

Abstract
Molecular modeling techniques were applied to the design, synthesis and optimization of a new series of xanthine based adenosine A2A receptor antagonists. The optimized lead compound was converted to a PEG derivative and a functional in vitro bioassay used to confirm efficacy. Additionally, the PEGylated version showed enhanced aqueous solubility and was inert to photoisomerization, a known limitation of existing antagonists of this class.
Keywords: Adenosine receptor Xanthine, A2A, Hypoxia KW-6002, Synthesis
1. Introduction
The adenosine receptors are a class of GPCR’s divided into four subtypes, A1, A2A, A2B, and A3, differentiated based on their pathways of signal transduction.1 Activation of A1R and A3R induces coupling to Gi pertussis-toxin sensitive proteins causing downregulation of adenylate cyclase while activation of the A2AR and the A2BR results in Gs protein coupling and subsequent upregulation of adenylate cyclase.1 These mechanisms result in control of intracellular cAMP levels. Additionally, adenosine receptors, when bound to other G proteins, play a role in the control of ion channels via protein kinases and phospholipase C (PLC) activity.1a,c Until recently, homology models of the four AR subtypes (based on the X-ray structure of rhodopsin) represented the principal means of probing receptor function and binding modes. However, in 2008 Jaakola reported the X-ray crystal structure of the human A2AR bound to the high-affinity antagonist ZM241385 (Fig. 1).2
Figure 1.
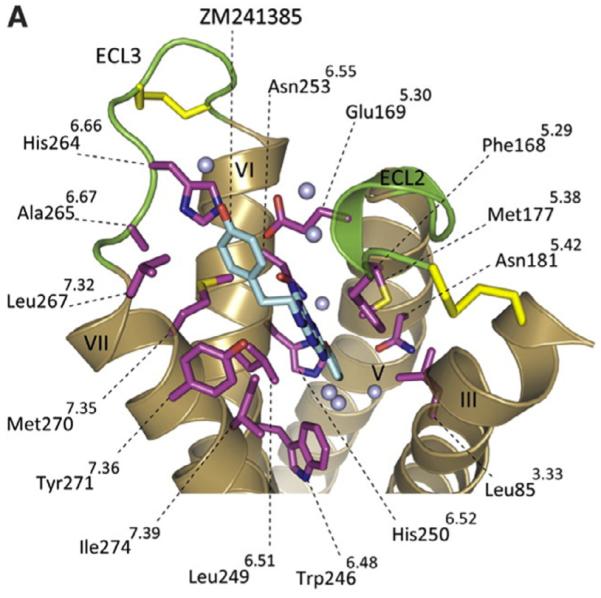
Crystal structure of the human A2AR bound to ZM241385 antagonist.
This report prompted considerable research to identify additional antagonists, as A2ARs are implicated in several diseased states offering the potential to impact neurodegenerative disorders, cardiac regulation, inflammation and cancer,1b,3 the latter a focus of interest to these laboratories.
In normal tissues, pathogen exposure causes accumulation of extracellular adenosine and triggers ‘adenosinergic’ signaling, mediated by A2AR and A2BR, which in turn induces increased production of cAMP.4 Increased levels of cAMP then function to inhibit overactive immunosuppressive T cells from attacking healthy tissues.4 However, this response is also demonstrated in the hypoxic tumor microenvironment, and results in misguided protection of cancerous cells. It is important to note that the effectiveness of the immune response is dependent on the densities of the receptor.5 This means that if the number of receptors has decreased by half, twice as many agonist ligands would need to be present to activate the receptor. Differences in receptor affinity can also be observed across the four subtypes.6 Agonist binding varies widely between A1, A2A, and A3 with Kd values of 0.6, 20, and 6 nM, respectively. Ohta et al.7 reported that based on these mechanisms, ZM241385 significantly delays the rapid growth of CL8-1 melanoma in mice with endogenously developed antitumor T cells. Based on this promising result, we embarked on an effort to ascribe structure with function in a series of synthetic antagonists. However, based on the limited circulating half-life of ZM241385 in vivo (<60 min) we sought a more robust platform and elected to investigate the xanthine class. Caffeine, a prototypical xanthine owes its CNS activity to adenosine receptor antagonism and both it and its structural analog theophylline, demonstrate antagonism in the micromolar range for A2AR.1b The 1, 3, 7, and 8-positions of the xanthine scaffold have been most widely exploited in pursuit of A2AR selective antagonists. Substitution at N1 is most important for both A1R and A2AR binding. The A2AR tolerates smaller alkyl substituents, that is, methyl, allyl, propargyl, ethyl, propyl, and cyclopentyl, while the A1R binding pocket is able to accommodate larger substituents at this position.8 Small alkyl chains are also most commonly found at N3, including methyl, ethyl, propyl, and 3-hydroxypropyl, but larger substituents, including phenoxy groups are also tolerated.9
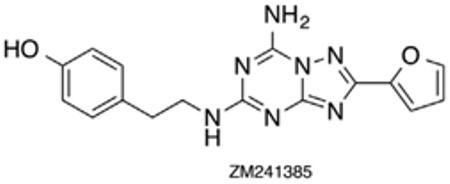
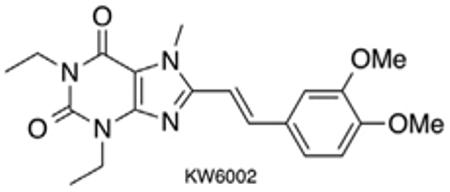
Methyl substitution at N7 appears to impart the A2AR specific activity of caffeine and paraxanthine analogs. 7-Methylation was found to reduce affinity for both A1R and A2AR in the case of 8-substituted xanthines, with one exception; 8-styrylxanthines. 7-Methyl-8-styrylxanthines demonstrated increased selectivity and either comparable or improved potency at the A2AR.8 The first highly potent and selective A2AR antagonist, 54 nM with 529-fold selectivity, CSC, bears a m-chlorostyryl group at C8. The discovery of CSC provided a thrust for further exploration of the SAR around this class of compounds.1b Meta- and para-substitution of the styryl phenyl group is generally well tolerated. Meta-halogens and meta/para-methoxy substituents impart improved selectivity of analogs with small alkyl chains at the 1 and 3 positions of the xanthine core. Replacement of the phenyl group with thienyl (DMPTX), furyl, pyridyl and pyrazolyl groups yield compounds inferior to the styryl derivatives.8 These studies eventually resulted in the clinical candidate KW-6002 (Istradefylline), with meta- and para-methoxy substituents, which was submitted to phase III clinical trials for the treatment of PD. As KW-6002 displays a favorable t1/2 of ~2 h (rat), we conducted preliminary experiments using a tumor immunotherapy model which confirmed its potential.10,11
Unfortunately, KW-6002 and related 8-styrylxanthines are plagued with issues of vinyl photoisomerization in dilute solution and have been reported to undergo [2+2] cycloaddition in the solid form. The Z-isomers are significantly less potent and selective than the E-isomers, and hence replacements for the vinylene linker have been sought. Significant loss of affinity is observed when vinylene is replaced with an alkynyl, cyclopropyl, and 2-naphthyl linkers and also when tricyclic constraint is indroduced.1b Azo derivatives retain selectivity, but not affinity for A2AR, while the imine analogs are both more potent and selective, but are significantly less stable due to their Schiff base structure.1b,8
Based on the promising data supporting use of KW-6002 in immunotherapy models,4a we proposed to use molecular modeling guided synthesis of a library of analogs and optimize for chemical stability and target specificity. Specific goals were to:
Produce a readily synthetically accessible library of compounds.
Control biodistribution by minimizing bbb penetration.
Reduce photo-isomerization and other degradative pathways.
Improve water solubility.
