. Arthralgia, diarrhoea, fatigue, alopecia and occurrence of other skin lesions tend to occur over weeks and months. While photosensitivity continues with drug use, other AEs including rash, skin lesions and arthralgia can regress and be less problematic after the first few months.
Table 1.
Comparative toxicities of vemurafinib, dabrafenib and trametinib reported in different randomized trials.
| Vemurafenib*$ | Dabrafenib‡ | Trametinib§ | Dabrafenib + trametinib‖¶# | |
|---|---|---|---|---|
| Rash | 41 (9) | 30 (0) | 57 (8) | 27 (0) |
| Cutaneous SCC | 19 (19) | 10 (4) | 0 | 7 (5) |
| Diarrhoea | 25 (<1) | NR | 43 (0) | 36 (2) |
| Pyrexia | NR | 16 (3) | NR | 51 (6) |
| Arthralgia | 56 (6) | 19 (<1) | NR | 24 (0) |
| Fatigue | 46 (3) | 18 (1) | 26 (4) | 53 (4) |
| Cardiac | NR | NR | 7 (1) | 9 (0) |
| ILD/pneumonitis | NR | NR | 2 (2) | 1 |
| Ophthalmologic | NR | NR | 9 (<1) | 2 (2) |
| Hypertension | NR | 4 (0) | 15 (12) | 9 (2) |
| Hyperglycaemia | NR | 49 (2)# | NR | 58 (5) |
| Liver laboratory abnormalities: | 36 (11) | 26 (2) # | 24 (2) | 60 (2) |
| Alkaline phosphatase | 11 (0) # | 39 (3) | 42 (4) | |
| Alanine aminotransferase | 0 (0) # | NR | 15 (0) | |
| Bilirubin | 60 (2)# | 60 (2) | 60 (5) | |
| Aspartate aminotransferase |
Toxicities are expressed as percentage of all CTC grades (CTC grade 3/4). The data in this table are summarized from different trials and do not represent direct comparisons.
Data are a composite of phase I/II and III trial data since limited data are available from the phase III trial.
SCC, squamous cell carcinoma; ILD, interstitial lung disease; NR, not reported.
A second BRAFi, dabrafenib (Tafinlar, recommended dose 150 mg twice daily), was subsequently developed by GlaxoSmithKline (GSK, Middlesex, UK) and compared in the registration BREAK-3 trial with standard dacarbazine. In BREAK-3, 53% of patients receiving dabrafenib experienced grade 2 or higher treatment-related AEs (Table 1) compared with 44% who received dacarbazine [Hauschild et al. 2012, 2013]. The most common dabrafenib-induced AEs were hyperkeratosis (39%), headache (35%), arthralgia (35%) and pyrexia (32%). Other skin toxicities included keratocanthoma and SCC (10%), but unlike vemurafenib, photosensitivity was extremely rare. Grade 3–4 AEs were uncommon, but pyrexia was identified as a new and potentially serious toxicity occurring in some 5% of treated patients, not previously seen with vemurafenib. Dose reduction of dabrafenib was needed in 28% of trial patients, but only 3% discontinued entirely because of drug intolerance. Accumulating experience with the two BRAFi suggests that although some toxicities such as skin toxicity, arthralgia and pyrexia are common to both inhibitors, the type and severity of these toxicities vary considerably and may influence choice of drug. For example, photosensitivity is common with vemurafenib but rare with dabrafenib, while fever and chills are common with dabrafenib but rare with vemurafenib.
Characteristic of treatment with BRAFi is the emergence of drug resistance via activation of parallel signalling pathways [Sosman et al. 2012]. Inhibition of MEK, downstream of BRAF, has been tested as a strategy to bypass resistance. In the METRIC trial [Flaherty et al. 2012b], the MEK-1 and MEK-2 inhibitor, trametinib (Mekinist; GSK, recommended dose: 2 mg once daily) was reported to be superior to standard dacarbazine, although indirect comparisons suggest that the benefit in terms of response rate and progression-free survival is less than with BRAFi. The toxicity profile of trametinib differs from BRAFi (Table 1). Rash, diarrhoea, fatigue, peripheral oedema and acneiform dermatitis are the most common side effects. Cardiac (decreased ejection fraction or ventricular dysfunction), ocular and interstitial lung disease (ILD) or pneumonitis events were also observed in 7%, 9% and 2% of patients treated in the METRIC trial, which are not recognized side effects of BRAFi. However, secondary skin neoplasms do not occur with the MEKi.
