

Abstract
Alcoholism is a complex disorder with genetic and environmental risk factors. The presence of withdrawal symptoms is one criterion for alcohol dependence. Genetic animal models have followed a reductionist approach by quantifying various effects of ethanol withdrawal separately. Different ethanol withdrawal symptoms may have distinct genetic etiologies, and therefore differentiating distinct neurobiological mechanisms related to separate signs of withdrawal would increase our understanding of various aspects of the complex phenotype. This study establishes motor incoordination as a new phenotype of alcohol withdrawal in mice. Mice were made physically dependent on ethanol by exposure to ethanol vapor for 72 h. The effects of ethanol withdrawal in mice from different genetic backgrounds were measured on the accelerating rotarod, a simple motor task. Ethanol withdrawal disrupted accelerating rotarod behavior in mice. The disruptive effects of withdrawal suggest a performance rather than a learning deficit. Inbred strain comparisons suggest genetic differences in magnitude of this withdrawal phenotype. The withdrawal-induced deficits were not correlated with the selection response difference in handling convulsion severity in selectively bred Withdrawal Seizure-Prone and Withdrawal Seizure-Resistant lines. The accelerating rotarod seems to be a simple behavioral measure of ethanol withdrawal that is suitable for comparing genotypes.
Keywords: dependence, ethanol, inbred strains, motor, mouse, rotarod, selected lines, withdrawal, Withdrawal Seizure-Prone, Withdrawal Seizure-Resistant
Introduction
Alcoholism is a complex trait. Individual risk arises from the interaction of multiple genes of small effect that seem to influence susceptibility to alcohol (ethanol; EtOH) dependence (Crabbe, 2002a; Sher et al., 2005). EtOH dependence cannot be measured directly, but is inferred from the presence of withdrawal symptoms upon cessation of EtOH administration. Withdrawal from chronic EtOH administration is characterized by central and autonomic nervous system hyperexcitability symptoms, which build to a peak and then dissipate over time. Alcohol withdrawal consists of both physiological (e.g. convulsions, tremor, nausea, electrolyte disturbances, and liver disease) and psychological (e.g. anxiety, stress, depression, hallucinations, agitation, and craving for alcohol) symptoms (Valdez and Koob, 2004). Chronic alcohol affects multiple neurotransmitter receptor subtypes and electrophysiological, pharmacological, and genetic studies have led to the elucidation of various neurobiological mechanisms underlying the separate signs of withdrawal.
Preclinical tests in rodents have characterized various in vivo effects of EtOH withdrawal, such as the handling-induced convulsion (HIC), anxiety-like behaviors, and decreased locomotor activity. The most extensively characterized EtOH withdrawal sign in mice is the HIC (Goldstein and Pal, 1971; Crabbe et al., 1991), a very sensitive behavioral index of withdrawal severity. The HIC is readily quantified, can be tested repeatedly using a within-subjects experimental design, and effectively measures dose (Goldstein, 1972) and genotype-dependent effects (Goldstein, 1973). However, not all EtOH withdrawal signs characterized in rodents are as straightforward. For example, attempts to detect increased anxiety-like behaviors during withdrawal in rodent assays based on exploration of a novel environment are often potentially confounded by reductions in locomotor activity (see review by Kliethermes, 2005). Moreover, the separate signs of EtOH withdrawal may be genetically distinct (i.e. genetically uncorrelated traits). Genetic rodent models have successfully followed a reductionist approach of attempting to model the various contributing features of the complex trait of alcohol withdrawal separately. Classical genetic methods focused on alcohol dependence include studies of inbred strains and selectively bred lines (Crabbe, 2002b).
All same sex members of an inbred strain (e.g. C57BL/6J mice) are essentially genetic clones produced through many generations of brother–sister matings, and each possesses two copies of the same allele in all genes. Therefore, individual differences in responses within an inbred strain must be because of environmental influences (including genotype–environment interactions). If many strains are compared for a trait in a controlled environment, then mean differences among the strains can be attributed largely to genetic differences, provided that the strain differences are substantially greater than the within-strain differences. The testing of inbred strains allows for the identification of genetically correlated responses that can be compared across laboratories and time (Bogue and Grubb, 2004; Wahlsten et al., 2006).
Elevated HIC during withdrawal has been extensively characterized genetically. The acute withdrawal reaction to a single intraperitoneal injection of 4.0 g/kg EtOH in mice has been quantified using this sensitive behavioral index (Crabbe et al., 1991). Strain differences in withdrawal HIC demonstrate substantial genetic correlations among 15 strain means in severity of acute withdrawal convulsions from EtOH, pentobarbital, and diazepam (Metten and Crabbe, 1994, 1999). Other studies have demonstrated a negative genetic correlation between severity of withdrawal from EtOH and voluntary consumption of 10% EtOH (Metten et al., 1998). A more recent study compared 15 strains exposed to EtOH vapor for 72 h (Metten and Crabbe, 2005). Comparisons of these data with other published strain differences supported previously reported genetic associations of high EtOH withdrawal with several traits: low drinking, high conditioned taste aversion, low tolerance to EtOH-induced hypothermia, and high stimulated activity after low dose EtOH, as well as the positive genetic relationships among EtOH, barbiturate, and benzodiazepine withdrawal severity. Conversely, a number of other alcohol-related traits were shown to be uncorrelated with withdrawal severity among inbred strains (Metten and Crabbe, 2005).
Another classical genetic model uses selective breeding (i.e. artificial selection), one of the oldest and most powerful methods in behavioral genetics for examining potential genetic correlations (Crabbe, 2002b). The development of selectively bred lines consists of testing animals from a genetically heterogeneous stock on the trait of interest and subsequently mating the extreme-scoring animals (i.e. selection of high-response and low-response lines). Nonselected lines serve as controls. For example, the Withdrawal Seizure-Prone (WSP) and Withdrawal Seizure-Resistant (WSR) mouse lines were selectively bred (in replicate) for EtOH withdrawal differences as measured by the HIC. WSP and WSR mice were exposed to chronic continuous (72 h) EtOH vapor inhalation using a procedure based on the method by Goldstein and Pal (1971). Mice from both replicates of the WSP line demonstrated 10-fold greater withdrawal severity than WSR mice by selection generation 11, whereas Withdrawal Seizure-Control (WSC) scores were intermediate (Crabbe et al., 1985). Relaxation of selection produced little change in withdrawal scores for either line, which implies that the genes mediating the respective withdrawal intensities were fixed in the homozygous state by selection (Phillips et al., 1989; Crabbe et al., 1990a; Crabbe and Phillips, 1993).