According to the above criteria it was evident that considerable structural modifications would be required. KW-6002 was designed for use in Parkinson’s disease, its lipophilicity advantageous for localization in the brain striatum environment, as confirmed by subsequent PET imaging studies with an [11C] analog.1b An appropriate strategy for our purposes appeared to be to derivitize an optimized lead compound as a PEGylated bioconjugate, enhancing its hydrophilicity and by virtue of molecular weight diminishing its blood–brain-barrier crossing ability, based on consideration of Lipinski’s rules.
2. Results and conclusions
Previous work in our laboratory has focused on the development of an efficient method for synthesis of 8-substituted xanthines from 5,6-diaminouracils and carboxaldehydes using bromodimethylsulfonium bromide.12 Our first synthesis candidate was an ortho substituted aryl ester, which would be subsequently converted to the PEGylated derivative for evaluation. Diaminouracil was coupled to methyl-2-formylbenzoate under BDMS conditions to afford xanthine benzoate 3 after filtration. Subsequent xanthine methylation and saponification of the benzoate yielded the key methylxanthine benzoic acid 5 for esterification (Scheme 1).
Scheme 1.

Synthesis of ester-linked polyethylene glycol conjugates.
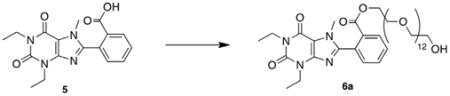
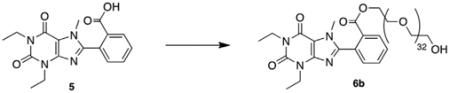
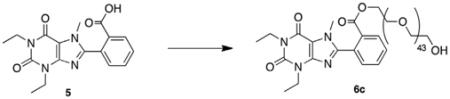
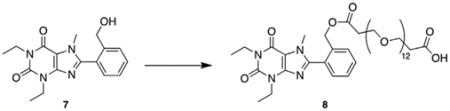
To demonstrate the accessibility of a range of PEGylated analogs, three PEG conjugates of different lengths were synthesized using EDCI mediated coupling conditions viz. a 600, 1500 and 2000 MW (6a–c). In addition to the desired conjugates, amounts of bis-xanthine conjugates were detected, which proved difficult to remove from the desired adducts. Remedy was found by allowing a pre-incubation period of 2 h with the xanthine and coupling agent prior to addition of the PEG-OH. To extend synthetic versatility, a benzyl ester derivative 8 was also prepared (Scheme 2), accompanied by quantities of the bis-conjugate 9. Unfortunately attempts to remove all traces of the dimer chromatographically were unsuccessful.
Scheme 2.
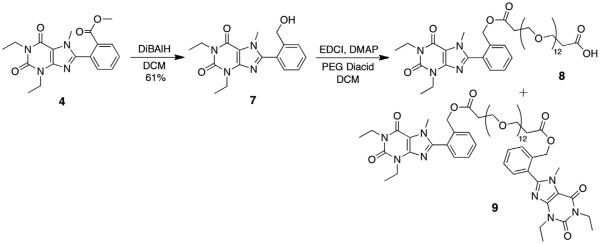
Synthesis of PEG-diacid conjugate from the xanthine benzoate.
With quantities of PEG-ylated derivatives in hand, we initiated preliminary in vitro assays to determine A2A affinity. However, though the derivatives were freely water soluble and essentially stable in solution, affinity for A2A was considerably inferior to control (KW-6002), and molecular modeling studies were initiated to guide identification of additional analogs for synthesis. Initial docking studies were performed in Yet Another Scientific Artificial Reality Application (YASARA) Structure which includes a derivative of AutoDock 4.013 and in Schrodinger’s GLIDE14 to asses affinity of potential analogs for the active site of the human A2AR (PDB file: 3EML)2 based on predicted theoretical binding energies (Fig. 2). The first important interaction of ZM241385 is the aromatic stacking of Phe168. The second is a hydrogen bond interaction of Asn253 to N15 atom of ZM241385.
Figure 2.
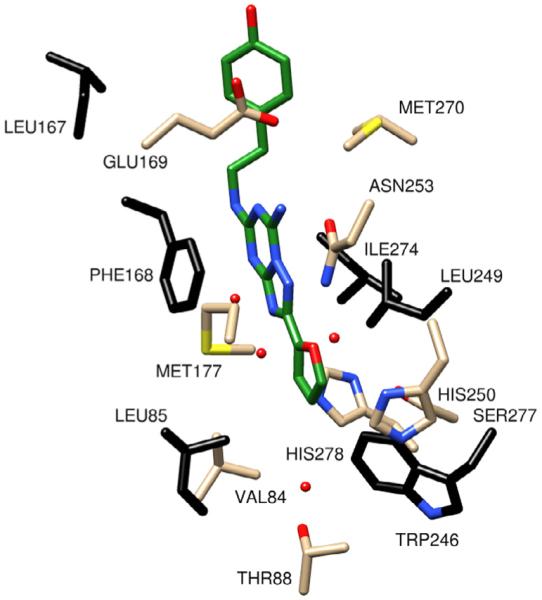
Scrutinized residues (in black) and water molecules (in red) in the active site of A2AR. A2AR from PDB ID 3EML bound to ZM241385 (in green) represents a conformation for the receptor in an inactive state.
A family of arylxanthine isomers derivitized as ortho-, meta-, and para-methyl esters were scrutinized as surrogates for the PEG-ylated versions, as the PEG chain was not well tolerated in AutoDock due to the number of rotatable bonds. As depicted in Figure 3, the o-methyl ester derivative was not planar, and the distortion of the arene-xanthine bond resulted in a collision with Phe168 and a positive theoretical binding energy. A similar distortion was observed with the p-methyl ester arylxanthine. The highly positive predicted binding energy was presumably due to poor orientation of the xanthine core. The m-methyl ester arylxanthine had a signiifcantly improved binding energy over the ortho- and para-isomers resulting from π-stacking between the arene and Phe168, which induced planarity in the molecule. However the predicted energy of this compound was not a highly negative value; therefore, it was concluded that the arylxanthine series was not the optimal scaffold for conjugation and further explains the poor outcome in the preliminary binding assays.
Figure 3.

Models of the o, m and p-methyl ester arylxanthines with corresponding theoretical binding energies.
The second extracellular loop (ECL2, residues Gln148 to Ser156) was missing from the two structures 3EML due to weak experimental electron density in that region. The homology modeling module in YASARA15 was used to build the missing loop in the 3EML structure. To evaluate model quality, the structures were analyzed using SWISS-MODEL Workspace PROCHECK (see methods).16 The docking studies revealed the ECL2 provides important interactions to the ligands and affects ligand confirmation.
Closer examination of ZM241385 docking provides insight into an additional issue with the arylxanthines. In the binding of ZM241385, Phe168 underwent π-stacking with the triazolotriazine core of the molecule, and not with the phenyl moiety. This difference suggested that the arylxanthines were positioned too deeply within the active site, and did not extend through the pocket sufficiently to have a high affinity. Accordingly, analogs with high structural similarity to KW-6002 were designed wherein the styryl moiety could provide appropriate spacing. Two compounds of immediate interest were styrylxanthines that maintained one methoxy group of KW-6002, at the meta- or para-position of the arene, and were conjugated to PEG in the respective para- or meta-position (Fig. 4).
Figure 4.