The combination of dabrafenib and trametinib was tested first in a phase I/II trial and a randomized component identified improved efficacy compared with dabrafenib alone [Flaherty et al. 2012a]. Remarkably, the combination regimen using full doses of both agents appeared to generate fewer skin AEs compared with BRAFi monotherapy. Interim results from the COMBI-d double-blind placebo-controlled randomized phase III trial which compared dabrafenib plus trametinib with dabrafenib alone confirmed that the combination of BRAFi/MEKi was superior to dabrafenib alone [Long et al. 2014]. COMBI-d recruited 423 patients and also confirmed a lower frequency of malignant and hyperproliferative skin lesions associated with combined BRAFi/MEKi (2% incidence) compared with dabrafenib monotherapy (9% incidence). There were no new, unexpected toxicities associated with the combination regimen (Table 1). AEs which occurred more frequently with combination therapy were fever (51%) and chills (30%), fatigue (35%), diarrhoea (24%), hypertension (22%) and vomiting (20%). Alopecia was reported less often in the combination arm. Of note, in the phase I/II trial, the most frequently occurring grade 3 or 4 AE in the full-dose combination arm was neutropenia in 11% of patients, with one case of febrile neutropenia. In the phase III COMBI-d trial, the most common serious AEs thought to be drug induced were fever (31% subjects), chills (8%) and decrease in left ventricular ejection fraction (5%).
This unique observation of dual therapy being less toxic to the skin than monotherapy has a biological explanation. The enhanced rate of secondary skin malignancies seen during BRAFi treatment alone is due to BRAFi-induced proliferation of cells possessing secondary mutations (Su et al. 2012). BRAFi do not initiate tumourigenesis but rather accelerate the progression of preexisting subclinical cancerous lesions, with paradoxical MAPK pathway activation, which is inhibited by treatment with a MEKi. Rare cases of mutant-Ras induced leukaemia [Callahan et al. 2012] and K-Ras mutated colon cancer [Andrews et al. 2013] have been reported in patients with advanced melanoma treated with BRAFi alone. It is postulated that these are driven by the same mechanism of paradoxical MAPK pathway activation.
Many of the characteristic side effects experienced by patients with BRAFi and MEKi are relatively novel compared with cytotoxic chemotherapy previously used to treat advanced melanoma. In contrast to cytotoxic chemotherapy, the common toxicities are generally not life threatening, but optimal use of these agents does require active and effective side-effect management since dose interruptions and modifications are often required, following which patients can remain on treatment for many months, with controlled disease. Dermatological toxicity management algorithms were published recently [Sinha et al. 2012]. No other consensus toxicity management guidelines exist, so the following recommendations based on published evidence and clinical experience offer consensus strategies to manage other common and serious side effects associated with BRAFi and MEKi therapy.
Management of common side effects of BRAFi and MEKi
Basic principles
Life-threatening toxicities associated with BRAFi and MEKi toxicities are extremely rare. There are two main stages to managing patients on these agents: those reactions which occur within days of starting the drug and those which are associated with their chronic use over many months of exposure. Prior to commencing treatment and in the first few weeks of therapy, good patient education, written information and specialist nurse support are essential. As a rule of thumb, drug treatment should continue in the presence of mild toxicities and other pharmacological agents be considered to assist in ameliorating symptoms. Moderate and severe toxicities warrant treatment interruption and retreatment should be considered once the reaction has resolved. Standard dose reductions are recommended by manufacturers (Table 2), although it may be possible to maintain a dose by managing toxicities with other interventions. Anecdotal and evidence from limited case reports suggests that temporary cessation of BRAFi or MEKi for limited periods is unlikely to negatively influence disease control and importantly any disease growth can be reversed on restarting the BRAFi/MEKi [Koop et al. 2014; Seghers et al. 2012]. Indeed, one study undertaken in mice reported improved survival on an interrupted vemurafenib dosing schedule [Das Thakur et al. 2013], providing a rationale for future testing of intermittent dosing as a strategy both to improve survival as well as to mitigate toxicities.
Table 2.
Recommended dose reductions for licensed BRAF and MEK inhibitors.
| Drug | Dose reduction for first occurrence of toxicity | Dose reduction for second occurrence of toxicity | Dose adjustment for third occurrence of toxicity |
|---|---|---|---|
| VemurafenibStandard dose: 960 mg twice daily | Reduce to 720 mg twice daily | Reduce to 480 mg twice daily | Doses less than 480 mg twice daily unlikely to be pharmacologically active, so consider switching to dabrafenib or intermittent dosing |
| Dabrafenib*Standard dose: 150 mg twice daily | Reduce to 100 mg twice daily | Reduce to 75 mg twice daily | Reduce to 50 mg twice daily |
| Trametinib*Standard dose: 2 mg once daily | Reduce to 1.5 mg once daily | Reduce to 1 mg once daily | Stop trametinib |
Standard doses of dabrafenib and trametinib are used in combination. Dose modifications should be made according to the more likely cause of adverse events.