This tactic can indicate neurobiological mechanisms that may be shared between the selected trait and another trait of interest. The WSP and WSR lines have been tested for many traits other than the selection response (HIC). Other behavioral measures of EtOH tolerance and withdrawal, sensitivity to EtOH intoxication, severity of withdrawal from other drugs, sensitivity to proconvulsant and anticonvulsant agents (in the naive and/or EtOH-withdrawn states), as well as various neurochemical differences, have been examined between the selected lines (Metten and Crabbe, 1996). As with findings in inbred strains, these data from WSP and WSR selected lines imply that the genes controlling EtOH withdrawal HIC are largely independent of those mediating many other effects of EtOH. As EtOH withdrawal is influenced by multiple genetic factors, different behavioral domains related to withdrawal are likely to involve different genes. This suggests that distinct neural pathways and brain regions underlie the various symptoms of EtOH withdrawal.
The behavioral domain of motor performance (broadly construed) is a frequent endpoint for studies of the intoxicating effects of EtOH. Inbred strain differences in several behavioral assays of intoxication have been reported (Crabbe et al., 2005). Motor coordination, however, has been virtually unexplored during EtOH withdrawal. The rotarod is one behavioral assay used to assess motor incoordination (Dunham and Miya, 1957; Jones and Roberts, 1968a, b; Watzman and Barry, 1968; Bogo et al., 1981). Basal performance on the rotarod task is sensitive to the effects of genetic background in mice, and mice from different genetic backgrounds also often differ in their response to the intoxicating effects of EtOH as demonstrated in studies characterizing inbred strains (Crabbe et al., 1994; Tarantino et al., 2000; Rustay et al., 2003), selected lines (Schafer and Crabbe, 1996; Rustay et al., 2001), and genetically engineered mutant mice (Boehm et al., 2000, 2003; Lalonde et al., 2005; Lalonde and Qian, 2007).
The current studies assessed the effects of EtOH withdrawal on performance and improvement of the accelerating rotarod task in mice from an outbred stock, seven standard inbred strains, and lines selectively bred for severe or mild EtOH withdrawal HIC. Physical dependence was induced via 72 h of chronic continuous EtOH vapor inhalation exposure. Mice were tested for performance and improvement of the accelerating rotarod task at various periods of withdrawal after EtOH exposure and compared with air-exposed controls.
Methods
Subjects
Male mice were used for all experiments. The genetically heterogeneous, outbred mouse stock (WSC-2) was derived from HS/Ibg originally obtained from the Institute for Behavioral Genetics (Boulder, Colorado, USA). The HS/Ibg stock was derived by intercrossing eight inbred mouse strains. WSC-2 mice were bred and maintained at the Veterinary Medical Unit, Department of Veterans Affairs Medical Center (Portland, Oregon, USA).
The inbred strains were selected from the Mouse Phenome Project’s (www.jax.org/phenome) A and B lists based on criteria such as use in gene-targeting studies (e.g. 129, FVB, and C57), widespread usage for other studies, and allelic diversity. The seven strains 129S1/SvImJ, A/J, BALB/cByJ, C3H/HeJ, C57BL/6J, DBA/2J, and FVB/NJ were obtained at 4–6 weeks from The Jackson Laboratory (Bar Harbor, Maine, USA) and the WSP and WSR lines were selectively bred in replicate for severe or mild HIC, respectively, during chronic EtOH withdrawal (Crabbe et al., 1985). WSP (1 and 2) and WSR (1 and 2) mice from selected generation 26 (filial generations 102–103) were bred at the Portland, VAMC. The WSP and WSR lines also were derived from the genetically heterogeneous HS/Ibg strain.
Husbandry and testing procedures for all experiments were in accordance with the US Department of Agriculture, the US Public Health Service guidelines and were approved by the Institutional Animal Care and Use Committee. Mice were housed three to four per polysulfone cage (28 × 17 × 11.5 cm). The cages were lined with Bedocob bedding (Green Products Company, Conrad, Iowa, USA) and changed once weekly. Food (Purina 5001, PMI International, St Louis, Missouri, USA) and water were freely available. The colony was maintained on a 12-h light/dark cycle with lights on at 06.00 h. Colony and procedure rooms were maintained at 22 ± 1°C. Studies were carried out in drug-naive male mice, 50–140 days old at testing in all experiments. Behavioral testing occurred between 08.00 and 18.00 h. Numbers of mice per group are given in figure captions and tables.
Ethanol dependence induction
The details of the EtOH vapor inhalation procedure have been published previously (Metten and Crabbe, 2005). Briefly, mice in the EtOH groups were initially injected intraperitoneally with EtOH [Pharmco, Brookfield, Connecticut, USA; 1.5 g/kg, 20% (vol/vol) in saline] to raise blood EtOH concentration (BEC) to approximately 1.50 mg EtOH/ml blood. The alcohol dehydrogenase inhibitor pyrazole HCl (Sigma-Aldrich, St Louis, Missouri, USA; 68.1 mg/kg) was administered in the EtOH solution to inhibit EtOH metabolism and stabilize BECs. Physical dependence on EtOH was induced via 72 h of chronic continuous EtOH vapor inhalation exposure. Inbred strains used in these studies differ in EtOH elimination rates (Crabbe et al., 2003), so different EtOH vapor concentrations were chosen based on earlier studies from this laboratory on a strain-specific basis to achieve approximately equal BECs in all mice (Metten and Crabbe, 2005). The WSP and WSR selected lines do not experience significantly different BEC during acute EtOH exposure (Kosobud and Crabbe, 1986), but have been shown to differ during chronic exposure (Terdal and Crabbe, 1994). Different EtOH vapor concentrations were maintained as stably as possible in each of two to three separate chambers (in practice, ± 3 – 12%) throughout the 72 h exposure. EtOH-treated mice were removed from the chambers at 24 and 48 h, given pyrazole in saline (68.1 mg/kg, intraperitoneally) and placed back in the chambers. Control mice were injected with pyrazole in saline daily, and were placed in identical inhalation chambers but were exposed only to air throughout the 72 h procedure.
Blood ethanol concentration assessment
After 24 and 48 h of EtOH vapor inhalation exposure (before the time of the daily pyrazole injection), mice were removed from the chambers in small groups and gently restrained; a 20-μl blood sample was drawn from the end of the nicked tail or periorbital sinus route with a capillary tube for BEC determinations. This allowed us to assess the actual EtOH doses experienced by each group of mice from different genetic backgrounds. After 72 h of chronic EtOH vapor inhalation exposure (and 24 h after the last pyrazole injection), the final blood samples were obtained from all mice. BEC was determined using a gas chromatographic assay detailed previously (Rustay and Crabbe, 2004). Sham blood sampling procedures were conducted on control mice. BECs are presented for each experimental group as an average ± standard error of the mean (SEM). Animals retained for behavioral analyses were matched for final BEC within the experiment to eliminate potential EtOH exposure differences.
Apparatus
The Accurotor Rota Rod (Accuscan Instruments, Columbus, Ohio, USA) was used for all accelerating rotarod tests. Our modified apparatus has a 63 cm fall height and 6.5 cm diameter rotating dowel. Dowel surfaces were covered with 320 grit wet/dry sandpaper to provide a uniform surface and reduce slipping. It is important to minimize the seam for the glued sandpaper as mice can use any seam to grasp the rod and increase their latency to fall (Wahlsten et al., 2003).
