

Abstract
Introduction
Late-onset Tay-Sachs disease (LOTS) is a lysosomal storage disease caused by deficient Beta-hexosaminidase A activity.
Methods
We describe a 53-year-old woman who presented with adult-onset leg weakness, and whose initial diagnosis was progressive muscular atrophy without identifiable etiology. Development of cerebellar ataxia in mid-life prompted reassessment.
Results
Beta-hexosaminidase A quantification assay demonstrated absence of the isozyme. Genetic testing identified compound heterozygous mutations in the HEXA gene, confirming the diagnosis of LOTS.
Conclusions
The phenotypic spectrum of LOTS includes motor neuronopathy, ataxia, choreoathetosis, neuropathy, and psychiatric symptoms in various combinations. This patient highlights the emergence of different clinical features over many years and emphasizes the need to consider LOTS in the differential diagnosis of progressive muscular atrophy.
Keywords: late-onset Tay-Sachs disease, hexosaminidase, progressive muscular atrophy, ataxia, cerebellum
Late-onset Tay-Sachs disease (LOTS) is an autosomal recessive lysosomal storage disease due to compound heterozygous or homozygous mutations in HEXA.1 These lead to decreased Beta-hexosaminidase A activity and subsequent intracellular accumulation of CNS gangliosides.2 Patients may present in childhood, adolescence, or early adulthood. Initial features may include weakness due to motor neuron disease, neuropathy, dysarthria, spasticity, dystonia, tremor, ataxia or psychosis.3 We present a woman who was first seen by us at age 53 with a diagnosis of spinal muscular atrophy (SMA) which had been symptomatic since her 20s. The emergence of additional features suggested the involvement of central systems and prompted diagnostic reevaluation.
CASE REPORT
This 53-year-old woman had leg cramps and difficulty running since childhood but was otherwise in excellent health until her early 20s when she had difficulty arising from a chair and stepping from a curb. At age 28 she developed difficulty climbing stairs and a wide-based, waddling gait, prompting her first neurological evaluation. She was of Ashkenazi Jewish descent and did not have a family history of weakness or gait disorder. There was no known consanguinity. Records from her evaluation at age 28 reported symmetric proximal arm and leg weakness. Electrodiagnostic studies (EDX) suggested “chronic neuropathy,” but further details were unavailable. Muscle biopsy revealed nonspecific changes, including a preponderance of type I fibers with increased oxidative activity in the subsarcolemmal region in many fibers and a slight increase in interfascicular connective tissue. Brain MRI was reportedly unremarkable.
In her mid-30s, climbing stairs and standing from a chair became more difficult, and she began to fall every few months. There was no myalgia or muscle atrophy. Repeat EDX presumably indicated motor neuron disease (although a formal report was not available), and the possibility of a variant of SMA was raised. Genetic testing was not pursued.
In her early 40s she developed intermittent hand tremor and decreased dexterity, which affected her handwriting. Speech became slurred and had a staccato quality. By her late 40s, balance problems led to falls and injuries, and by age 51 she used a walker intermittently.
She was reevaluated by a neurologist at age 52. Brain MRI now showed isolated, prominent cerebellar atrophy (Fig. 1), and EDX were reported as consistent with “chronic, generalized motor axonopathy.” Comprehensive spinocerebellar ataxia (SCA) and recessive ataxia panels revealed no mutations in the genes coding for SCAs 1, 2, 3, 5, 6, 7, 8, 10, 12, 13, 14, 17, 28 or dentatorubral-pallidoluysian atrophy, nor in the FXN, APTX, SETX, POLG1, SIL1, or TTPA genes.
FIGURE 1.
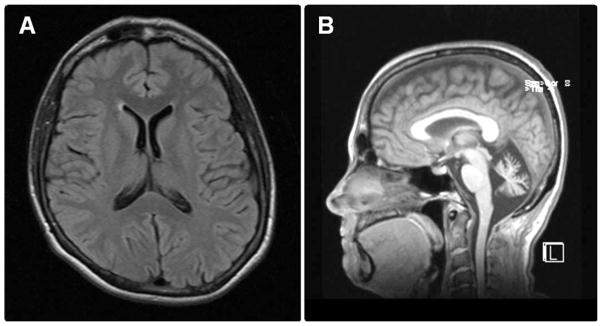
Brain MRI, age 52. Axial T2-weighted (A) and sagittal (B) T1-weighted brain MRI shows marked prominence of the cerebellar and vermian sulci with enlargement of the 4th ventricle consistent with cerebellar volume loss. Notice the normal appearance of the brainstem and the supratentorial ventricles, sulci, and brain parenchyma.