It should also be noted that several side effects of BRAFi and MEKi – fatigue, rash, diarrhoea, deranged liver function, pneumonitis – are common to those of immune checkpoint inhibitors such as ipilimumab and anti-programmed death receptor (PD)-1 inhibitors now also entering routine clinical practice. These two classes of agents are currently primarily being used in sequence, but even so, the potential for overlapping toxicity when switching patients between treatments should be considered. The range of toxicities associated with these new agents indicates the need to ensure local specialist support services are available in dermatology, ophthalmology, cardiology, respiratory and acute oncology.
Skin toxicities
Skin toxicities are the most common AEs associated with BRAFi, occurring in up to 57% of patients (Table 1). Rash, photosensitivity, pruritis, dry skin, papilloma, alopecia, keratoacanthoma and SCC are the signature of BRAFi monotherapy, with photosensitivity being primarily associated with vemurafenib. Excellent practical advice on diagnosis, prevention and management of the main BRAFi-induced skin effects has previously been published by Sinha et al. (2012) and are summarized in Fig. 1(a, b). Skin reactions occur within days of commencing these targeted agents. Photosensivity associated with vemurafenib requires that sunscreen with sun protection factor at least 30 is used routinely and can be prescribed for patients. Rapid onset of rash requires that patients have easy access to the clinical team in the first few weeks of treatment for advice on whether or not to interrupt treatment or modify the dose. With close communication and dose adjustments, most severe reactions can be avoided and an optimal drug dose identified within the first 1–2 months of starting treatment. The absence of photosensitivity with dabrafenib and lower frequency of skin AEs overall with dabrafenib compared with vemurafenib means that dabrafenib is an appropriate alternative treatment option for those patients who are intolerant of vemurafenib due to skin toxicity. Monitoring of cutaneous eruptions associated with BRAFi can be undertaken by the prescribing team. Assessment by a trained dermatologist is not mandatory, but referrals should be individualized based on the nature of individual skin reactions. Rapidly growing lesions, keratoacanthomata or any lesion suspicious for skin malignancy warrant urgent referral for intervention. The incidence of skin lesions generally subsides after the first few months of treatment.
Figure 1.

(a) Suggested management of skin toxicities (based on algorithms suggested in Sinha et al. [2012]). (b) Suggested management of macular/papular rash or perifollicular eruptions. KI, kinase inhibitor; SCC, squamous cell carcinoma; SPF, sun protection factor; UVA, ultraviolet A; UVB, ultraviolet B. Adapted with written permission from Sinha et al. [2012].
Diarrhoea
Diarrhoea is a relatively common side effect associated with BRAF targeted therapy: 25% incidence with vemurafenib, 14% with dabrafenib and 24% with the dabrafenib/trametinib combination [Chapman et al. 2011; Long et al. 2014]. Being usually mild to moderate in severity, diarrhoea may be managed symptomatically on an outpatient basis with immediate initiation of loperamide, dose interruption and restarting treatment at a lower dose as appropriate (Table 2). Both pharmacological and nonpharmacological treatments should be used to control the symptoms and infective aetiology should always be considered if symptoms persist despite intervention or there are other risk factors. Patients continuing on treatment with chronic but tolerable loose bowel motions should also be advised of dietary modifications, including eating small frequent meals, introduction of a ‘BRAT’ diet (Bananas, Rice, Apples, Toast), stopping lactose-containing products and maintaining fluid intake with electrolyte replacement (Figure 2).
Figure 2.

Suggested management of diarrhoea. ADL, activity of daily living; KI, kinase inhibitor.
Pyrexia
Fever is specifically associated with dabrafenib rather than vemurafenib treatment. In BREAK-3, the incidence of pyrexia associated with dabrafenib was 26%, while it was considerably higher (51%) with dabrafenib/trametinib combined [Long et al. 2014]. In BREAK-3, the median time to initial onset of fever of any severity was 11 days (range 1–202 days), and the median duration of fever was 3 days (range 1–129 days). Uncomplicated pyrexia can usually be managed with good patient education and supportive care. In some instances, pyrexia may be accompanied by hypotension or general malaise and these patients may require hospitalization. Furthermore, while myelosuppression is extremely rare, risk of neutropenic sepsis should be considered and full blood count checked. This is an important aspect of educating acute oncology teams, who are used to associating fever in a patient with cancer on treatment with risk of neutropaenic sepsis. While this risk is low for patients with melanoma on targeted therapies, grade 3 neutropenia was reported in one patient on dabrafenib in the BREAK-3 trial, and in up to 11% of patients taking combination therapy (including at least one case of febrile neutropenia). It is therefore relevant to exclude sepsis in patients with pyrexia who have recently started targeted therapy, or in those experiencing new symptoms of fever despite having been on treatment for some time. In those patients who are pyrexial and unwell but not septic, dose interruption, use of paracetamol or low-dose prednisolone may be effective and allow reintroduction of drug on recovery. For some patients, intermittent fever is a pattern of their chronic therapy. In the absence of complicating factors or sepsis, these systemically well patients can remain at home and manage their fever with paracetamol, steroids and/or temporary drug dose interruption (Figure 3).