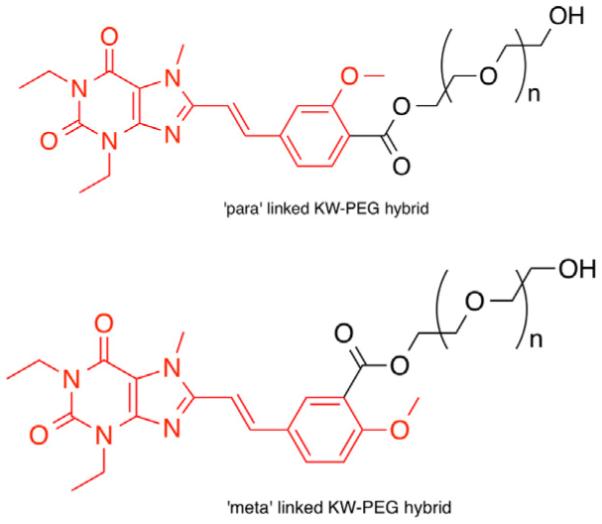
Styrylxanthine analogs selected for additional docking studies. The portion of the compound shown in red is analogous to KW-6002.
At this point, the tolerance of AutoDock 4.0 and Schrodinger’s GLIDE for rotatable bonds was expanded so that more authentic modeling studies could be accomplished. This exercise allowed for the addition of a diethylene glycol chain to the docked compounds and analysis of the placement of the oligomer in the binding pocket (Fig. 5). Both the meta- and para-PEG derivatives demonstrated significantly improved binding energies over the arylxanthine compounds, −8.194 kcal/mol and −9.20 kcal/mol, respectively in AutoDock and Glide Score −7.723 and −7.960, respectively. Critical π-stacking between the xanthine core and Phe168 and hydrogen bonding between xanthine C2-carbonyl and Asn253 contribute to the high affinity of these conjugates.2 Additionally, the incorporation of the vinyl linker extended the molecule so that the arene was stabilized by hydrophobic contact with Met270, which is a contact observed in the binding of the phenyl pharmacophore in ZM241385.2
Figure 5.

Molecular models of styryl xanthines n = 0, bound to the A2AR crystal structure. (Left) The para-PEG derivative docked into the 3EML structure (light brown), GLIDE pose (orange) and AutoDock pose (pink) have similar docking poses and (right) the meta-PEG derivative docked into the 3EML structure (light brown), GLIDE (orange) and Autodock pose (pink) have similar results to a docking pose.
Since neither analog displayed a significant advantage over the other in binding at the xanthine core or the styryl pharmacophore, attention was directed to the conformation of the oligoethylene glycol moiety. Upon conjugate binding to the receptor, the polymeric carrier should not disrupt the binding of the antagonist. Therefore, the PEG chain should lie outside of the active site, as it does with the para-PEG conjugate. The docking of the meta-PEG derivative shows the diethylene glycol folded back into the pocket, and although this did not effect the conjugate’s theoretical binding energy, it is indicative of an interference that may occur with a longer ethylene glycol chain (n ≥ 4). Unfortunately, docking of a PEG chain with n ≥ 4 was not possible with AutoDock 4.0 or Schrodinger, and would most likely have been unreliable. Based on these observations, the para-PEG analog of KW-6002 was selected for development.
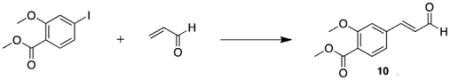
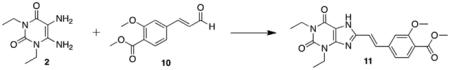
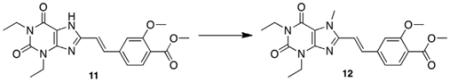
Synthesis of the styrylxanthine began with Heck coupling of acrolein to iodobenzoate under phosphine-free conditions (Scheme 3). This reaction proceeded with full conversion to the desired cinna-maldehyde product 10, and neither aryl–aryl homocoupling nor iodide elimination was observed. Compound 10 was then coupled to diaminouracil 2 in the presence BDMS and the product 11 isolated in high purity after recrystallization from DMSO/water. Xanthine methylation and subsequent ester saponification yielded the functionalized styrylxanthine 13 for polymer conjugation.
Scheme 3.

Synthesis of styrylxanthine-PEG conjugate.
The styrylxanthine compound was conjugated to PEG monomethyl ethers (PEG-Me) to alleviate the bis-coupling issues observed with derivatization to the PEG-diols. Two different length chains, octaethylene glycol-Me and PEG750-Me (14a,b) were selected with the ultimate goal of comparing their physiochemical properties in vitro and in vivo. Condensation was initially attempted under the conditions used for the arylxanthine compound, but the reactions were low yielding, <20%. Several alternative procedures were investigated, including use of Mitsonobu conditions, the Mukaiyama reagent, and alumina to little benefit.17 Ultimately, modification of the EDCI coupling (additional 1.1 equiv EDCI and 10 mol % DMAP with overnight reflux) produced the octaethylene glycol–Me conjugate and the PEG750-Me conjugate in 40% and 27% isolated yields, respectively. Noteworthy in the route to 13 is that chromatography is not required at any stage prior to PEG coupling, allowing efficient scale up.
Given there is a correlation between the CNS penetration of a molecule and its octanol/water partition coefficient,18,19 (log P) thus we elected to determine this for the PEG-esters using a modified HPLC method, based on the EPA Product Properties Test Guidelines (OPPTS 830.7570).20
Using multiple reference compounds an R2 = 0.993 was obtained and the experimental values compared to theoretically derived values (ChemAxon) (Table 1).21
Table 1.
Calculated lipophilicities of KW-6002 and synthetic variants 14
| Compound | Logk | Experimental logP
(HPLC) |
Theoretical logP
(ChemAxon) |
|---|---|---|---|
| KW-6002 | 0.162 | 2.975 | 2.42 |
| KW-octaethylene glygol-Me |
0.145 | 2.802 | 2.20 |
| KW-PEG750-Me | 0.143 | 2.775 | 1.83 |
A marginal decrease in log P, approximately 0.2 logP units, is observed for the PEG conjugates versus KW-6002, and while a relatively mild impact, the nearly twofold increase in the hydrophilicity of the compounds bodes well for development, with KW-6002 partitioning in a 944:1 ratio between the lipophilic and hydrophilic layers, and the conjugates partitioning in a 633:1 (octaethylene glycol–Me) and a 596:1 ratio (PEG750-Me) respectively. We also assessed the degree of photo-stability of the PEG analogs versus KW-6002 using an HPLC assay. Whereas freshly prepared KW6002 underwent photo-isomerization to the Z isomer (less than 10% abundance in solution within 24 h, and >50% abundance within 7 days), no isomerization of the PEGylated variants was detected even after 2 weeks in solution, as shown in Figure 6. While the precise mechanism of this protective function is unknown, it is tempting to hypothesize that the steric constraints in the PEG has an impact, favoring the E isomer.
Figure 6.

Photoisomerization data of PEG conjugates and KW-60002. (A) KW-octaethylene glycol-Me sample after 2 weeks. (B) KW-PEG750-Me sample after 2 weeks. (C) KW-6002 after 8 h. (D) KW-6002 after 2 weeks.
3. Functional bioassay
Bioassays of the PEG analog 14a was conducted using two functional models that assess A2AR binding-dependent signaling through A2AR on the surface of T cells. In the first series of experiments, the functional efficacy of this antagonist (‘KW-PEG’) was tested by determining the extent of inhibition of A2AR-induced intracellular cAMP accumulation in the A2AR-expressing lymphocytes. The activation of A2AR in these assays was accomplished by using the classical agonist, CGS 21680. It is shown (Fig. 7) that both KW-6002 and KW-PEG are able to prevent CGS 21680-mediated signaling. Similar, or even stronger, antagonism was observed by KW PEG treatment when compared to KW-6002, and is directly attributable to the PEG chain as routine control experiments showed the methyl ester of 13 to have near identical activity to KW-6002 itself.
Figure 7.

