

SUMMARY
During persistent antigen stimulation, CD8+ T cells show a gradual decrease in effector function, referred to as exhaustion, which impairs responses in the setting of tumors and infections. Here we demonstrate that the transcription factor NFAT controls the program of T cell exhaustion. When expressed in cells, an engineered form of NFAT1 unable to interact with AP-1 transcription factors diminished T cell receptor (TCR) signaling, increased the expression of inhibitory cell surface receptors, and interfered with the ability of CD8+ T cells to protect against Listeria infection and attenuate tumor growth in vivo. We defined the genomic regions occupied by endogenous and engineered NFAT1 in primary CD8+ T cells, and showed that genes directly induced by the engineered NFAT1 overlapped with genes expressed in exhausted CD8+ T cells in vivo. Our data show that NFAT promotes T cell anergy and exhaustion by binding at sites that do not require cooperation with AP-1.
INTRODUCTION
The transcription factor nuclear factor of activated T cells (NFAT) is well established as a key regulator of T cell activation (Crabtree and Olson, 2002; Hogan et al., 2003; Macian, 2005; Rao et al., 1997). Of the five members of the NFAT family, NFAT1-4 (also known as NFATc1-c4) are regulated by Ca2+-calcineurin signaling; of these, NFAT1, NFAT2 and NFAT4 are expressed in cells of the immune system and have important roles in T cell development and function. All NFAT proteins make very similar contacts with DNA, but can have distinct expression patterns and functions as judged by the non-overlapping phenotypes of mice deficient in individual NFAT family members (Crabtree and Olson, 2002; Hogan et al., 2003; Macian, 2005). NFAT proteins interact with structurally unrelated Fos-Jun (AP-1) transcription factors to form cooperative NFAT: AP-1 complexes (Chen et al., 1998) that are critical for the induction of cytokine genes and other activation-associated genes (Macian et al., 2000). Adding to its versatility, NFAT forms dimers on palindromic κB-like sequence elements and can bind DNA as a monomer ((Chen et al., 1998; Giffin et al., 2003; Jin et al., 2010; Stroud et al., 2002); it also forms cooperative complexes with FOXP3, a transcription factor central to T regulatory function (Bandukwala et al., 2011; Chen et al., 1998; Macian et al., 2000; Wu et al., 2006). The ability to participate in multiple transcriptional complexes allows NFAT to contribute to different transcriptional programs depending on the cell type and signalling context in which it is activated (Hogan et al., 2003).
We previously linked NFAT not only to T cell activation, but also to T cell “tolerance” and “anergy” (Fehr et al., 2010; Heissmeyer et al., 2004; Macian et al., 2002), hyporesponsive states induced in T cells exposed to activating signals through the T cell receptor (TCR) in the absence of positive or presence of negative costimulatory signals (Nurieva et al., 2011). Another hyporesponsive state, termed CD8+ T cell “exhaustion”, is induced in antigen-specific cytolytic T cells (CTLs) exposed to persistent antigen stimulation, for instance in the context of chronic viral infections and cancer (Schietinger and Greenberg, 2014; Wherry, 2011). Exhausted CD8+ T cells display a transcriptional program distinct from that of functional effector or memory CD8+ T cells (Wherry et al., 2007), characterized, for example, by the expression of several inhibitory cell surface receptors including PD-1, LAG3, TIM3, TIGIT and CTLA4 (Schietinger and Greenberg, 2014; Wherry, 2011). However the key transcription factor(s) responsible for establishment of CD8+ T cell exhaustion have not yet been identified (Wherry, 2011).
Here we show that NFAT elicits two parallel programs of CD8+ T cell activation and exhaustion. To separate the NFAT-dependent programs of T cell activation and exhaustion, we generated an engineered version of NFAT1 that is incapable of AP-1 cooperation and therefore elicits no effector response. Expression of this engineered protein blunts T cell receptor (TCR) responses and elicits CD8+ T cell exhaustion in mouse models of tumor growth and bacterial infection. The alternative NFAT-dependent transcriptional program is not limited to this simplified experimental situation, however, since NFAT-deficient CD8+ T cells fail to express either effector cytokines or the inhibitory cell surface receptors PD-1, LAG3 and TIM3 that are characteristic of exhausted CD8+ T cells. Using genome-wide analyses, we have defined the DNA elements functionally occupied by endogenous and engineered NFAT1 proteins, and have correlated occupancy with gene expression and in vivo function. Our results elucidate the transcriptional programs of hyporesponsiveness (anergy and exhaustion) in both CD4+ and CD8+ T cells, and show that NFAT proteins have a primary role.