Figure 3.

Suggested management of pyrexia. BP, blood pressure; FBC, full blood count; KI, kinase inhibitor; NSAID, nonsteroidal anti-inflammatory drug.
When prescribing antibiotics, potential drug interactions should be considered. Vemurafenib is primarily metabolized by cytochrome P450 (CYP)-3A4, and dabrafenib by CYP2C8 and CYP3A4. Therefore, the following commonly used antibiotics are contraindicated: ciprofloxacin, moxifloxacin, erythromycin, clarithromycin, azithromycin (and similar drugs in these classes), in addition to some antifungal agents including fluconazole, ketoconazole, itraconazole and voriconazole (and other drugs in this class). In cases where these agents need to be used, advice is to temporarily omit the targeted agent.
Arthralgia
Arthralgia (defined as marked discomfort in one or more joints) is a common side effect of treatment with BRAFi which may develop in the first few months of treatment. The incidence and severity of arthralgia is probably higher with vemurafenib compared with dabrafenib: 56% versus 35% incidence overall and 21% versus 6% moderate–severe pain was reported in the BRIM-3 [Chapman et al. 2011] and BREAK-3 [Hauschild et al. 2012, 2013] trials. Arthralgia is not characteristic of MEKi and events were similar in both arms of the COMBI-d trial [Long et al. 2014]. Any joints may also be affected and the pain may be intermittent or constant. Arthralgia accompanied by panniculitis was reported as the presenting AEs in two patients treated with BRAFi [Zimmer et al. 2012] and both conditions improved with simple analgesia and anti-inflammatory drugs, enabling the patients to continue on BRAFi, albeit with a dose modification in one case. Severe polyarthritis which developed in a melanoma patient after 7 days of treatment with vemurafenib [Babacan et al. 2014] was reported to be effectively treated with dose interruption, nonsteroidal anti-inflammatory drugs and low-dose corticosteroids and reintroduction of vemurafenib at a lower dose. Arthralgia may be self-limiting and modified doses of BRAFi can be increased at a later date if symptoms abate. While most patients respond to conventional medications, patients with debilitating symptoms or joint effusions should be referred for rheumatology specialist advice (Figure 4).
Figure 4.
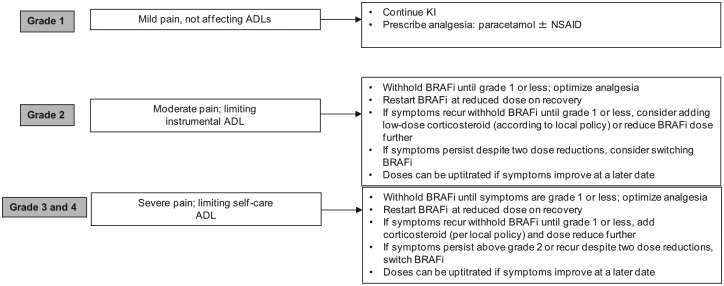
Suggested management of arthralgia. ADL, activity of daily living; BRAFi, BRAF inhibitor; KI, kinase inhibitor; NSAID, nonsteroidal anti-inflammatory drug.
Fatigue
Fatigue is a well recognized problem with many oral kinase inhibitors. Its mechanism is not well understood. The significance of fatigue is frequently downplayed by healthcare professionals, yet chronic fatigue negatively affects patients’ quality of life. In some cases, it may be managed with dose modifications or low-dose steroids. It is important that other causes of fatigue such as disease progression, infection, haematological and biochemical abnormalities (including anaemia, electrolyte and endocrine abnormalities) are considered and acted upon if identified.