Accelerating rotarod test
Four animals were placed on the stationary rod facing the wall opposite the experimenter, one each in the four lanes divided by Plexiglas rounds. Once all animals were placed on the rod, the motor was turned on and the rod was continuously accelerated from 0 rpm at a constant rate of acceleration of 20 rpm/m. After all mice had fallen from the dowel, they were placed back on the apparatus at 0 rpm for each consecutive trial. There was a minimum of 30 s between trials. The maximum speed of 99.9 rpm allowed us to obtain a measure of peak performance, as no mouse achieved this speed (i.e. all mice eventually fell off the rod). Latency to fall was recorded in seconds. Experiments 1–3 evaluated procedural parameters for the purpose of designing the experiments testing mice of different genetic backgrounds (experiments 4 and 5). Mice were not tested for HICs and were handled during rotarod testing so as not to elicit an HIC inadvertently.
Experiment 1
This study was conducted to evaluate the effects of duration of EtOH withdrawal on performance and improvement on the accelerating rotarod task. WSC-2 mice were examined at 8, 10, 12, and 24 h after withdrawal using a within-subjects design. EtOH-exposed and air-exposed mice were given three trials on the accelerating rotarod at 8, 10, and 12 h and then 10 trials at 24 h.
Experiment 2
The results from experiment 1 (see Results and Fig. 1) could potentially have been because of air-exposed control mice receiving more extended training during the initial trials at 8, 10, or 12 h than EtOH-treated mice, which tended to have shorter latencies to fall at these earlier time points and, therefore, less practice. To control for potential practice effects, experiment 2 was conducted using an air-exposed control mouse yoked to each EtOH-exposed mouse. This procedure equated amount of practice time between two pairs of EtOH-treated and air-treated groups (one pair tested at 8 h and another tested at 12 h) by gently removing an air-control mouse from the accelerating rotarod whenever the matched EtOH-treated mouse fell. Mice were given five trials at either 8 or 12 h. All four groups of mice were subsequently tested on 10 trials at 24 h.
Fig. 1.
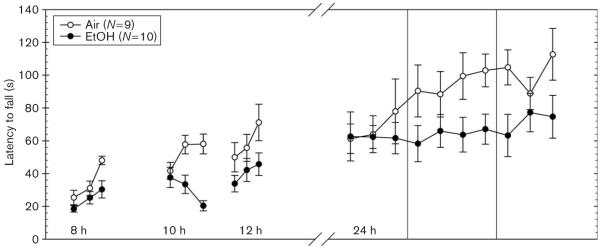
Average latency to fall from an accelerating rotarod after 72 h chronic continuous ethanol vapor inhalation exposure (closed circles) or air (open circles) at 8, 10, 12, and 24 h. EtOH, ethanol.
Experiment 3
Withdrawal effects on accelerating rotarod behavior could be because of disrupted performance or impaired learning of the task. We hypothesized that if these deficits were because of performance effects, then they would not be evident by 1 week, after acute effects of withdrawal had presumably dissipated. Alternatively, if these deficits were because of disrupted ability to learn the task, then they should still be evident after 1 week (i.e. alcohol-treated mice would show less memory savings). Four groups of air-exposed or EtOH-exposed mice were given five trials on the accelerating rotarod using the yoked-control procedure at 8 h. Half of the mice were subsequently tested at 24 h and the other half were tested 1 week later.
Experiment 4
To assess potential genetic influences on the EtOH withdrawal-induced accelerating rotarod deficit, male mice from inbred strains were tested for performance and improvement of the accelerating rotarod task at 8 or 12 h after EtOH vapor or air exposure.
Experiment 5
Naive, selectively bred lines of WSP and WSR mice of both replicates were tested for performance and improvement on the accelerating rotarod task at 8 h after EtOH vapor or air exposure. This study was conducted to assess whether or not EtOH withdrawal-induced deficits measured by the accelerating rotarod task were genetically correlated with the selection response, HIC severity.
Data analysis
Systat (Chicago, Illinois, USA) version 12 was used for statistical analyses. Data were analyzed using analysis of variance (ANOVA). After matching groups within an experiment by equating groups for BEC, rotarod data were examined for missing latencies to fall on each trial. Two ways by which missing data for a rotarod trial occurred were (i) the photocell beam was blocked and thus no latency to fall was recorded (a very rare event) and (ii) the mouse jumped from the rod during the trial. This latter problem was the most pronounced in the EtOH-withdrawing animals. Animals were retained despite jumping if there were no jumps on adjacent trials (e.g. trials 3 and 4), if there were fewer than three trials missing, and if at least two of the first three and two of the last three trials were not missing. Few WSC-2 mice failed these criteria (2, 1, and 0 in the experiments 1–3, respectively). For the genetic studies (experiments 4 and 5), there were somewhat more mice failing criteria (15–17%). Mice passing criteria were included in analyses by averaging the two trials surrounding the missing trial (e.g. if trial 6 was missing, the latency to fall was estimated as the average latencies of trials 5 and 7). These values in all cases were comparable with the rest of the trials as verified by visual inspection within animal.
Repeated measures comparisons were used to assess between-group differences in the latency to fall across trials. To provide simpler indices, ‘performance’ of the task was indexed by the average latency to fall on all trials. ‘Improvement’ on the task was indexed as the difference between the average latency on the last three trials (or two, depending on the experiment) and the average on the first three (or two) trials. When significant interactions were present, follow-up analyses were performed by separate ANOVAs on between-group factors or Tukey’s honestly significant difference post-hoc tests. For the genetic studies, genotypes differ in basal performance. Therefore, to create indices of performance and improvement decrements owing to EtOH withdrawal, we calculated difference scores for each measure, by subtracting the air group mean value for its strain or selected line from each EtOH-exposed animal’s individual performance (or improvement) score. Statistical differences were considered significant at P value of less than 0.05. We also assessed the genetic relationship among inbred strain mean values between scores on the accelerating rotarod task and HIC after EtOH withdrawal using Spearman’s rank-order correlation, using HIC data published previously by Metten and Crabbe (2005). Estimates of heritability for strain data were h2 = SS Between strains/SS Total from the one-way ANOVA on strain.
Results
Experiment 1: withdrawal impairment (Fig. 1)
EtOH-exposed WSC-2 mice averaged 1.54 ± 0.10 mg/ml BEC at the time withdrawal commenced. As the tests during the early withdrawal hours reflected very limited training (three trials each), we first analyzed the performance during the first 12 h. Each animal’s performance was taken as the average of the three trials at each time point. A repeated-measures ANOVA showed that performance during withdrawal was significantly different from control mice [F(1,17) = 5.79; P < 0.05]. Animals improved over time [F(2,34) = 10.52; P < 0.001] and there was no significant interaction of treatment and time [F(2,34) = 0.57; NS]. We analyzed the 10 trials at the 24-h withdrawal duration separately. Analyses of latency for performance (i.e. average of all trials) at 24 h of withdrawal did not show a significant difference between EtOH-exposed (65.69 ± 9.78 s) and air-exposed (89.05 ± 10.88 s) controls across the 10 trials [F(1,17) = 2.56; NS]. EtOH-exposed mice (9.49 ± 6.99 s), however, did show significantly less improvement than air controls (34.37 ± 8.30 s) across the 10 trials.