RESULTS
We generated an engineered version of NFAT1, CA-RIT-NFAT1, that is constitutively nuclear and therefore constitutively active (CA) (Okamura et al., 2000) (Figure S1A), and also unable to interact with AP-1 (“RIT” refers to three residues – R468, I469 and T535 in mouse NFAT1 – that have been mutated to interfere selectively with the NFAT:AP-1 interaction (Macian et al., 2002; Macian et al., 2000)). The engineered CA-RIT-NFAT1 elicits no effector response, and so was a convenient tool for the genome-wide analysis. However, all three NFAT proteins present in T cells contribute to the negative regulatory program, as described below.
CA-RIT-NFAT1-expressing cells display defective TCR signaling
We used a bicistronic (IRES-GFP) retrovirus to introduce CA-RIT-NFAT1 into in vitro-activated P14+ TCR transgenic T cells that also bore a deletion of the TCR Cα region (Tcra−/−); the P14 TCR transgene recognizes a peptide from the gp33 protein of mouse lymphocytic choriomeningitis virus (LCMV) presented on H-2Db. Naïve T cells were stimulated with anti-CD3 and anti-CD28, infected with IRES-GFP retrovirus expressing CA-RIT-NFAT1 or CA-RIT-NFAT1 with four point mutations in the DNA binding loop that abolish DNA binding (DBDmut-CA-RIT-NFAT1) (Jain et al., 1995) or empty vector (mock), and expanded with interleukin-2 (IL-2) to generate effector CTL (Pipkin et al., 2010). Expression of CA-RIT-NFAT1 in CD8+ T cells substantially decreased IL-2 production in response to secondary stimulation (Figure S1B, S1C), even though CA-RIT-NFAT1 was expressed at amounts comparable to or lower than endogenous NFAT1 (Figure S1D, S1E).
To ask whether CA-RIT-NFAT1 expression impaired Ca2+ influx, we transduced CD4+ T cells with CA-RIT-NFAT1 or empty vector (mock), labeled them with the Ca2+ indicators Fluo-4 or Fura-2, and quantified Ca2+ influx by flow cytometry (Figure S1F) or single-cell imaging (Figure S1G). Compared to cells transduced with empty vector, cells expressing CA-RIT-NFAT1 displayed increased basal Ca2+ (Figure S1F) as well as reduced Ca2+ influx upon TCR stimulation (Figure S1F; Figure S1G, left panel). In contrast, Ca2+ influx was not diminished when TCR signaling was bypassed with thapsigargin treatment, which depletes endoplasmic reticulum (ER) Ca2+ stores by inhibiting the SERCA Ca2+-ATPase (Figure S1G, right panel). Moreover, the increased phosphorylation of both ZAP-70 and PLCγ1 observed in control cells within minutes of re-stimulation with anti-CD3 and anti-CD28 was strongly impaired in cells expressing CA-RIT-NFAT1 (Figure S1H). Thus CA-RIT-NFAT1 expression affects two of the earliest steps of TCR signaling upstream of Ca2+ entry; other steps in the signaling window between TCR stimulation and ER store depletion could potentially also be impaired (Heissmeyer et al., 2004).
CA-RIT-NFAT1-expressing cells display impaired function in vivo
To test the biological effects of expressing CA-RIT-NFAT1 in CD8+ T cells, we utilized an in vivo Listeria protection assay (modified from (Kaech et al., 2003)) (Figures 1A, S1I). Naïve P14+ TCR transgenic CD8+ T cells were stimulated with anti-CD3 and anti-CD28 and transduced one day later with CA-RIT-NFAT1, DBDmut-CA-RIT-NFAT1 or empty vector, then expanded with a low concentration of IL-2 in vitro to generate “memory-like” CD8 T cells (Pipkin et al., 2010). Transduced GFP+ cells were then sorted by flow cytometry and transferred into naïve recipient mice; one day later, the mice were infected with genetically-modified Listeria monocytogenes expressing gp33 peptide (Figures 1A, S1I). Consistent with induction of an effective immune response against the Listeria-gp33, mice receiving mock-transduced gp33-specific T cells showed a significant reduction in bacterial colony-forming units (CFU) per spleen at 3 and 5 days after infection, compared to mice that did not receive any cells, whereas mice receiving cells transduced with CA-RIT-NFAT1 did not control Listeria infection effectively (Figures 1B, S1J). Thus CA-RIT-NFAT1 expression blunted the secondary immune response of CD8 T cells in vivo; some protection was still evident, however, indicating that T cell function was strongly diminished but not completely eliminated.
Figure 1. CTLs expressing a constitutively active NFAT1 incapable of cooperating with AP-1 have impaired in vivo function.