Cardiac complications
Left ventricular systolic ejection fraction dysfunction
The early trials of trametinib reported 8% of patients experienced left ventricular ejection fraction (LVEF) dysfunction (defined as an absolute decrease of >10% in LVEF compared with baseline with the ejection fraction below the institution’s lower limit of normal). However, few events were thought to be specifically related to trametinib and a recorded drop in LVEF was rarely symptomatic. LVEF dysfunction is less commonly associated with BRAFi: in COMBI-d, decrease in LVEF was reported as a serious AE in 5% of patients in both arms of the trial. Thirty-one percent of patients receiving combination dabrafenib/trametinib were reported to experience significant peripheral oedema [Flaherty et al. 2012a], although this was not reported in COMBI-d. Peripheral oedema is characteristic of MEKi; however, since serial echocardiography (ECHO) was not performed routinely in the combination trial, it is difficult to fully assess whether a decrease in LVEF contributed. In the absence of known cardiac history, routine ECHO is probably not justified. However, cases of peripheral oedema or symptoms suggestive of heart failure should be investigated using standard cardiac workup, including electrocardiogram (ECG), ECHO and referral to a cardiologist if appropriate [Figure 5(a)].
Figure 5.

(a) Suggested management of reduced left ventricular ejection fraction (LVEF). (b) Suggested management of prolonged QTc. ADL, activity of daily living; BRAFi, BRAF inhibitor; KI, kinase inhibitor; LLN, lower limit of normal; LVEF, left ventricular ejection fraction; QTc, corrected QT interval.
QTc prolongation
Exposure-dependent QTc prolongation is rarely associated with BRAFi [Chapman et al. 2011; Ascierto et al. 2013; Larkin et al. 2014]. QTc prolongation (measured using Bazett’s formula) of more than 60 ms was observed in 52 (2%) vemurafenib-treated patients, of whom two concurrently developed cardiac arrhythmia [Larkin et al. 2014]. Both patients had predisposing cardiac risk factors (hypertension and ischaemic heart disease). Median time to development of the first incidence of QTc prolongation was 1.9 months. QTc prolongation of more than 60 ms was also observed in 3% of dabrafenib-treated patients (with one >500 ms in the integrated safety population) [Ascierto et al. 2013]. Trametinib is not associated with increased risk of QTc prolongation as a single agent, and the risk of QTc prolongation remained at 3% when combined with dabrafenib [Flaherty et al. 2012a, 2012b].
Treatment with either dabrafenib or vemurafenib is therefore not recommended in patients with uncorrectable electrolyte abnormalities (including low magnesium), long QT syndrome or those who are taking medicinal products known to prolong the QT interval. Initiation of treatment with dabrafenib or vemurafenib is also not recommended in patients with QTc greater than 500 ms. It is recommended that ECG and electrolytes are measured in all patients before starting treatment with a BRAFi. Prescribing information recommends repeating these tests after 1 month of treatment and after any dose modification made for prolonged QTc. Close monitoring is also recommended in patients with moderate to severe hepatic impairment which may impact on drug metabolism [Figure 5(b)].
Pericarditis
Pericarditis has not been reported with dabrafenib or trametinib use to date. However, seven cases of vemurafenib-induced pericarditis have been reported, two with effusion and related tamponade [Mahoney et al. 2013]. Early recognition of this very rare complication of treatment is important, since continuation of vemurafenib treatment in the presence of pericarditis led to life-threatening tamponade, which was reversible on drug discontinuation. Additionally, after recovery, Mahoney and colleagues demonstrated that it is safe to restart vemurafenib at a reduced dose, without reoccurrence of the side effect and with ongoing good control of the patient’s melanoma.
Respiratory complications
ILD or pneumonitis
Respiratory complications are extremely rare with vemurafenib or dabrafenib monotherapy. However, ILD or pneumonitis occurred in 2.4% of patients (5/211) treated with trametinib monotherapy [Flaherty et al. 2012b] and all patients required hospitalization. The median time to first presentation of pneumonitis was 160 days (range 60–172 days). Cough was reported in 29% of the phase I/II dabrafenib/trametinib trial compared with 21% of patients treated with single agent dabrafenib, although no specific cases of ILD or pneumonitis were confirmed to explain the cough associated with dabrafenib-treated patients [Flaherty et al. 2012a]. The possibility of pneumonitis in patients on trametinib who develop cough, shortness of breath, or abnormal chest signs should be investigated with plain chest X-ray or chest computed tomography scan, and treatment should be halted at least temporarily if pneumonitis is suspected (Figure 6).
Figure 6.
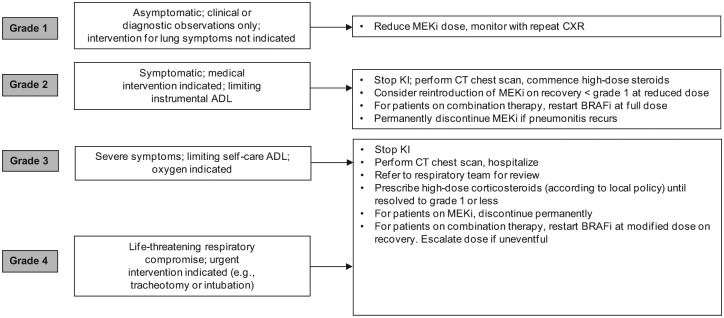
Suggested management of pneumonitis. ADL, activity of daily living; CT, computed tomography; CXR, chest X-ray; KI, kinase inhibitor; MEKi, MEK inhibitor.