Experiment 2: elimination of the role of practice (Fig. 2)
Fig. 2.
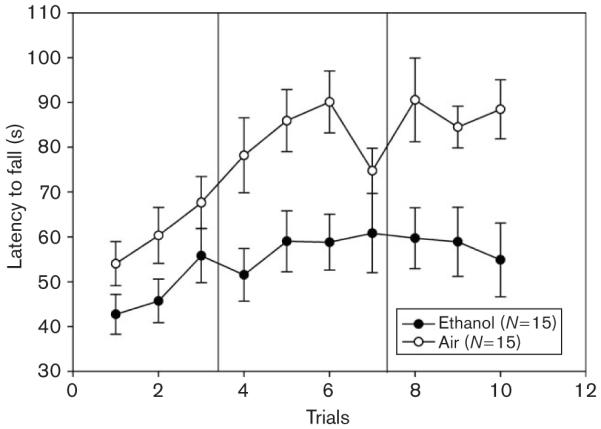
Ethanol-exposed (closed circles) and air-exposed control mice (open circles) were given five initial trials on the accelerating rotarod using a yoked-control procedure to equate rotarod training exposure at 8 or 12 h into withdrawal (data not shown) then tested for an average latency to fall from an accelerating rotarod at 24 h after.
EtOH-exposed WSC-2 mice averaged 1.76 ± 0.09 mg/ml BEC and this was not significantly different between animals tested on the accelerating rotarod at 8 h (1.74 ± 0.10 mg/ml) or 12 h (1.78 ± 0.16 mg/ml) of withdrawal [F(1,13) = 0.06; NS]. The two groups of EtOH-exposed animals did not differ during their initial test at 8 or 12 h in accelerating rotarod performance [data not shown; F(1,24) = 0.28; NS]. Therefore, the two EtOH-treated groups initially tested at 8 versus 12 h were combined and the effect of 24 h of EtOH withdrawal across the 10 trials was compared with combined air-treated groups.
EtOH-exposed animals exhibited disrupted performance compared with air-exposed control animals [F(1,28) = 10.33; P < 0.01]. The mean ± SEM latency for performance in EtOH-exposed and air-exposed animals was 54.83 ± 4.99 s and 77.45 ± 4.97 s, respectively. EtOH-withdrawn animals also exhibited significantly less improvement than control animals [F(1,28) = 5.73; P < 0.05]. The mean ± SEM increase in latency across trials in EtOH-exposed and air-exposed animals was 9.77 ± 6.13 s and 27.12 ± 3.87 s, respectively.
Experiment 3: performance versus learning (Fig. 3a and b)
Fig. 3.
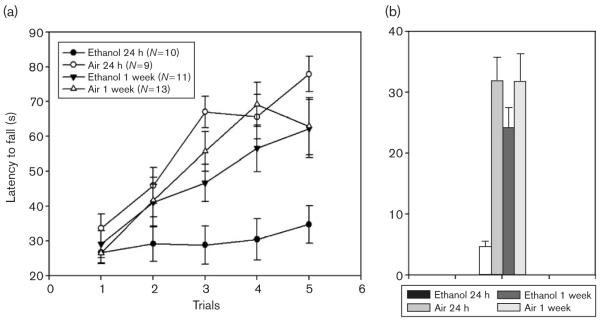
Ethanol-exposed and yoked, air control Withdrawal Seizure Control-2 mice had equivalent accelerating rotarod experience at 8 h (data not shown). Ethanol-withdrawing mice were subsequently tested at 24 h or 1 week of withdrawal compared with controls. (a) Performance on the rotarod over five trials. (b) Improvement across trials (average of the last two trials minus the average of the first two trials).
EtOH-exposed WSC-2 mice averaged 1.88 ± 0.09 mg/ml BEC. Mice to be tested at 24 h versus 1 week did not differ in BEC [1.90 ± 0.10 mg/ml vs. 1.87 ± 0.15 mg/ml, respectively; F(1,19) = 0.17; NS]. All mice were initially tested at 8 h using the yoked-control procedure. The mean ± SEM latencies to fall for the two EtOH-exposed groups of animals did not differ across the five trials at 8 h [F(1,19) = 0.58, NS] (data not shown). The mean ± SEM at 8 h for the 24-h withdrawal group was 44.00 ± 4.16 s, and for the 1-week withdrawal group was 49.2 ± 3.57 s.
Animals were then tested on the accelerating rotarod at either 24 h or 1 week of withdrawal (Fig. 3a). EtOH-exposed animals demonstrated a significant main effect of treatment on performance compared with control animals [F(1,39) = 11.92; P < 0.01], no significant main effect of withdrawal duration [F(1,39) = 1.23], but a significant interaction of treatment and withdrawal duration [F(1,39) = 6.67; P < 0.05]. A post-hoc analysis revealed that the EtOH-exposed animals tested at 24 h performed significantly worse than those tested at 1 week [F(1,19) = 6.80; P < 0.05] and that the air-exposed animals tested at 24 h were no different than those tested at 1 week [F(1,20) = 1.09; NS].
EtOH-exposed animals also demonstrated significantly less improvement than control animals [F(1,39) = 13.35; P < 0.01] (Fig. 3b), 14.94 ± 4.06 s versus 31.93 + 3.16 s, respectively. A significant effect of withdrawal duration [F(1,39) = 4.20; P < 0.05] and a significant interaction of treatment and withdrawal duration on improvement were observed [F(1,39) = 4.23; P < 0.05] (Fig. 3b). EtOH-exposed animals tested at 24 h improved significantly less than those tested at 1 week [F(1,19) = 7.84; P < 0.05], whereas air-exposed animals tested at 24 h were no different than those tested at 1 week [F(1,20) = 0.00; NS].
Experiment 4: inbred strains (Fig. 4)
Fig. 4.

Ethanol (EtOH)-exposed and air-exposed inbred strains (a–g) were tested on the accelerating rotarod. Refer to Table 2 for strain and treatment numbers.
To create groups with equal exposure, we selected mice from all strains that averaged 1.48 ± 0.03 mg/ml BEC. Mean BEC (mg/ml) and the range values for individual strains are given in Table 1. Two-way ANOVA detected no significant differences in BEC values between groups of animals tested at different withdrawal durations [8 vs. 12 h; F(1,141) = 0.02; NS], among strains [F(6,141) = 1.73; NS] or interaction of withdrawal duration and strain [F(6,141) = 0.93; NS]. Thus, after selecting mice to match BECs, strain differences in withdrawal could not be attributed to differential exposure to EtOH.
Table 1.