Cyclic-AMP levels in lymphocytes after incubation with vehicle (VEH), 1 μM CGS (CGS), 1 μM CGS plus 0.5–10 μm KW (KW), and 1 μM CGS plus KW-PEG (KW-PEG) is shown. The intracellular cAMP levels were determined 15 min following stimulation using quantitative cAMP ELISA and are expressed as pmols/million cells. Data shown represent mean ± SEM of triplicate samples.
In addition, we used a biological assay of cytokine secretion that is considered to be sensitive in the determination of functional effects of A2AR signaling. In these assays, immediately following TCR activation by CD3 ligand, T cells from splenocytes were incubated with CGS 21680 since it was shown to prevent IFN-gamma secretion due to increases in A2AR-induced immunosuppressive intracellular cAMP. It is shown (Fig. 8) that the activated T cells produced IFN-gamma, but were significantly inhibited by engagement of immunosuppressive cAMP-elevating A2AR signaling. Importantly, repeating the same assay in the presence of increasing quantities of KW-6002 and the PEG-ylated version restored IFN-gamma secretion.
Figure 8.
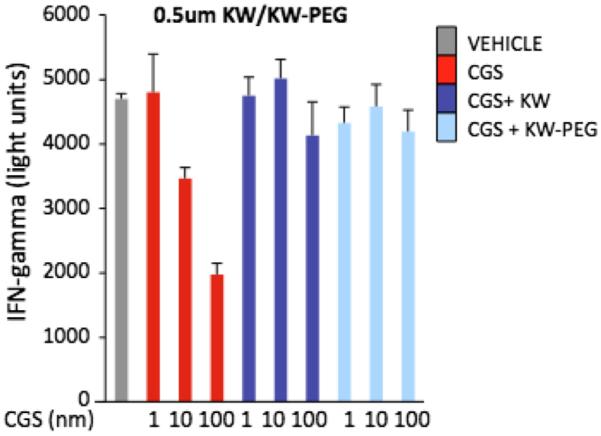
The INF-gamma production by lymphocytes after activation with 0.1 μg/ml mAB-CD3 and when treated with vehicle (VEH), 1–100 nm CGS (CGS), 1–100 nm CGS plus −0.5 μm KW (KW), and 1–100 nm CGS plus −0.5 μm KW-PEG (KW-PEG) is shown. The IFN-γ levels were determined in the supernatant one day following stimulation using quantitative ELISA and are expressed as pg/mL. Data shown represent mean ± SEM of triplicate samples.
The activity of the PEGylated version was nearly identical to KW-6002, noteworthy given that substantial structural modification has been made. This result also vindicated the molecular modeling strategy used in the design (Fig. 5).
These findings are noteworthy, and confirm that efficient chemical synthesis guided by computational analysis can be used for the identification of functional xanthine analogs with desirable physical and chemical properties. With the functional in vitro assays complete we are now engaged in the design of in vivo tumor regression studies using hypoxic models, the details of which will be reported in due course.
4. Conclusion
A computationally directed approach to the molecular design of improved adenosine A2A receptor antagonists based on KW-6002 has been employed leading to identification of a promising lead compound. Synthesis of the styrylxanthine analog and its PEGylated derivatives was accomplished using an efficient method which allows facile modification of molecular weight and thus lipophilicity and water solubility. The lead compound did not undergo photoisomerization even after extended periods in solution. Preliminary functional bioassay suggest the compound has a comparable antagonist profile to KW-6002, rendering them suitable for studies as cancer immunotherapy agents, which will now be pursued.
5. Experimental procedures
5.1. General synthetic methods
1H NMR and 13C NMR spectra were obtained on either a Varian Mercury 400 (400 MHz) or a Varian Inova 500 (500 MHz) spectrometer and are reported in ppm downfield relative to the residual solvent peak. 1H NMR data is reported followed by multiplicity, coupling constant, and number of protons. High resolution mass spectrometry and MALDI were conducted at the mass spectrometry facility at the University of Illinois, Urbana-Champaign. HPLC was conducted on an Agilent Technologies 1120 Compact LC equipped with a 100-tray autosampler, heated column compartment, 100 μL variable volume injection loop, and single pump with solvent selection valve. An Agilent ZORBAX Eclipse XDB-C18 column (4.6 × 150 mm, SN: USRK025565) was used. Signals were detected using a variable wavelength UV detector (serial no.: DE80700574, part no.: G4290A) and a standard UV ?ow cell (part no.: G1314-600086). High-resolution TEM analyses were performed on JEOL JEM-1010 and the images were captured using a Xr41B 4-megapixel, bottom-mount camera purchased from Advanced Microscopy Techniques. Analytical TLC was carried out using silica gel 60 F254 precoated plates (Scientific Adsorbents, Inc.) and visualized using a 254 nm/ 366 nm UV lamp, phosphomolybdic acid or ninhydrin stains, or an iodine chamber. Preparative TLC was carried out using silica gel GF plates (Analtech, Inc.). Flash chromatography was performed using SiliaFlash P60 (230– 240 mesh) silica gel (Silicycle).
5.2. Synthetic procedures
5.2.1. 2-(1,3-Diethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)benzoate (3)
BDMS (0.101 g, 0.451 mmol, 0.9 equiv) was added to a stirring solution of 1,3-diethyl-5,6-aminouracil 2 (0.100 g, 0.504 mmol,1.0 equiv) and methyl 2-formylbenzoate (0.070 mL, 0.504 mmol, 1.0 equiv) in acetonitrile (10 mL). The reaction was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo and the resulting residue was purified via silica gel chromatography (9:1 hexanes:ethyl acetate to 1:1 hexanes:ethyl acetate) to afford the product as a white crystalline solid (0.099 g, 57%). Mp = 198–201 °C; 1H NMR (500 MHz, DMSO-d6): δ 13.86 (br s, 1H), 7.86 (d, J = 7.0 Hz, 1H), 7.71 (t, J = 8.0 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.61 (t, J = 8.0 Hz, 1H), 4.03 (q, J = 7.0 Hz, 2H), 3.95 (q, J = 7.5 Hz, 2H), 1.24 (t, J = 7.0 Hz, 3H), 1.15 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 168.8, 154.6, 152.6, 152.4, 150.8, 148.1, 132.9, 131.8, 130.0, 129.6, 108.5, 52.9, 38.7, 36.5, 13.9, 13.8; HRMS (ESI) m/z C17H18N4O4 (M+H)+ calcd 343.1403, obsd 343.1406.
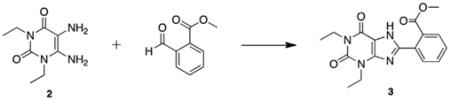
5.2.2. Methyl 2-(1,3-diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)benzoate (4)
To a solution of methyl 2-(1,3-diethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)benzoate 3 (1.13 g, 3.29 mmol, 1.0 equiv) in DMF (25 mL) was added K2CO3 (1.52 g, 11.0 mmol, 3.0 equiv) followed by iodomethane (0.686 mL, 11.0 mmol, 3.0 equiv). The reaction mixture was heated to 60 °C overnight. The solution was diluted with water (100 mL) and extracted with chloroform (2 × 100 mL). The combined organic extracts were washed with water (2 × 100 mL) and brine (100 mL), dried over MgSO4, and concentrated in vacuo to afford a yellow oil. The residue was purified by silica gel chromatography (9:1 hexanes:ethyl acetate to 1:1 hexanes:ethyl acetate, pre-neutralized with 19:1 hexanes:triethylamine) to afford the product as a yellow solid (0.699 g, 60%). Mp = 270 °C (decomposition); 1H NMR (500 MHz, CDCl3):δ 8.05 (d, J = 7.5 Hz, 1H), 7.62 (t, J = 7.5 Hz, 1H), 7.55 (t, J = 7.5 Hz, 1H), 7.41 (d, J = 7.0 Hz, 1H), 4.09 (q, J = 7.0 Hz, 2H), 4.02 (q, J = 7.0 Hz, 2H), 1.26 (t, J = 7.0 Hz, 3H), 1.19 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 166.2, 155.5, 151.7, 151.0, 147.8, 132.9, 131.7, 131.0, 130.8, 130.1, 108.2, 38.7, 36.6, 32.9, 13.7, 13.6; HRMS (ESI) m/z C18H20N4O4 (M+H)+ calcd 357.1551, obsd 357.1563.