Ophthalmologic complications
A number of ophthalmologic complications have been observed during treatment with BRAF targeted agents, most are associated with MEKi. Ocular side effects of targeted agents and their management have recently been reviewed [Renouf et al. 2012]. The most common ocular toxicity associated with BRAFi is uveitis (including iritis), occurring in 1% (6/586) of patients treated with dabrafenib across clinical trials [Hauschild et al. 2012], and in 2.1% (7/336) of patients receiving vemurafenib [Chapman et al. 2011]. When it occurs, it tends to develop over weeks and months of drug exposure. In general it is easily managed with temporary dose interruption, ophthalmology review, a course of topical steroids and in most cases a dose reduction (Figure 7).
Figure 7.

Suggested management of ocular complications. ADL, activity of daily living; BRAFi, BRAF inhibitor; KI, kinase inhibitor; MEKi, MEK inhibitor; RVO, retinal vein occlusion; RPED, retinal pigment epithelial detachment.
In the phase I trial of trametinib, 15% of patients experienced ocular toxicities, including three patients with central serous retinopathy (CSR) at dose levels higher than 2 mg daily, and one retinal vein occlusion (RVO) at 2 mg daily [Infante et al. 2012]. However, the incidence of RVO and retinal pigment epithelial detachments (RPEDs) across 1749 patients treated with trametinib in trials is 0.2% (4 cases) and 0.8% (14 cases), respectively. RPEDs were usually bilateral and multifocal, occurring in the macular region of the retina. RPEDs led to reduction in visual acuity that resolved after a median of 11.5 days (range 3–71 days) after stopping trametinib, although abnormalities observed using ocular coherence tomography persisted beyond a month. No cases of RVO were reported in the COMBI-d trial [Long et al. 2014].
While CSR reportedly recovers after drug cessation, unfortunately, RVO does not appear to be reversible, although significant improvement in visual acuity was achieved after administration of intraocular bevacizumab [Infante et al. 2012]. CSR, RVO and RPED are likely class effects of MEK inhibition. Patients describing ocular symptoms such as blurred vision or loss of vision, or colour spots should stop trametinib, undergo urgent ophthalmologic review, and if symptoms abate, in the absence of RVO, can be retreated with a lower drug dose. In addition, prior to initiating MEKi treatment, a risk assessment should be undertaken and treatment avoided in patients with preexisting ocular conditions such as glaucoma.
Vascular complications
Hypertension
Hypertension was one of the most common serious AEs observed in patients receiving trametinib: 12% experienced grade 3/4 hypertension in the METRIC trial [Flaherty et al. 2012b]. In COMBI-d, frequency of any grade hypertension was reported in 14% of patients in the dabrafenib arm and 22% in the dabrafenib/trametinib arm. It is therefore advisable to measure and monitor blood pressure in patients being considered for or taking BRAFi and MEKi (Figure 8).
Figure 8.

Suggested management of hypertension. ADL, activity of daily living; BP, blood pressure; KI, kinase inhibitor; MEKi, MEK inhibitor; ULN, upper limit of normal; WNL, within normal limits.
Hyperglycaemia
Asymptomatic hyperglycaemia has been reported in clinical trials of BRAFi and MEKi. The incidence of grade 3 hyperglycaemia based on laboratory values is in the range of 2–6%. The significance of this observation is probably confined only to diabetics on therapy: 5 of 12 patients in the METRIC trial with a history of diabetes required more intensive hypoglycaemic intervention while taking trametinib. Therefore it is recommended that serum glucose levels are monitored, as clinically appropriate, in patients with preexisting diabetes or hyperglycaemia. No dose modifications of BRAFi or MEKi are required unless hyperglycaemia is refractory to intervention.
Liver abnormalities
Liver function abnormalities accounted for 13% of AEs recorded in the vemurafenib safety study [Larkin et al. 2014]. They included elevations in alkaline phosphatase (ALP), bilirubin, transaminases as well as other biliary and liver impairment; 5% of cases were graded 3 or 4. Although largely asymptomatic in practice, the risk of potential harm to patients should not be discounted. Indeed, a phase I trial testing the combination of vemurafenib and the anti-CTLA-4 antibody ipilimumab was discontinued due to hepatotoxicity [Ribas et al. 2013] and there have been reports of fatal hepatotoxicity when vemurafenib is used in combination with radiotherapy [Anker et al. 2013]. Therefore, liver blood tests (transaminases, ALP and bilirubin) should be monitored before initiation of treatment and monthly during treatment with vemurafenib, or as clinically indicated. For patients embarking on a course of radiotherapy, it is generally advised to omit targeted therapy and restart on completion.