Blood ethanol concentrations (BEC) from experiment 4
| 129S1/SvImJ | 1.39 ±0.10 (0.70–2.29) |
| A/J | 1.35 ± 0.07 (0.85–2.00) |
| BALB/cByJ | 1.51 ±0.10 (0.74–2.28) |
| C3H/HeJ | 1.65 ±0.05 (1.28–1.97) |
| C57BL/6J | 1.55 ±0.05 (1.07–1.94) |
| DBA/2J | 1.41 ± 0.06 (0.70–2.15) |
| FVB/NJ | 1.57± 0.08 (1.10–2.26) |
Strain BECs are reported as the mean ± SEM and the range.
EtOH-exposed animals had shorter latencies to fall across trials compared with air-exposed controls [F(1,322) = 188.02; P < 0.001], there was no significant main effect of withdrawal duration [F(1,322) = 0.16; NS] and strains differed [F(6,322) = 28.61; P < 0.001] in latencies to fall across accelerating rotarod trials. A significant interaction of treatment and strain was observed [F(6,322) = 2.50; P < 0.05], but no significant interactions of treatment with withdrawal duration [F(1,322) = 0.26; NS], strain and withdrawal duration [F(6,322) = 1.20; NS] or treatment, strain and withdrawal duration [F(6,322) = 1.51; NS]. As there were no significant main or interaction effects of withdrawal duration with any independent variable tested (including BEC), we collapsed on this variable to simplify subsequent analyses of strain differences.
Data are shown in Fig. 4 and Table 2. EtOH-exposed animals exhibited poorer performance than air-exposed animals [F(1,336) = 189.32; P < 0.001], strains performed differentially [F(6,336) = 28.12; P < 0.001], and there was a significant interaction of treatment and strain [F(6,336) = 2.57; P < 0.05]. Separate ANOVAs showed that EtOH exposure led to a significant withdrawal performance decrement in all strains tested [Table 2; all F(1,40–51) ≥ 14.96; P < 0.001]. The heritability estimate (strain effect size, estimated as SS between strains/ST total from each ANOVA) derived from the withdrawal performance decrement, was 0.23.
Table 2.
Accelerating rotarod performance and improvement compared with handling-induced convulsion severity in air- and ethanoltreated mice
| Strain | N (A) | N (E) | Performance (A) | Performance (E) | Withdrawal performance decrement |
Improvement (A) | Improvement (E) | Withdrawal improvement decrement |
|---|---|---|---|---|---|---|---|---|
| 129S1/SvImJ | 29 | 22 | 38.12 ±1.98 | 18.86 ±1.60 | 19.26 ± 1.60 | 25.24 ± 3.52 | 3.98± 1.67 | 21.25 ± 1.67 |
| A/J | 28 | 23 | 37.64 ±1.34 | 21.92 ±1.41 | 15.72± 1.41 | 9.26 ± 2.50 | − 1.49± 1.37 | 10.76± 1.37 |
| BALB/cByJ | 28 | 25 | 35.77 ±1.85 | 25.94 ±1.72 | 9.83 ± 1.72 | 15.12 ± 1.98 | 6.79± 1.47 | 8.33 ± 1.47 |
| C3H/HeJ | 26 | 16 | 35.69 ±1.48 | 26.67 ±1.18 | 9.02 ± 1.18 | 10.64± 2.49 | 4.41 ± 3.07 | 6.22 ± 3.07 |
| C57BL/6J | 26 | 24 | 51.87 ±2.70 | 38.01 ±2.02 | 13.86± 2.02 | 30.95 ± 4.18 | 21.99± 2.55 | 8.97± 2.55 |
| DBA/2J | 23 | 27 | 35.38 ±3.01 | 16.82 ±0.77 | 18.55 ± 0.77 | 11.61 ± 2.04 | − 0.47± 1.13 | 12.08± 1.13 |
| FVB/NJ | 30 | 23 | 28.21 ±1.80 | 18.14 ±1.46 | 10.07± 1.46 | 4.59± 1.43 | 1.77± 1.61 | 2.82 ± 1.61 |
| Strain | N (A) | N (E) | Δ Area 25** | |||||
| 129S1/SvImJ | 9 | 14 | 47.22 ±1.06 | |||||
| AKR | 8 | 8 | 53.19 ±9.60 | |||||
| BALB/cByJ | 8 | 6 | 31.25 ±2.21 | |||||
| C3H/HeJ | 9 | 9 | 59.67 ±7.19 | |||||
| C57BL/6J | 9 | 19 | 25.47 ±4.74 | |||||
| DBA/2J | 10 | 12 | 75.81 ±2.49 | |||||
| FVB/NJ. | 13 | 16 | 53.1 ±2.52 |
Upper half: Performance and improvement values are given as the mean ±standard error of the mean (s) and were defined in Data analysis section. Withdrawal performance and withdrawal improvement decrement indices were derived as described in the Data analysis section.
Lower half: Δ Area 25** = area under the 25-h handling-induced convulsion (HIC) curve for each strain expressed as a difference score for the strain mean control area 25 values (analogous to withdrawal decrement scores for rotarod variables above). Data are from Metten and Crabbe (2005).
A, air controls; E, ethanol-withdrawing mice.
EtOH-withdrawing animals also improved less [F(1,336) = 59.84; P < 0.001] than air-exposed controls, strains exhibited different degrees of improvement [F(6,336) = 23.21; P < 0.001] and there was a significant interaction of treatment and strain [F(6,336) = 2.88; P < 0.01]. Table 2 also shows improvement and withdrawal improvement decrement scores for the strains. EtOH-withdrawing mice of the 129S1/SvImJ, A/J, BALB/cByJ, and DBA/2J strains showed significantly less improvement compared with their air controls [all F(1,48–51) ≥ 10.97; P < 0.01], whereas the C3H/HeJ, C57BL/6J, or FVB/NJ strains did not show significant treatment effects [all F(1,40–51) ≤ 3.22; P ≥ 0.08]. The heritability estimate for withdrawal improvement decrement was 0.28.
The correlation between strain mean values for withdrawal-induced deficits in performance and improvement was highly significant (r = 0.89, P < 0.01). Neither withdrawal performance nor improvement decrement, however, was significantly correlated with the withdrawal effects on HIC scores from Metten and Crabbe (2005) (both r < 0.05; NS).
Experiment 5: Withdrawal Seizure-Prone and Withdrawal Seizure-Resistant mice (Fig. 5)
Fig. 5.
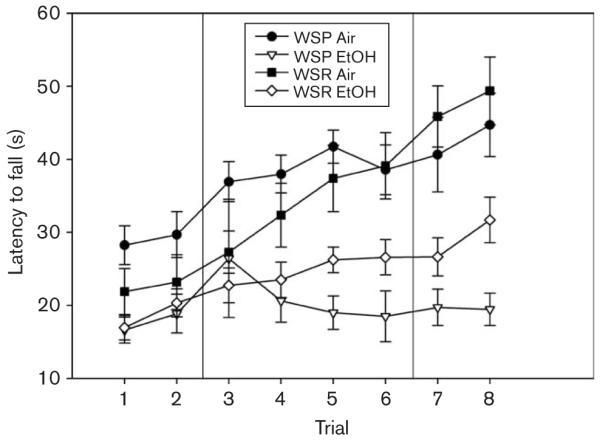
Ethanol (EtOH)-exposed and air-exposed Withdrawal Seizure-Prone (WSP) and Withdrawal Seizure-Resistant (WSR) lines were tested on the accelerating rotarod.