5.2.3. 2-(1,3-Diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)benzoic acid (5)
A 1 M aqueous LiOH solution (5.27 mL, 6.0 equiv) was added to a solution of methyl 2-(1,3-diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)benzoate 4 (0.313 g, 0.878 mmol, 1.0 equiv) in THF (20 mL) and the resulting solution was stirred at 50 °C for 3 h. The reaction mixture was diluted with water (50 mL) and extracted with ethyl acetate (2 × 50 mL). The combined organics were discarded, and the aqueous layer was acidified to pH 2 with 5% v/v aqueous HCl and then extracted with ethyl acetate (3 × 50 mL). The combined organic extracts were dried over MgSO4 and concentrated in vacuo to afford the product as a white crystalline solid (0.189 g, 65%). Mp = 193–195 °C; 1H NMR (500 MHz, CDCl3): δ 11.35 (br s, 1H), 8.06 (d, J = 7.5 Hz, 1H), 7.60 (t, J = 7.5 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 7.38 (d, J = 7.0 Hz, 1H), 4.06 (q, J = 7.0 Hz, 2H), 4.01 (q, J = 7.0 Hz, 2H), 1.21 (t, J = 7.0 Hz, 3H), 1.17 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 168.1, 155.6, 152.1, 151.0, 147.5, 133.1, 131.8, 131.6, 131.0, 129.6, 108.3, 39.0, 36.9, 33.0, 13.6, 13.5; HRMS (ESI) m/z C17H18N4O4 (M+H)+ calcd 343.1409, obsd 343.1406.
5.3. Representative procedure for arylxanthine PEG conjugation
5.3.1. 2-(1,3-Diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)benzyl ester polyethylene glycol 600 (6a)

EDCI·HCl (0.013 g, 0.065 mmol, 1.1 equiv), and DMAP (0.001 g, 0.006 mmol, 0.1 equiv) was added to a solution of 2-(1,3-diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)benzoic acid 5 (0.020 g, 0.059 mmol, 1.0 equiv), in dichloromethane. After 2 h, Poly(ethylene glycol) 600 (0.039 g, 0.065 mmol, 1.1 equiv) was added to the solution. The reaction mixture was stirred overnight at room temperature. The reaction mixture was concentrated in vacuo and the residue was purified using preparative silica gel TLC (9:1 dichloromethane:methanol) to afford the title compound as a clear, colorless oil (0.008 g, 14%). 1H NMR (400 MHz, CDCl3): d 8.18 (d, J = 7.0, 1H), 7.70 (t, J = 7.2, 1.0 Hz, 1H), 7.64 (t, J = 7.2 Hz, 1H), 7.47 (d, J = 7.2, 1H), 4.32 (m, 2H), 4.18 (q, J = 7.0 Hz, 2H), 4.10 (q, J = 7.0 Hz, 2H), 3.78–3.57 (m, 50H), 1.32 (t, J = 7.0 Hz, 3H), 1.28 (t, J = 7.0 Hz, 3H); MALDI C17H18N4O4-PEG: calcd 915.47, obsd 915.34.
5.3.2. 2-(1,3-Diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)benzyl ester PEG-1500 (6b)
The title compound was isolated following the general procedure (0.049 g, 46% as a clear, colorless oil). 1H NMR (400 MHz, CDCl3): δ 8.19 (dd, J = 7.0, 1.0 Hz, 1H), 7.70 (dt, J = 7.0, 1.0 Hz, 1H), 7.64 (dt, J = 7.0, 1.0 Hz, 1H), 7.48 (dd, J = 7.2, 1.0 Hz, 1H), 4.32 (m, 2H), 4.18 (q, J = 7.0 Hz, 2H), 4.11 (q, J = 7.0 Hz, 2H), 3.71 (s, 3H), 3.70–3.58 (m, 134H), 1.34 (t, J = 7.0 Hz, 3H), 1.28 (t, J = 7.0 Hz, 3H); MALDI C17H18N4O4-PEG: calcd 1840.02, obsd 1840.33.
5.3.3. 2-(1,3-Diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)benzyl ester PEG-2000 (6c)
The title compound was isolated following the general procedure (0.070 g, 51% as a clear, colorless oil). 1H NMR (500 MHz, CDCl3): δ 8.18 (dd, J = 7.0, 1.0 Hz, 1H), 7.70 (dt, J = 7.5, 1.0 Hz, 1H), 7.67 (dt, J = 7.5, 1.0 Hz, 1H), 7.49 (dd, J = 8.0, 1.5 Hz, 1H), 4.32 (m, 2H), 4.17 (q, J = 7.0 Hz, 2H), 4.11 (q, J = 7.0 Hz, 2H), 3.80–3.38 (m, 178H), 1.35 (t, J = 7.0 Hz, 3H), 1.28 (t, J = 7.0 Hz, 3H); MAL-DI C17H18N4O4-PEG: calcd 2324.31, obsd 2325.39.
5.3.4. 1,3-Diethyl-8-(2-(hydroxymethyl)phenyl)-7-methyl-1H-purine-2,6(3H,7H)-dione (7)
DiBAlH (1 M in hexanes, 1.67 mL, 1.67 mmol, 3.0 equiv) was slowly added to a solution of methyl 2-(1,3-diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)benzoate 4 (0.199 g, 0.558 mmol, 1.0 equiv) in dichloromethane (20 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 4 h then warmed to room temperature overnight. The reaction was quenched with methanol (1 mL), diluted with 1% v/v aqueous HCl (50 mL) and extracted with ethyl acetate (3 × 25 mL). The combined organic extracts were washed with brine (50 mL), dried over MgSO4, and concentrated in vacuo to afford an orange solid. The solid was purified via silica gel chromatography (9:1 hexanes:ethyl acetate to 3:7 hexanes:ethyl acetate) to afford the product as a white oily solid (0.112 g, 61%). 1H NMR (500 MHz, CDCl3): d 7.50 (d, J = 7.5 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 7.38 (m, 2H), 4.91 (t, J = 7.0 Hz, 2H), 4.39 (d, J = 7.0 Hz, 2H), 4.07 (q, J = 7.5 Hz, 2H), 4.02 (q, J = 7.0 Hz, 2H), 3.91 (s, 3H), 1.27 (t, J = 7.5 Hz, 3H), 1.19 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3): d 155.4, 151.1, 150.7, 147.0, 142.1, 131.2, 131.2, 130.1, 128.0, 127.5, 108.8, 64.1, 38.9, 36.7, 34.1, 13.6, 13.5; HRMS (ESI) m/z C17H20N4O3 (M+H)+ calcd 329.1605, obsd 329.1614.