Laboratory liver abnormalities are also observed with dabrafenib and trametinib treatment, particularly elevated ALP (26% and 60% of all grade toxicities with dabrafenib alone or dabrafenib and trametinib in combination, respectively) [Flaherty et al. 2012a]. No grade 4 liver toxicities have been reported. Laboratory abnormalities should be managed with dose reduction, treatment interruption, or with treatment discontinuation. Additionally, since liver laboratory abnormalities are less common during treatment with dabrafenib, consideration should be given to switching patients from vemurafenib to dabrafenib if liver abnormalities are encountered (Figure 9).
Figure 9.

Suggested management of liver function abnormalities. ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, asparate aminotransferase; BRAFi, BRAF inhibitor; CMV, cytomegalovirus; EBV, Epstein Barr virus; INR, international normalized ratio; KI, kinase inhibitor; LFTs, liver function tests; MEKi, MEK inhibitor; ULN, upper limit of normal.
Other potential treatment-related complications
A number of other less common complications have been reported during use with BRAFi and MEKi, including thromboembolic events and haemorrhage. They should be managed according to standard local or national guidelines. It should also be noted that dabrafenib contains a sulfonamide moiety, and as such, confers a potential risk of haemolytic anaemia in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Dabrafenib is therefore not recommended in patients with known G6PD deficiency.
At risk groups
Older patients
Both clinical and trial experience suggest that risk of severe vemurafenib toxicity increases in an older patient population. The vemurafenib safety study included 257 patients aged 75 years or more. These patients experienced higher rates of severe toxicity: 59% versus 43% grade 3 and 4% versus 3% grade 4 AEs, compared with patients under the age of 75 years [Larkin et al. 2014]. Therefore, the authors recommend that a starting dose for vemurafenib of 720 mg twice daily should be considered in these patients, particularly if they are relatively frail or have multiple medical comorbidities. The dose may be escalated depending on treatment tolerance over the first 1–2 months.
Patients with deranged renal or liver function
Limited studies suggest that clearance of BRAFi and MEKi in patients with mild and moderate renal or liver impairment is similar to that in patients with normal renal or liver function. However, the manufacturers recommend initiating treatment at modified doses (Table 2).
Summary
The development of BRAF-targeted therapies has revolutionized treatment for BRAF mutant metastatic melanoma by improving outcomes for those patients treated. The specific BRAF mutation mutation inhibitors, vemurafenib and dabrafenib, and the MEKi, trametinib, are now licensed for routine clinical use. As with other targeted therapies, acute and chronic exposure to these drugs is associated with predictable patterns of AEs. Simple algorithms have been designed to assist in the management of the more common and serious AEs. Use of these alongside high-quality patient education and support should serve to avoid unnecessary drug discontinuation and so maximize the opportunity for patients to benefit from prolonged disease control.
Footnotes
Funding: This work received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Conflict of interest statement: SJW has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. PC has received consultancy and advisory board honoraria and research funding from Roche and GSK. No writing assistance was utilized in the production of this manuscript.