Only four WSP-2 mice were available for testing. Therefore, we collapsed across replicates for all analyses. EtOH-exposed WSP mice in this experiment averaged 1.68 ± 0.07 mg/ml BEC, and ethanol-exposed WSR mice averaged 1.71 ± 0.08 mg/ml. One-way ANOVA revealed no significant line difference [F(1,33) = 0.32; NS].
EtOH-exposed mice exhibited significant disruptions in performance compared with air-exposed controls [F(1,52) = 35.80; P < 0.001], and there was no main effect of line [F(1,52) = 0.13; NS] or interaction of line and treatment [F(1,52) = 2.42; NS] on performance. For improvement, EtOH-exposed mice improved significantly less than air-exposed controls [F(1,52) = 19.94; P < 0.001], the WSR line improved significantly more than the WSP line [F(1,52) = 11.48; P < 0.01] but there was no interaction of treatment with line [F(1,52) = 0.21; NS].
Discussion
This study demonstrates that EtOH withdrawal impairs performance and/or improvement across trials on the accelerating rotarod task in mice. This new EtOH withdrawal phenotype differs in severity among genotypes. The characterization of this withdrawal sign in mice may help focus research on novel neurobiological mechanisms for the selective drug treatment of alcoholism. The precise description of this phenotype will ultimately allow us to map the relevant genes so that their pleiotropic range of influence on the multiple phenotypes constituting the alcohol withdrawal syndrome can be determined.
Neurotransmitter receptor systems that modulate the acute effects of EtOH on rotarod performance may provide clues as to which receptor mechanisms are important for these withdrawal-induced effects. For example, it has been shown that cerebellar adenosine modulates EtOH-induced motor incoordination (Dar, 1997). There appear to be functionally opposing roles for glutamate N-methyl-d-aspartate and adenosine A1 receptors (Manzoni et al., 1994), which together may comodulate cerebellar structures important for EtOH-induced motor incoordination (Dar, 2002). Nicotine has been shown to antagonize EtOH-induced motor impairment (Dar et al., 1993, 1994), possibly influenced by nitric oxide (Al-Rejaie and Dar, 2006a) and the cerebellar nitric oxide-guanylyl cyclase messenger system (i.e. cGMP) (Al-Rejaie and Dar, 2006b). There also seems to be a crucial role for glutamate in the functional interaction of the antagonist effects of nicotine on EtOH-induced motor impairment (Al-Rejaie and Dar, 2006c). These cerebellar mechanisms underlying EtOH-induced motor impairment offer a useful initial target for studies of EtOH withdrawal-induced motor impairment. Additionally, differences in receptor densities among these systems may be important for the differential effects of withdrawal among inbred strains.
Genotypic differences could provide important clues for underlying mechanisms of withdrawal severity. In this study, the C57BL/6J and 129S1/SvImJ strains exhibited more improvement across trials on the accelerating rotarod task than all other strains tested. After EtOH exposure, the rank of the C57BL/6J strain remained the highest but the 129S1/SvImJ strain rank fell to fourth, suggesting more pronounced withdrawal severity in the 129S1/SvImJ relative to C57BL/6J. Comparison of the 129S1/SvImJ with C57BL/6J or another strain showing relatively small withdrawal-induced improvement decrement using the measure of improvement may yield important differences in receptor systems (e.g. adenosine) possibly related to EtOH withdrawal. Furthermore, experimenters may want to choose a particular strain to maximize the probability of detecting the treatment effect of interest. For example, the 129S1/SvImJ strain may provide a genotype especially suited to detect depressant-like drug withdrawal effects hypothesized to impair motor skills and to test putative drug treatments hypothesized to attenuate these withdrawal signs. Conversely, the FVB/NJ strain control animals showed very little improvement in the accelerating rotarod task. This lack of improvement on the task may have produced a floor effect, thus rendering it improbable that a disruptive effect could be detected using the accelerating rotarod task. This potential confound may possibly be avoided by changing the parameters of the accelerating rotarod task (i.e. slowing the acceleration rate to make the task easier).
In this study, we accelerated the rotarod at a rate of 20 rpm/m, a rate used earlier in this laboratory (Rustay et al., 2003). Mean baseline performance in this study showed remarkable consistency with the performance rank order of strains tested earlier under these conditions. C57BL/6J (1), BALB/cByJ (2), C3H/HeJ (3), and DBA/2J (6) had identical ranks in both studies, the 129S1/SvlmJ switched from (4) in the earlier study to (5) in the current one, FVB/NJ from (5) to (7) and the A/J from (7) to (4), respectively. It should be noted that the BTBR T+ tf/J inbred strain was initially included in this study. This strain has a propensity to jump from the rotarod rather than run on top of it and was excluded before analyses because of low sample size. Data that were collected (not shown), however, indicated that this strain performs markedly better than all other strains tested, an observation that was also noted previously (Rustay et al., 2003). Therefore, it may be useful to characterize the BTBR T+ tf/J in other behavioral assays related to motor impairment.
Aspects that may influence improvement include fatigue, motivation, and/or ability to learn the task. Learning versus performance of motor tasks may involve distinct neural circuits. Results in experiment 3 argue against a learning deficit during withdrawal and instead suggest a physical inability to perform the task. EtOH-exposed and air-exposed mice receiving equal partial training time on the accelerating rotarod (yoked control procedure) at 8-h demonstrated treatment group differences at 24 h (EtOH-treated mice performed worse). No treatment group differences were, however, exhibited when testing was delayed for 1 week (these animals were not tested at 24 h). This suggests that EtOH-exposed and air-exposed mice were able to learn an equivalent amount about the task during the few trials at the 8-h time point. Disruption of rotarod behavior at 24 h must then be because of physical impairment, because a separate group of animals (receiving the same training period) showed no deficit after 1 week, presumably after withdrawal-induced motor effects had dissipated.
Similar to the conclusion based on inbred correlations, data from WSP and WSR lines tested during withdrawal on the accelerating rotarod task did not support a genetic correlation with the HIC response (Crabbe et al., 1990b). Thus, we suspect that no genetic correlation exists between the rotarod and HIC indices of EtOH withdrawal severity. Differences in EtOH withdrawal-induced impairment in the rotarod task performance and improvement during the test are heritable traits and our data suggest that the traits are genetically independent of HIC severity during withdrawal. On account of this, we hypothesize that independent neural pathways and brain regions may be responsible for mediating EtOH withdrawal-induced motor deficits and HIC susceptibility. Studies with rats and mice have shown that some physical signs of EtOH withdrawal largely dissipate within 24 h (Crabbe et al., 1990a; Guppy et al., 1995). Thus, it is not unreasonable that withdrawal signs detected in the accelerating rotarod task had dissipated at 1 week after EtOH exposure. Further studies are needed to compare inbred strains for withdrawal-induced deficits in accelerating rotarod behavior after protracted withdrawal (i.e. 24 h or more). Therefore, continuing to explore new EtOH withdrawal phenotypes provides an important tool for dissecting the genetic contributions to alcohol dependence.