We first evaluated her at age 53. Cognition was excellent, and she had monotonic but pressured and fluent speech. Her extraocular movements were full in all directions, with normal saccades, optokinetic nystagmus, and no square wave jerks. Other cranial nerve functions were normal. Triceps, iliopsoas, and quadriceps muscle strength was graded as 4-/5 bilaterally, and other muscles demonstrated full strength. Tone was normal. Reflexes were 3+ in the arms and 1+ in the legs with no Babinski signs. Gowers sign was present. Occasional mild chorea was seen, mostly in the form of fidgetiness while seated, although some subtle upper trunk movements were also present. A mild amplitude, medium frequency finger tremor was evident bilaterally when performing the finger-nose-finger maneuver. There was no bradykinesia, although rapid alternating movements were clumsy. Sensation was normal for pinprick, temperature, vibration, and position. There was overshoot and dysmetria in the arms and legs. The gait was ataxic and wide-based, there were slow, cautious turns, and she used a walker. She was unable to tandem walk and had poor postural stability.
Vitamin E levels and thyroid function tests were normal. While total hexosaminidase quantification was normal (13.6 U/L, reference range 10.4–23.8), hexosaminidase A activity was absent. Genetic testing identified the TATCins1278 and Gly269Ser Tay-Sachs disease mutations. Of note, the latter is known to be associated with LOTS in the homozygous state, or in compound heterozygosity with a null allele.1 Null alleles are associated with the classic, acute infantile variant of Tay-Sachs disease when present in the homozygous state.
DISCUSSION
The differential diagnosis of progressive muscular atrophy (PMA) beginning as cramping and difficulty running in childhood includes both hereditary and sporadic childhood onset disorders.3 Juvenile-onset progressive weakness and motor neuronopathy may be due to SMA types III or IV,4 juvenile-onset amyotrophic lateral sclerosis (ALS),3 the GM2 gangliosidoses, and Fazio-Londe syndrome. The exclusively proximal pattern of weakness present in the patient is not particularly suggestive of Fazio-Londe syndrome5 or a juvenile form of ALS but is observed in SMA variants and GM2 gangliosidoses. Hence, assessing hexosaminidase levels would have been justified at the time of initial presentation.
The late onset of tremor, decreased dexterity, speech changes, and frequent falls suggested cerebellar pathology, which was likely either absent or very subtle at the time of initial evaluation. The presence of ataxia suggests an SCA or an autosomal recessive ataxia as an additional consideration. Although cerebellar atrophy would be expected as a feature in either of these conditions, it is possible that it was not prominent early in the course of her disease. Some SCAs, as well as autosomal recessive ataxias, may overlap with motor neuron disease: SCA 2 may present with progressive ataxia, parkinsonism and motor neuropathy6; SCA 3 typically affects the cerebellar, pyramidal, extrapyramidal, motor neuron, and oculomotor systems7; SCA 36 may show adult-onset truncal and limb ataxia, dysarthric ataxia, hyperreflexia, fasciculations, and atrophy8; and in SCAR8 upper and lower motor involvement may precede the development of cerebellar ataxia9 by years.10 Of interest, ataxia-telangiectasia may include pure distal SMA in the absence of ataxia.11
As with PMA, the differential for cerebellar ataxia also includes GM2 gangliosidoses, particularly if patients belong to an ethnic group in which the mutation carrier state is known to be highly prevalent. The GM2 gangliosidoses include Sandh-off and Tay-Sachs disease, and the former is caused by mutations in the HEXB gene.2,12 Patients with late-onset Sandhoff disease usually present with either a cerebellar syndrome or lower motor neuron disease, and a few show isolated autonomic dysfunction.12 Neuronopathy and axonopathy that are either mild or only detectable by EDX have also been reported.12
Given the patient’s ethnic background, the preferential weakness of the triceps and quadriceps muscles, and the presence of tremor and subtle chorea, LOTS is the more likely clinical diagnosis, as was determined by enzyme assessment and further confirmed by genetic testing. The isolated cerebellar atrophy on MRI and generalized motor axonopathy on EDX are also consistent with the diagnosis. The initial presentation of motor neuron disease followed years later by cerebellar ataxia attests to the phenotypic progression that may be seen in individuals throughout their lifetimes. It is not clear when the tremor and chorea began, as they were undoubtedly mild initially.