5.3.5. 1,3-Diethyl-8-(2-(hydroxymethyl)phenyl)-7-methyl-1H-purine-2,6(3H,7H)-dione polyethylene glycol carboxylic acid (8)
1,3-Diethyl-8-(2-(hydroxymethyl)phenyl)-7-methyl-1H-purine-2,6(3H,7H)-dione 7 (0.020 g, 0.061 mmol, 1.0 equiv) was added to a solution of PEG diacid (0.048 g, 0.070 mmol, 1.1 equiv), ED-CI-HCl (0.013 g, 0.070 mmol, 1.1 equiv), and DMAP (0.09 g, 0.070 mmol, 1.1 equiv) in dichloromethane (4 mL) and the reaction stirred at room temperature overnight. The reaction mixture was concentrated in vacuo and purified via preparative silica gel TLC (19:1 dichloromethane:methanol) to afford the product as a light yellow oil (0.063 g, 14%). 1H NMR (400 MHz, CDCl3): δ 7.58 (d, J = 8.0 Hz, 1H), 7.53 (dt, J = 7.2, 1.2 Hz, 1H), 7.46 (dt, J = 7.2, 1.6 Hz, 1H), 7.35 (dd, J = 7.6, 1.2 Hz), 5.14 (s, 2H), 4.17 (q, J = 7.2 Hz, 2H), 4.10 (q, J = 7.2 Hz, 2H), 3.68 (t, J = 6.8 Hz, 2H), 3.68–3.54 (m, 52H), 2.54 (t, J = 6.8 Hz, 2H), 1.34 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.6 Hz, 3H). MALDI C17H20N4O3-PEG: calcd 1001.51, obsd 1001.66 and 1311.56.
5.3.6. (E)-Methyl 2-methoxy-4-(3-oxoprop-1-en-1-yl)benzoate (10)22
A solution of 4-iodo-2-methoxybenzoate (10.5 g, 36.0 mmol, 1.0 equiv), tetrabutylammonium bromide (14.3 g, 44.2 mmol, 1.2 equiv), and K2CO3 (14.9 g, 108 mmol, 3.0 equiv) in DMF (100 mL) was degassed for 1 h. Palladium(II) acetate (0.404 g, 5 mol %) was added followed by 2-propenal (5.60 mL, 83.8 mmol, 2.3 equiv) and the reaction stirred overnight at 60 °C. The reaction mixture was taken up in chloroform (300 mL) and washed with water (3 × 200 mL) and brine (2 × 200 mL). The organic extract was dried over MgSO4 and concentrated in vacuo. The resulting residue was passed through a short plug of silica gel (1:1 hexanes:ethyl acetate) to afford the title compound as an orange crystalline solid (3.07 g, 39%). Mp = 109–114 °C; 1H NMR (500 MHz, CDCl3): δ 9.61 (d, J = 7.5 Hz, 1H), 7.97 (d, J = 2.5 Hz, 1H), 7.65 (dd, J = 9.0, 2.5 Hz, 1H), 7.37 (d, J = 16.0 Hz, 1H), 6.99 (d, J = 9.0 Hz, 1H), 6.59 (d, J = 16.0, 7.5 Hz, 1H), 3.91 (s, 3H), 3.87 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 193.6, 166.0, 161.4, 151.5, 133.6, 1332.6, 127.7, 126.5, 120.8, 112.8, 56.5, 52.5; HRMS (ESI) m/z C12H12O4 (M+Na)+ calcd 243.0633, obsd 243.0644.
5.3.7. (E)-Methyl 4-(2-(1,3-diethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)vinyl)-2-methoxybenzoate (11)
BDMS (7.32 g, 33.0 mmol, 1.5 equiv) was added to a solution of 1,3-diethyl-5,6-aminouracil 2 (4.36 g, 22.0 mmol, 1.0 equiv) and (E)-methyl 2-methoxy-4-(3-oxoprop-1-en-1-yl)benzoate 10 (6.22 g, 28.0 mmol, 1.3 equiv) in acetonitrile (150 mL). The reaction mixture was stirred at room temperature for 48 h, filtered and the yellow precipitate collected. Pure product, a light yellow crystalline solid, was obtained via recrystallization from DMSO/water (1.94 g, 22%). Mp = 285–287 °C; 1H NMR (500 MHz, DMSO-d6): δ 13.70 (br s, 1H), 7.70–7.66 (m, 2H), 7.42 (s, 1H), 7.26–7.21 (m, 2H), 4.05 (q, J = 7.5, 2H), 3.94 (q, J = 7.0 Hz, 2H), 3.89 (s, 3H), 3.79 (s, 3H), 1.26 (t, J = 7.0 Hz, 3H), 1.14 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 165.8, 158.7, 153.7, 150.2, 149.0, 147.9, 140.6, 134.0, 131.4, 119.8, 118.6, 118.2, 111.1, 107.7, 72.3, 56.0, 51.9, 38.1, 35.8, 13.2; HRMS (ESI) m/z C20H22N4O5 (M+H)+ calcd 399.1668, obsd 399.1678.
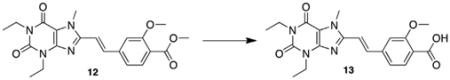
5.3.8. (E)-Methyl 4-(2-(1,3-diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)vinyl)-2-methoxybenzoate (12)
To a solution of (E)-methyl 4-(2-(1,3-diethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)vinyl)-2-methoxybenzoate 11 (1.22 g, 3.06 mmol, 1.0 equiv) in DMF (40 mL) was added K2CO3 (1.27 g, 9.19 mmol, 3.0 equiv) followed by iodomethane (0.572 mL, 9.19 mmol, 3.0 equiv). The reaction mixture was heated to 50 °C overnight. The solution was diluted with water (100 mL) and extracted with chloroform (2 × 100 mL). The combined organic extracts were washed with water (2 × 100 mL) and brine (1200 mL), dried over MgSO4, and concentrated in vacuo to afford a light yellow crystalline solid (0.911 g, 72%). Mp = 212–215 °C; 1H NMR (400 MHz, CDCl3): δ 7.82 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 15.6 Hz, 1H), 7.22 (dd, J = 8.0, 1.6 Hz, 1H), 7.09 (s, 1H), 6.97 (d, J = 15.2 Hz, 1H), 4.19 (q, J = 7.2, 2H), 4.06 (q, J = 6.4 Hz, 2H), 4.07 (s, 3H), 3.96 (s, 3H), 3.89 (s, 3H), 1.36 (t, J = 7.2 Hz, 3H), 1.24 (t, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 166.4, 159.8, 155.3, 150.9, 149.4, 148.3, 140.9, 137.0, 132.5, 120.6, 118.7, 113.9, 111.5, 108.7, 56.4, 52.4, 38.7, 36.7, 31.8, 13.7, 13.6; HRMS (ESI) m/z C21H23N4O5 (M+H)+ calcd 413.1825, obsd 413.1834.
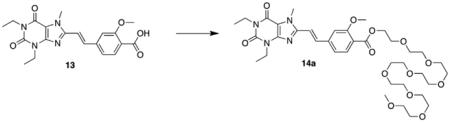
5.3.9. (E)-4-(2-(1,3-Diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)vinyl)-2-methoxybenzoic acid (13)
Aqueous LiOH solution (1 M, 13.3 mL, 6.0 equiv) was added to a solution of (E)-methyl 4-(2-(1,3-diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)vinyl)-2-methoxybenzoate 12 (0.911 g, 2.21 mmol, 1.0 equiv) in THF (50 mL) and the resulting solution stirred at 50 °C overnight. The reaction mixture was diluted with water (200 mL) and extracted with dichloromethane (2 × 100 mL). The combined organics were discarded, and the aqueous layer acidified to pH 2 with 5% v/v aqueous HCl and then extracted with dichloromethane (3 × 100 mL). The combined organic extracts were dried over MgSO4 and concentrated in vacuo to afford the product as a light yellow crystalline solid (0.647 g, 73%). Mp = 212.0–215.7 °C; 1H NMR (500 MHz, CDCl3): d 8.20 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 15.5 Hz, 1H), 7.40 (d, J = 8.0 Hz ,1H), 7.17 (s, 1H), 7.04 (d, J = 15.5, 1H), 4.21 (q, J = 7.0 Hz, 2H), 4.15 (s, 3H), 4.11 (s, 3H), 4.08 (d, J = 7.0 Hz, 2H), 1.38 (t, J = 7.0 Hz, 3H), 1.26 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 165.1, 158.6, 155.4, 150.9, 149.0, 148.3, 145.4, 142.5, 136.1, 134.5, 120.3, 118.0, 115.0, 111.3, 57.1, 38.7, 36.7, 31.9, 13.66, 13.6; HRMS (ESI) m/z C20H22N4O5 (M+H)+ calcd 399.1668, obsd 399.1668.