Acknowledgements
The authors thank Michelle Sorensen, Lauren Brown, Chia-Hua Yu, Stephanie Spence and Jason Schlumbohm for assistance in collecting the data, data analyses, and presentation; Michelle Bobo for data curation; Matt Lattal for helpful discussion; and Mark Rutledge-Gorman for assistance in electronic submission. This study was supported by NIH grants AA10760, AA13519 and the US Department of Veterans Affairs. S.D.P. is supported by T32AA07468.
References
- Al-Rejaie S, Dar MS. Possible role of mouse cerebellar nitric oxide in the behavioral interaction between chronic intracerebellar nicotine and acute ethanol administration: observation of cross-tolerance. Neuroscience. 2006a;138:575–585. doi: 10.1016/j.neuroscience.2005.11.034. [DOI] [PubMed] [Google Scholar]
- Al-Rejaie S, Dar MS. Antagonism of ethanol ataxia by intracerebellar nicotine: possible modulation by mouse cerebellar nitric oxide and cGMP. Brain Res Bull. 2006b;69:187–196. doi: 10.1016/j.brainresbull.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Al-Rejaie S, Dar MS. Behavioral interaction between nicotine and ethanol: possible modulation by mouse cerebellar glutamate. Alcohol Clin Exp Res. 2006c;30:1223–1233. doi: 10.1111/j.1530-0277.2006.00143.x. [DOI] [PubMed] [Google Scholar]
- Boehm SL, 2nd, Schafer GL, Phillips TJ, Browman KE, Crabbe JC. Sensitivity to ethanol-induced motor incoordination in 5-HT (1B) receptor null mutant mice is task-dependent: implications for behavioral assessment of genetically altered mice. Behav Neurosci. 2000;114:401–409. [PubMed] [Google Scholar]
- Boehm SL, 2nd, Peden L, Chang R, Harris RA, Blednov YA. Deletion of the fyn-kinase gene alters behavioral sensitivity to ethanol. Alcohol Clin Exp Res. 2003;27:1033–1040. doi: 10.1097/01.ALC.0000075822.80583.71. [DOI] [PubMed] [Google Scholar]
- Bogo V, Hill TA, Young RW. Comparison of accelerod and rotarod sensitivity in detecting ethanol- and acrylamide-induced performance decrement in rats: review of experimental considerations of rotating rod systems. Neurotoxicology. 1981;2:765–787. [PubMed] [Google Scholar]
- Bogue MA, Grubb SC. The Mouse Phenome Project. Genetica. 2004;122:71–74. doi: 10.1007/s10709-004-1438-4. [DOI] [PubMed] [Google Scholar]
- Crabbe JC. Genetic contributions to addiction. Annu Rev Psychol. 2002a;53:435–462. doi: 10.1146/annurev.psych.53.100901.135142. [DOI] [PubMed] [Google Scholar]
- Crabbe JC. Alcohol and genetics: new models. Am J Med Genet. 2002b;114:969–974. doi: 10.1002/ajmg.b.10984. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Phillips TJ. Selective breeding for alcohol withdrawal severity. Behav Genet. 1993;23:171–177. doi: 10.1007/BF01067422. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Kosobud A, Young ER, Tam BR, McSwigan JD. Bidirectional selection for susceptibility to ethanol withdrawal seizures in Mus musculus. Behav Genet. 1985;15:521–536. doi: 10.1007/BF01065448. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Merrill CM, Kim D, Belknap JK. Alcohol dependence and withdrawal: a genetic animal model. Ann Med. 1990a;22:259–263. doi: 10.3109/07853899009148937. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Phillips TJ, Kosobud A, Belknap JK. Estimation of genetic correlation: interpretation of experiments using selectively bred and inbred animals. Alcoho Clin Exp Res. 1990b;14:141–151. doi: 10.1111/j.1530-0277.1990.tb00461.x. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Merrill C, Belknap JK. Acute dependence on depressant drugs is determined by common genes in mice. J Pharmacol Exp Ther. 1991;257:663–667. [PubMed] [Google Scholar]
- Crabbe JC, Gallaher ES, Phillips TJ, Belknap JK. Genetic determinants of sensitivity to ethanol in inbred mice. Behav Neurosci. 1994;108:186–195. doi: 10.1037//0735-7044.108.1.186. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Cotnam CJ, Cameron AJ, Schlumbohm JP, Rhodes JS, Metten P, Wahlsten D. Strain differences in three measures of ethanol intoxication in mice: the screen, dowel and grip strength tests. [erratum appears in Genes Brain Behav 2004 3:252] Genes Brain Behavior. 2003;2:201–213. doi: 10.1034/j.1601-183x.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Metten P, Cameron AJ, Wahlsten D. An analysis of the genetics of alcohol intoxication in inbred mice. Neurosci Biobehav Rev. 2005;28:785–802. doi: 10.1016/j.neubiorev.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Dar MS. Mouse cerebellar adenosinergic modulation of ethanol-induced motor incoordination: possible involvement of cAMP. Brain Res. 1997;749:263–274. doi: 10.1016/s0006-8993(96)01263-2. [DOI] [PubMed] [Google Scholar]
- Dar MS. Mouse cerebellar adenosine-glutamate interactions and modulation of ethanol-induced motor incoordination. Alcohol Clin Exp Res. 2002;26:1395–1403. doi: 10.1097/01.ALC.0000030564.69414.74. [DOI] [PubMed] [Google Scholar]
- Dar MS, Li C, Bowman ER. Central behavioral interactions between ethanol, (−)-nicotine, and (−)-cotinine in mice. Brain Res Bull. 1993;32:23–28. doi: 10.1016/0361-9230(93)90314-2. [DOI] [PubMed] [Google Scholar]
- Dar MS, Bowman ER, Li C. Intracerebellar nicotinic-cholinergic participation in the cerebellar adenosinergic modulation of ethanol-induced motor incoordination in mice. Brain Res. 1994;644:117–127. doi: 10.1016/0006-8993(94)90354-9. [DOI] [PubMed] [Google Scholar]
- Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc. 1957;46:208–209. doi: 10.1002/jps.3030460322. [DOI] [PubMed] [Google Scholar]
- Goldstein DB. Relationship of alcohol dose to intensity of withdrawal signs in mice. J Pharmacol Exp Ther. 1972;180:203–215. [PubMed] [Google Scholar]
- Goldstein DB. Inherited differences in intensity of alcohol withdrawal reactions in mice. Nature. 1973;245:154–156. doi: 10.1038/245154a0. [DOI] [PubMed] [Google Scholar]