Although the phenotypic spectrum of the disease has been well characterized, atypical presentations have been described in patients with LOTS. Shapiro and Natowicz reported a case of childhood stutter that was later identified as LOTS.13 In 8 of a cohort of 30 patients with LOTS, Shapiro et al. also found evidence of a predominantly axonal polyneuropathy affecting distal nerve segments in the arms and legs.14 Godeiro-Junior et al. chronicled a 30-year-old Brazilian Caucasian man with young onset and slowly progressive spastic tetraparesis resembling primary lateral sclerosis whose brain MRI revealed abnormal signal in the corticospinal tracts, as seen in typical ALS, but there was no cerebellar atrophy.15 Hexosaminidase A quantification revealed reduced isozyme levels, but no genetic analysis was performed. Psychiatric features may also be the presenting symptom, as was reported in a boy with treatment-resistant catatonic schizophrenia who rapidly developed neuroleptic malignant syndrome after being exposed to neuroleptics16 and whose metabolic screening revealed a severe hexosaminidase A deficiency.
Although infantile Tay-Sachs disease is lethal, the prognosis is more variable in LOTS, making its accurate diagnosis important for different reasons. First, and as mentioned above, certain psychotropic medications (particularly haloperidol, chlorpromazine, and risperidone) may worsen the neurologic condition of patients with LOTS and should be avoided.2,16 Furthermore, while clinical trials, such as substrate deprivation therapy with miglustat17 or enzymatic rescue with pyrimethamine,18 did not demonstrate efficacy, there remains great promise for treatment of slowly evolving metabolic diseases such as LOTS. While our patient was of Ashkenazi descent and the mutation readily identified as confirmatory, in other populations, screening for the common mutations may be insufficient. Thus, measurement of enzyme activity should be performed as an initial step when the diagnosis is suspected. Finally, because LOTS manifests in compound heterozygotes, awareness of the carrier state has important implications in selected populations where the prevalence of infantile Tay-Sachs is known to be high. In LOTS, patients harbor 1 childhood onset mutation and 1 adult-onset mutation,1 and unless the adult-onset mutation is included in the screening, the risk for LOTS transmission may not be known. Therefore, family members of LOTS patients with identified mutations should have the adult-onset mutations included in their prenatal screen to ensure accurate genetic counseling.
Acknowledgments
Dr. Deik was supported by a Clinical Research Fellowship from the Dystonia Medical Research Foundation, and was a co-investigator on a grant from the Marcled Foundation. Dr. Saunders-Pullman is supported by NIH-NINDS K02-NS073836. She also received research funding from the Marcled Foundation, and currently receives funding from the Bigglesworth Foundation, the Empire Clinical Research Initiative Program, the Michael J. Fox Foundation for Parkinson’s Research, and the Bachmann-Strauss Dystonia and Parkinson’s Foundation.
We thank the patient for her careful assembling of her records. We also thank Dr. Lewis P. Rowland for his insightful review of the manuscript.