5.4. Representative procedure for styrylxanthine PEG conjugation
5.4.1. (E)-4-(2-(1,3-Diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)vinyl)-2-methoxybenzyl ester polyethylene glycol (8-mer) monomethyl ether (14a)
EDCI·HCl (0.792 g, 4.140 mmol, 2.2 equiv), and DMAP (0.046 g, 0.376 mmol, 0.2 equiv) was added to a solution of (E)-4-(2-(1,3-diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)vinyl)-2-methoxybenzoic acid 13 (0.750 g, 1.882 mmol, 1.0 equiv) in dichloromethane. The reaction mixture was refluxed for 3 h. Octaethylene glycol monomethyl ether (0.730 mL, 2.070 mmol, 1.1 equiv) was added and the solution was refluxed overnight. The reaction mixture was concentrated in vacuo and the residue was purified via silica gel chromatography (50:1 dichloromethane:methanol) to afford the title compound as a dark yellow oil (0.572 g, 40%). 1H NMR (400 MHz, CDCl3): d 7.87 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 15.6 Hz, 1H), 7.24 (d, J = 8.8 Hz, 1H), 7.11 (s, 1H), 6.99 (d, J = 16.4 Hz, 1H), 4.47–4.45 (m, 2H), 4.21 (q, J = 7.6 Hz, 2H), 4.10 (s, 3H), 4.09 (q, J = 7.6 Hz, 2H), 3.97 (s, 3H), 3.84–3.82 (m, 2H), 3.70–3.64 (m, 26H), 3.37 (s, 3H), 1.38 (t, J = 7.2 Hz, 3H), 1.26 (t, J = 7.6 Hz, 3H); HRMS (ESI) m/z C37H56N4O13 (M+H)+: calcd 765.3921, obsd 765.3922.
5.4.2. (E)-4-(2-(1,3-Diethyl-7-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)vinyl)-2-methoxybenzyl ester polyethylene glycol 750 monomethyl ether (14b)
The title compound was isolated following the general procedure (0.081 g, 27% as a waxy yellow solid). 1H NMR (400 MHz, CDCl3): δ 7.87 (d, J = 8.0 Hz, 1H), 7.78 (d, J = 15.6 Hz, 1H), 7.24 (d, J = 9.6 Hz, 1H), 7.11 (s, 1H), 6.99 (d, J = 16.0 Hz, 1H), 4.47–4.45 (m, 2H), 4.22 (q, J = 7.2 Hz, 2H), 4.10 (s, 3H), 4.09 (q, J = 7.2 Hz, 2H), 3.97 (s, 3H), 3.95–3.53 (m, 62H), 3.38 (s, 3H), 1.39 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H); MALDI C20H21N4O5-PEG: calcd 1116.59, obsd 1117.06.
5.5. Bioassay procedures
5.5.1. Measuring functionality of A2AR antagonism by cAMP assay
Stimulation of intracellular cAMP production and measurement of cAMP levels were performed as described previously.7,23 Lymphocytes were isolated from the spleen of C57/BL6 mice and treated with at 1 μM CGS 21680 (A2AR-specific agonist; from Tocris, Ellisville, MO) with or without a range of concentrations of KW or KW-peg (0.1–10 lM). The cells were incubated for 15 min at 37 °C, and the reaction was stopped by addition of 1 N hydrochloric acid. Levels of cAMP were determined by ELISA (Amersham Biosciences, Buckinghamshire, UK). All treatment groups were done in triplicate.
5.5.2. Cytokine release assay
Lymphocytes were isolated from the spleen of C57/BL6 mice and cultured with 0.1 μg/mL CD3 mAb to induce production of IFN-gamma. Immediately following the addition of mAb-CD3, the cells were treated with or without 1, 10, or 100 nM CGS 21680 agonist. To examine functionality of the antagonists, KW or KW-peg (0.5 μm) was added to the cells. After 24 h, supernatants were collected and the concentration of IFN-gamma was measured by ELISA using paired mAb and standard purchased from BD Pharmingen. All treatment groups were done in triplicate.
5.6. Molecular modeling procedures
5.6.1. Ligand preparation
For docking in YASARA AutoDock 4.0, the ligands were prepared using GlycoBioChem PRODRG2 Server24 and minimized in YASARA Structure. For docking in Schrodinger’s GLIDE, the ligands were prepared using the LigPrep of Maestro v9.3.515 in the Schrödinger Suite 2012.25 The ligands were pre-processed through LigPrep specifying a pH value of 7.0.
5.6.2. Homology modeling
Using the protein sequence of human A2AR (UniProtKB P29274), a model was built for a portion of the second extracellular loop (ECL2) residues Gln148 to Ser156. Residues were missing due to weak experimental electron density in that region. Homology modeling of the was performed with the YASARA.15 The quality of the model was examined using PROCHECK16 and was found to be of sufficiently good quality. The co-crystallized ligand, ZM241385, was re-docked into the homology model and the RMSD of the Cα backbone was less than 3 Å (Figs. 9–11).
Figure 9.

PROCHECK Ramachandran plot and statistics for the homology model that show the good quality in the most favored regions.
Figure 11.
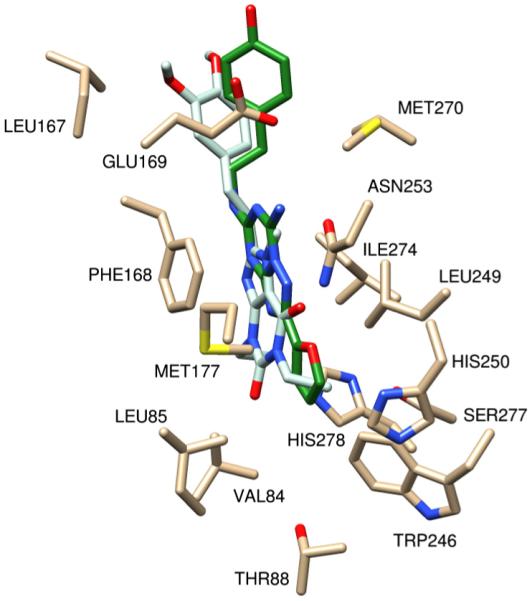
KW6002 (in light blue) is docked in 3EML and is shown with ZM241385 (in green). The important residues for contact for the xanthine core (KW6002) and the bicyclic triazolotriazine (ZM241385) are both in contact via aromatic stacking with Phe168. The hydrophobic interactions at the top and bottom of the cavity are seen with the methyl (KW6002) and furan (ZM241385). The AUTODOCK binding energy of ZM241385 is -8.99 kcal/mol and binding energy of KW6002 is −8.313 kcal/mol.