Contributor Information
Sarah J. Welsh, Cambridge Cancer Centre Department of Oncology, Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK
Pippa G. Corrie, Cambridge Cancer Centre Department of Oncology, Cambridge University Hospitals NHS Foundation Trust, Hills Road, Cambridge CB2 0QQ, UK
References
- Andrews M., Behren A., Chionh F., Mariadason J., Vella L., Do H., et al. (2013) BRAF inhibitor-driven tumor proliferation in a KRAS-mutated colon carcinoma is not overcome by MEK1/2 inhibition. J Clin Oncol 31: e448–e451. [DOI] [PubMed] [Google Scholar]
- Anker C., Ribas A., Grossmann A., Chen X., Narra K., Akerley W., et al. (2013) Severe liver and skin toxicity after radiation and vemurafenib in metastatic melanoma. J Clin Oncol 31: e283–e287. [DOI] [PubMed] [Google Scholar]
- Ascierto P., Minor D., Ribas A., Lebbe C., O’Hagan A., Arya N., et al. (2013) Phase II trial (BREAK-2) of the BRAFi dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol 31: 3205–3211. [DOI] [PubMed] [Google Scholar]
- Babacan T., Türkbeyler I., Balakan O., Pehlivan Y., Suner A., Kısacık B. (2014) A case of vemurafenib-induced polyarthritis in a patient with melanoma: how to manage it? Int J Rheum Dis 12 May [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Callahan M., Rampal R., Harding J., Klimek V., Chung Y., Merghoub T., et al. (2012) Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med 367: 2316–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman P., Hauschild A., Robert C., Haanen J., Ascierto P., Larkin J., et al. (2011) BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364: 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Thakur M., Salangsang F., Landman A., Sellers W., Pryer N., Levesque M., et al. (2013) Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494: 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty K., Infante J., Daud A., Gonzalez R., Kefford R., Sosman J., et al. (2012a) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367: 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty K., Robert C., Hersey P., Nathan P., Garbe C., Milhem M., et al. (2012b) METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 367: 107–114. [DOI] [PubMed] [Google Scholar]
- Hauschild A., Grob J., Demidov L., Jouary T., Gutzmer R., Millward M., et al. (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380: 358–365. [DOI] [PubMed] [Google Scholar]
- Hauschild A., Grob J., Demidov L., Jouary T., Gutzmer R., Millward M., et al. (2013) An update on BREAK-3, a phase III, randomized trial: dabrafenib (DAB) versus dacarbazine (DTIC) in patients with BRAF V600E-positive mutation metastatic melanoma (MM). J Clin Oncol 31(Suppl.): abstract 9013. [Google Scholar]
- Infante J., Fecher L., Falchook G., Nallapareddy S., Gordon M., Becerra C., et al. (2012) Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEKi trametinib: a phase 1 dose-escalation trial. Lancet Oncol 13: 773–781. [DOI] [PubMed] [Google Scholar]
- Koop A., Satzger I., Alter M., Kapp A., Hauschild A., Gutzmer R. (2014) Intermittent BRAF-inhibitor therapy is a feasible option: report of a patient with metastatic melanoma. Br J Dermatol 170: 220–222. [DOI] [PubMed] [Google Scholar]
- Larkin J., Del Vecchio M., Ascierto P., Krajsova I., Schachter J., Neyns B., et al. (2014) Vemurafenib in patients with BRAFV600 mutated metastatic melanoma: an open-label, multicentre, safety study. Lancet Oncol 15: 436–444. [DOI] [PubMed] [Google Scholar]
- Long G., Stroyakovsky D., Gogas H., Levchenko E., deBraud F., Larkin J., et al. (2014) COMBI-d: a randomized, double-blinded, phase III study comparing the combination of dabrafenib and trametinib to dabrafenib and trametinib placebo as first-line therapy in patients with unresectable or metastatic BRAF V600E/K mutation-positive cutaneous melanoma. J Clin Oncol 32(Suppl.): abstract 9011. [Google Scholar]
- Mahoney K., Ackerman A., Cho D., McDermott D., Peters T., Atkins M. (2013) Vemurafenib-induced cardiac tamponade: a rare but potentially life-threatening complication. J Clin Oncol 31: e364–e366. [DOI] [PubMed] [Google Scholar]
- Renouf D., Velazquez-Martin J., Simpson R., Siu L., Bedard P. (2012) Ocular toxicity of targeted therapies. J Clin Oncol 30: 3277–3288. [DOI] [PubMed] [Google Scholar]
- Ribas A., Flaherty K. (2011) BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol 8: 426–433. [DOI] [PubMed] [Google Scholar]
- Ribas A., Hodi F., Callahan M., Konto C., Wolchok J. (2013) Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med 368: 1365–1366. [DOI] [PubMed] [Google Scholar]
- Seghers A., Wilgenhof S., Lebbé C., Neyns B. (2012) Successful rechallenge in two patients with BRAF-V600-mutant melanoma who experienced previous progression during treatment with a selective BRAF inhibitor. Melanoma Res 22: 466–472. [DOI] [PubMed] [Google Scholar]
- Sinha R., Edmonds K., Newton-Bishop J., Gore M., Larkin J., Fearfield L. (2012) Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, prevention and management of the main treatment-related skin toxicities. Br J Dermatol 167: 987–994. [DOI] [PubMed] [Google Scholar]
- Sosman J., Kim K., Schuchter L., Gonzalez R., Pavlick A., Weber J., et al. (2012) Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 366: 707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F., Viros A., Milagre C., Trunzer K., Bollag G., Spleiss O., et al. (2012) RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 366: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer L., Livingstone E., Hillen U., Dömkes S., Becker A., Schadendorf D. (2012) Panniculitis with arthralgia in patients with melanoma treated with selective BRAF inhibitors and its management. Arch Dermatol 148: 357–361. [DOI] [PubMed] [Google Scholar]