Abbreviations
- AOA2
ataxia-oculomotor apraxia 2
- ALS
amyotrophic lateral sclerosis
- EDX
electrodiagnostic studies
- HEXA
gene coding for the α subunit of hexosaminidase
- HEXB
gene coding for the β subunit of hexosaminidase
- LOTS
late-onset Tay-Sachs
- PMA
progressive muscular atrophy
- SCAs
spinocerebellar ataxias
- SETX
senataxin
- SMA
Spinal muscular atrophy
References
- 1.Cao Z, Natowicz MR, Kaback MM, Lim-Steele JS, Prence EM, Brown D, et al. A second mutation associated with apparent beta-hexosaminidase A pseudodeficiency: identification and frequency estimation. Am J Hum Genet. 1993;53:1198–1205. [PMC free article] [PubMed] [Google Scholar]
- 2.Shapiro BE, Hatters-Friedman S, Fernandes-Filho JA, Anthony K, Natowicz MR. Late-onset Tay-Sachs disease: adverse effects of medications and implications for treatment. Neurology. 2006;67:875–877. doi: 10.1212/01.wnl.0000233847.72349.b6. [DOI] [PubMed] [Google Scholar]
- 3.Rowland LP. Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle Nerve. 2010;41:161–165. doi: 10.1002/mus.21565. [DOI] [PubMed] [Google Scholar]
- 4.D’Amico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orphanet J Rare Dis. 2011;6:71. doi: 10.1186/1750-1172-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bosch AM, Stroek K, Abeling NG, Waterham HR, Ijlst L, Wanders RJ. The Brown-Vialetto-Van Laere and Fazio Londe syndrome revisited: natural history, genetics, treatment and future perspectives. Orphanet J Rare Dis. 2012;7:83. doi: 10.1186/1750-1172-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishikawa N, Nagai M, Tsujii T, Tanabe N, Takashima H, Nomoto M. Three spinocerebellar ataxia type 2 siblings with ataxia, parkinsonism, and motor neuronopathy. Intern Med. 2011;50:1429–1432. doi: 10.2169/internalmedicine.50.5262. [DOI] [PubMed] [Google Scholar]
- 7.Bettencourt C, Lima M. Machado-Joseph disease: from first descriptions to new perspectives. Orphanet J Rare Dis. 2011;6:35. doi: 10.1186/1750-1172-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abe K, Ikeda Y. [Spinocerebellar ataxia type 36 (nicknamed asidan)] Brain Nerve. 2012;64:937–941. [PubMed] [Google Scholar]
- 9.Izumi Y, Miyamoto R, Morino H, Yoshizawa A, Nishinaka K, Udaka F, et al. Cerebellar ataxia with SYNE1 mutation accompanying motor neuron disease. Neurology. 2013;80:600–601. doi: 10.1212/WNL.0b013e3182815529. [DOI] [PubMed] [Google Scholar]
- 10.Gros-Louis F, Dupre N, Dion P, Fox MA, Laurent S, Verreault S, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. 2007;39:80–85. doi: 10.1038/ng1927. [DOI] [PubMed] [Google Scholar]
- 11.Hiel JA, van Engelen BG, Weemaes CM, Broeks A, Verrips A, ter Laak H, et al. Distal spinal muscular atrophy as a major feature in adult-onset ataxia telangiectasia. Neurology. 2006;67:346–349. doi: 10.1212/01.wnl.0000224878.22821.23. [DOI] [PubMed] [Google Scholar]
- 12.Delnooz CC, Lefeber DJ, Langemeijer SM, Hoffjan S, Dekomien G, Zwarts MJ, et al. New cases of adult-onset Sandhoff disease with a cerebellar or lower motor neuron phenotype. J Neurol Neurosurg Psychiatry. 2010;81:968–972. doi: 10.1136/jnnp.2009.177089. [DOI] [PubMed] [Google Scholar]
- 13.Shapiro BE, Natowicz MR. Late-onset Tay-Sachs disease presenting as a childhood stutter. J Neurol Neurosurg Psychiatry. 2009;80:94–95. doi: 10.1136/jnnp.2008.147645. [DOI] [PubMed] [Google Scholar]
- 14.Shapiro BE, Logigian EL, Kolodny EH, Pastores GM. Late-onset Tay-Sachs disease: the spectrum of peripheral neuropathy in 30 affected patients. Muscle Nerve. 2008;38:1012–1015. doi: 10.1002/mus.21061. [DOI] [PubMed] [Google Scholar]
- 15.Godeiro-Junior C, Felicio AC, Benites V, Chieia MA, Oliveira AS. Late-onset hexosaminidase A deficiency mimicking primary lateral sclerosis. Arq Neuropsiquiatr. 2009;67:105–106. doi: 10.1590/s0004-282x2009000100024. [DOI] [PubMed] [Google Scholar]
- 16.Rosebush PI, MacQueen GM, Clarke JT, Callahan JW, Strasberg PM, Mazurek MF. Late-onset Tay-Sachs disease presenting as catatonic schizophrenia: diagnostic and treatment issues. J Clin Psychiatry. 1995;56:347–353. [PubMed] [Google Scholar]
- 17.Shapiro BE, Pastores GM, Gianutsos J, Luzy C, Kolodny EH. Miglustat in late-onset Tay-Sachs disease: a 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet Med. 2009;11:425–433. doi: 10.1097/GIM.0b013e3181a1b5c5. [DOI] [PubMed] [Google Scholar]
- 18.Clarke JT, Mahuran DJ, Sathe S, Kolodny EH, Rigat BA, Raiman JA, et al. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants) Mol Genet Metab. 2011;102:6–12. doi: 10.1016/j.ymgme.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]