- Goldstein DB, Pal N. Alcohol dependence produced in mice by inhalation of ethanol: grading the withdrawal reaction. Science. 1971;172:288–290. doi: 10.1126/science.172.3980.288. [DOI] [PubMed] [Google Scholar]
- Guppy LJ, Crabbe JC, Littleton JM. Time course and genetic variation in the regulation of calcium channel antagonist binding sites in rodent tissues during the induction of ethanol physical dependence and withdrawal. Alcohol Alcohol. 1995;30:607–615. [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ. The quantiative measurement of motor inco-ordination in naive mice using an acelerating rotarod. J Pharm Pharmacol. 1968a;20:302–304. doi: 10.1111/j.2042-7158.1968.tb09743.x. [DOI] [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ. A rotarod suitable for quantitative measurements of motor incoordination in naive mice. Naunyn-Schmiedebergs Archiv fur Experimentelle Pathologie und Pharmakologie. 1968b;259:211. doi: 10.1007/BF00537801. [DOI] [PubMed] [Google Scholar]
- Kliethermes CL. Anxiety-like behaviors following chronic ethanol exposure. Neurosci Biobehav Rev. 2005;28:837–850. doi: 10.1016/j.neubiorev.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Kosobud A, Crabbe JC. Ethanol withdrawal in mice bred to be genetically prone or resistant to ethanol withdrawal seizures. J Pharmacol Exp Ther. 1986;238:170–177. [PubMed] [Google Scholar]
- Lalonde R, Qian S. Exploratory activity, motor coordination, and spatial learning in Mchr1 knockout mice. Behav Brain Res. 2007;178:293–304. doi: 10.1016/j.bbr.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Le Pêcheur M, Strazielle C, London J. Exploratory activity and motor coordination in wild-type SOD1/SOD1 transgenic mice. Brain Res Bull. 2005;66:155–162. doi: 10.1016/j.brainresbull.2005.04.015. [DOI] [PubMed] [Google Scholar]
- Manzoni OJ, Manabe T, Nicoll RA. Release of adenosine by activation of NMDA receptors in the hippocampus. Science. 1994;265:2098–2101. doi: 10.1126/science.7916485. [DOI] [PubMed] [Google Scholar]
- Metten P, Crabbe JC. Common genetic determinants of severity of acute withdrawal from ethanol, pentobarbital and diazepam in inbred mice. Behav Pharmacol. 1994;5:533–547. doi: 10.1097/00008877-199408000-00014. [DOI] [PubMed] [Google Scholar]
- Metten P, Crabbe JC. Dependence and withdrawal. In: Deitrich RA, Erwin VG, editors. Pharmacological effects of ethanol on the nervous system. CRC Press; Boca Raton, New York, London, Tokyo: 1996. pp. 269–290. [Google Scholar]
- Metten P, Crabbe JC. Genetic determinants of severity of acute withdrawal from diazepam in mice: commonality with ethanol and pento-barbital. Pharmacol Biochem Behav. 1999;63:473–479. doi: 10.1016/s0091-3057(99)00017-9. [DOI] [PubMed] [Google Scholar]
- Metten P, Crabbe JC. Alcohol withdrawal severity in inbred mouse (Mus musculus) strains. Behav Neurosci. 2005;119:911–925. doi: 10.1037/0735-7044.119.4.911. [DOI] [PubMed] [Google Scholar]
- Metten P, Phillips TJ, Crabbe JC, Tarantino LM, McClearn GE, Plomin R, et al. High genetic susceptibility to ethanol withdrawal predicts low ethanol consumption. Mamm Genome. 1998;9:983–990. doi: 10.1007/s003359900911. [DOI] [PubMed] [Google Scholar]
- Phillips TJ, Feller DJ, Crabbe JC. Selected mouse lines, alcohol and behavior. Experientia. 1989;45:805–827. doi: 10.1007/BF01954056. [DOI] [PubMed] [Google Scholar]
- Rustay NR, Crabbe JC. Genetic analysis of rapid tolerance to ethanol’s incoordinating effects in mice: inbred strains and artificial selection. Behav Genet. 2004;34:441–451. doi: 10.1023/B:BEGE.0000023649.60539.dd. [DOI] [PubMed] [Google Scholar]
- Rustay NR, Boehm SL, 2nd, Schafer GL, Browman KE, Erwin VG, Crabbe JC. Sensitivity and tolerance to ethanol-induced incoordination and hypothermia in HAFT and LAFT mice. Pharmacol Biochem Behav. 2001;70:167–174. doi: 10.1016/s0091-3057(01)00595-0. [DOI] [PubMed] [Google Scholar]
- Rustay NR, Wahlsten D, Crabbe JC. Assessment of genetic susceptibility to ethanol intoxication in mice. Proc Natl Acad Sci U S A. 2003;100:2917–2922. doi: 10.1073/pnas.0437273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer GL, Crabbe JC. Sensitivity to ethanol-induced ataxia in HOT and COLD selected lines of mice. Alcohol Clin Exp Res. 1996;20:1604–1612. doi: 10.1111/j.1530-0277.1996.tb01705.x. [DOI] [PubMed] [Google Scholar]
- Sher KJ, Grekin ER, Williams NA. The development of alcohol use disorders. Annu Rev Clin Psychol. 2005;1:493–523. doi: 10.1146/annurev.clinpsy.1.102803.144107. [DOI] [PubMed] [Google Scholar]
- Tarantino LM, Gould TJ, Druhan JP, Bucan M. Behavior and mutagenesis screens: the importance of baseline analysis of inbred strains. Mamm Genome. 2000;11:555–564. doi: 10.1007/s003350010107. [DOI] [PubMed] [Google Scholar]
- Terdal ES, Crabbe JC. Indexing withdrawal in mice: matching genotypes for exposure in studies using ethanol vapor inhalation. Alcohol Clin Exp Res. 1994;18:542–547. doi: 10.1111/j.1530-0277.1994.tb00907.x. [DOI] [PubMed] [Google Scholar]
- Valdez GR, Koob GF. Allostasis and dysregulation of corticotropin-releasing factor and neuropeptide Y systems: implications for the development of alcoholism. Pharmacol Biochem Behav. 2004;79:671–689. doi: 10.1016/j.pbb.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Wahlsten D, Metten P, Phillips TJ, Boehm SL, II, Burkhart-Kasch S, Dorow J, et al. Different data from different labs: lessons from studies of gene-environment interaction. J Neurobiol. 2003;54:283–311. doi: 10.1002/neu.10173. [DOI] [PubMed] [Google Scholar]
- Wahlsten D, Bachmanov A, Finn DA, Crabbe JC. Stability of inbred mouse strain differences in behavior and brain size between laboratories and across decades. Proc Natl Acad Sci U S A. 2006;103:16364–16369. doi: 10.1073/pnas.0605342103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watzman N, Barry H., 3rd Drug effects on motor coordination. Psychopharmacologia. 1968;12:414–423. doi: 10.1007/BF00401346. [DOI] [PubMed] [Google Scholar]