5.6.3. Docking studies
Initial docking studies were performed using AutoDock 4.013 using the default docking parameters supplied and point charges assigned according to the AMBER03 force field. The setup was done with the YASARA molecular modeling program. The binding energy calculated in YASARA calculates the binding energy of the ligand to protein in the force field AMBER03. The more negative the binding energy, the more favorable the interaction in context of the force field used. The model for A2AR (PDB 3EML) structure was prepared using the Maestro 9.3 protein preparation wizard (Schrodinger, LLC, 2012, New York, NY) before docking, bond orders were assigned and the orientation of hydroxyl groups, amide groups of the side chains of Asn and Gln, and the charge state of histidine residues were optimized. A restrained minimization of the protein structure was performed using the default constraint of 0.3 Å RMSD and the OPLS 2001 force field.26 The enclosing box (the centroid of the docked ligand around selected residues) dimension was set to 25 Å around selected residues of the homology models, using either Schrodinger Glide or YASARA implementation of Autodock. Both standard precision (SP) and extraprecision (XP) docking of ligands were carried out, XP docking scores are reported. Water molecules were not included in these docking studies due to the creation of the homology model.
Figure 10.
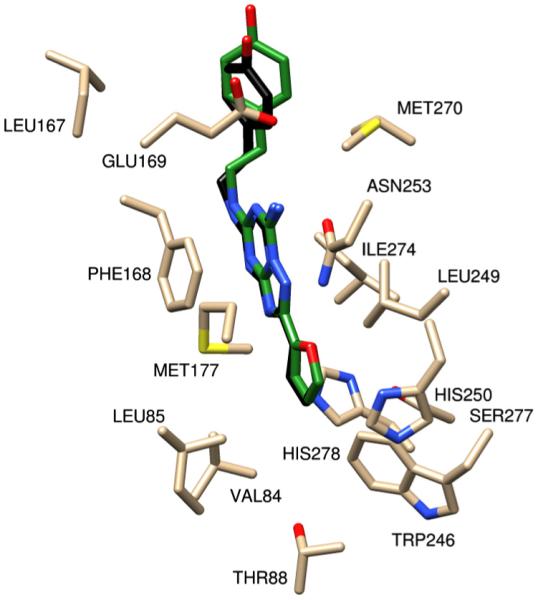
ZM241385 from the crystal structure (in green) and ZM241385 redocked into the homology model (in black), there is a slight variation with the 4-hydroxyphenyl group. The AUTODOCK binding energy of ZM241385 is −8.99 kcal/mol and Glide Score −6.99 kcal/mol.
References
- 1.(a) Jacobson KA, Gao Z-G. Nat. Rev. Drug Disc. 2006;5:247. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Adenosine Receptors in Health and Disease. Vol. 193. Springer; New York: 2009. [Google Scholar]; c Moro S, Gao Z-G, Jacobson KA, Spalluto G. Med. Res. Rev. 2006;26:131. doi: 10.1002/med.20048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaakola V-P, Griffith MT, Hanson MA, Cherezov V, Chen EYT, Lane JR, Ijzerman AP, Stevens RC. Science. 2008;322:1211. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bilkei-Gorzo A, Abo-Salem OM, Hayallah AM, Michel K, Muller CE, Zimmer A. Naunyn Schmiedebergs Arch. Pharmacol. 2008;377:65. doi: 10.1007/s00210-007-0252-9. [DOI] [PubMed] [Google Scholar]
- 4.(a) Sitkovsky M. Trends Immunol. 2009;30:102. doi: 10.1016/j.it.2008.12.002. [DOI] [PubMed] [Google Scholar]; b Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Mol. Pathways. 2008;14:5947. doi: 10.1158/1078-0432.CCR-08-0229. [DOI] [PubMed] [Google Scholar]; c Lukashev D, Sitkovsky M, Ohta A. Purinergic Signal. 2007;3:129. doi: 10.1007/s11302-006-9044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fredholm BB. Cell Death Differ. 2007;14:1315. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- 6.Klotz K. Naunyn Scmiedebergs Arch. Pharmacol. 2000:362. [Google Scholar]
- 7.Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, Huang X, Caldwell S, Liu K, Smith P, Chen J-F, Jackson EK, Apasov S, Abrams S, Sitkovsky M. Proc. Natl. Acad. Sci. 2006;103:13132. doi: 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azam F, Ibn-Rajab IA, Alruiad AA. Pharmazie. 2009;12:771. [PubMed] [Google Scholar]
- 9.Morella S, Sorrentino R, Pinto A. J. Recept., Ligand Channel Res. 2009;2:11. [Google Scholar]
- 10.Yang M, Soohoo D, Soelaiman S, Kalla R, Zablocki J, Chu N, Leung K, Yao L, Diamond I, Belardinelli L, Shryock JC. Naunyn Schmiedebergs Arch. Pharmacol. 2007;375:133. doi: 10.1007/s00210-007-0135-0. [DOI] [PubMed] [Google Scholar]
- 11.Kase H. Neurology. 2003;61:S97. doi: 10.1212/01.wnl.0000095219.22086.31. [DOI] [PubMed] [Google Scholar]
- 12.(a) Ma D, Sitkovsky M, Kallmerten AE, Jones GB. Tetrahedron Lett. 2008;49:4633–4635. [Google Scholar]; b LaBeaume P, Dong M, Sitkovsky M, Jones EV, Thomas R, Sadler S, Kallmerten AE, Jones GB. Org. Biomol. Chem. 2010;8:4155. doi: 10.1039/c003382k. [DOI] [PubMed] [Google Scholar]
- 13.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J. Comput. Chem. 1998;19:1639. [Google Scholar]
- 14.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J. Med. Chem. 2006;49:6177. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 15.Krieger E, Joo K, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K. Proteins. 2009;77:114. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arnold K, Bordoli L, Kopp J, Schwede T. Bioinformatics. 2006;22:195. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 17.(a) Sharghi H, Sarvari MH. Tetrahedron. 2003;59:3627. [Google Scholar]; b Brunel JM, Salmi C, Letourneux Y. Tetrahedron Lett. 2005;46:217. [Google Scholar]; c Matsugi M, Suganuma M, Yoshida S, Hasebe S, Kunda Y, Hagihara K, Oka S. Tetrahedron Lett. 2008;49:6573. [Google Scholar]
- 18.Abraham MH, Jr., Acree WE, Leo AJ, Hoekman D, Cavanaugh JE. J. Pharm. Sci. 2009:99. doi: 10.1002/jps.22010. [DOI] [PubMed] [Google Scholar]
- 19.Leo A, Hansch C, Elkins D. Chem. Rev. 1971:71. [Google Scholar]
- 20.Yousefi G, Foroutan SM, Zarghi A, Shafaati A. Chem. Pharm. Bull. 2010;58:147. doi: 10.1248/cpb.58.147. [DOI] [PubMed] [Google Scholar]
- 21.(a) Viswanadhan VN, Ghose AK, Revankar GR, Robins RK. J. Chem. Inf. Model. 1989:29. [Google Scholar]; b Klopman G, Li J-Y, Wang S, Dimayuga M. J. Chem. Inf. Model. 1994:34. doi: 10.1021/ci00022a014. [DOI] [PubMed] [Google Scholar]
- 22.Meijere AD, Kuchuk I, Sokolov VV, Labahn T, Rauch K, Es-Sayed M, Kramer T. Eur. J. Org. Chem. 2003;6:985. [Google Scholar]
- 23.Apasov S, Chen JF, Smith P, Sitkovsky M. Blood. 2000;95:3859. [PubMed] [Google Scholar]
- 24.Schuttelkopf AW, van Aalten DM. Acta Crystallogr., D Biol. Crystallogr. 2004;60:1355. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 25.Greenwood JR, Calkins D, Sullivan AP, Shelley JC. J. Comput. Aided Mol. Des. 2010;24:591. doi: 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- 26.Shivakumar D, Williams J, Wu YJ, Damm W, Shelley J, Sherman W. J. Chem. Theory Comput. 2010;6:1509. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]