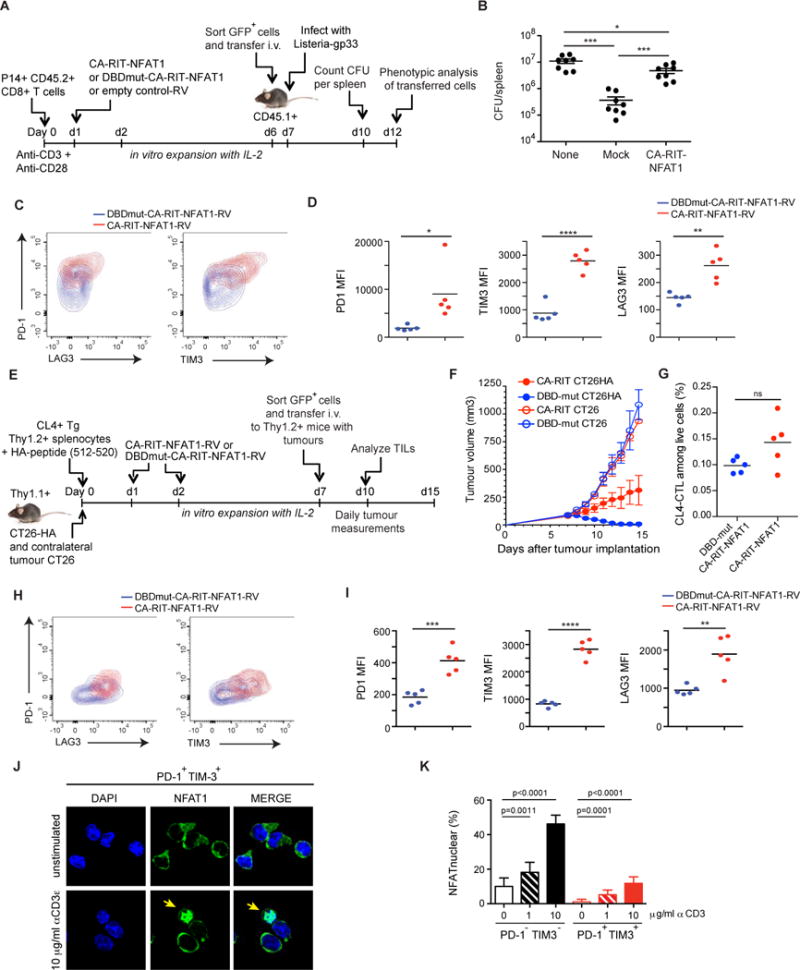
(A–D) GFPhi P14+ CD8+ T cells expressing CA-RIT-NFAT1 or its DNA-binding mutant were transferred to CD45.1+ congenic mice, which were infected one day later with gp33-expressing Listeria monocytogenes. (B) Three days after infection, total bacterial colony-forming units (CFU) per spleen were determined (each dot represents a mouse; mean ± SEM. *, p < 0.05; ***, p < 0.001 using t test). A representative experiment of two is shown. (C) Five days after infection, co-expression of PD-1 and LAG3 or PD-1 and TIM3 on transferred cells (CD45.2+) was determined by flow cytometry. Representative contour plots are shown. (D) The mean fluorescence intensity (MFI) for each receptor is shown. p values were calculated using t-test. *, p < 0.05; **, p < 0.005; ****, p< 0.0001. (E-I) GFPhi Thy1.2+ CL4+ TCR transgenic CD8+ T cells expressing CA-RIT-NFAT1 or its DNA-binding mutant were transferred to Thy1.1+ congenic mice, injected seven 7 days previously with CT26 and CT26HA tumor cells in contralateral flanks. (F) Tumor growth was determined on a daily basis after cell transfer. (G–I) Characterization of transferred cells 3 days after transfer. (G) Frequency of tumor-infiltrating CA-RIT-NFAT1- and DBDmut-CA-RIT-NFAT1-transduced cells in live gate. (H–I) Co-expression of inhibitory receptors on transferred cells determined by flow cytometry. Representative contour plots are shown. (I) MFI for each receptor is shown. p values were calculated using t-test. **, p < 0.005; ***, p < 0.0005; ****, p< 0.0001. (J–K) NFAT nuclear translocation in exhausted cells. Single cell suspensions from 10 day-old CT26HA tumors were stimulated ex vivo with increasing doses of plate-bound anti-CD3 and immediately fixed. (J) Confocal images of ex vivo-stimulated exhausted cells (CD45+CD8+PD-1+Tim-3+) purified by cell sorting and stained for endogenous NFAT1. Arrows highlight cells with nuclear NFAT1. Scale bar 10 μm. (K) NFAT nuclear translocation in exhausted (PD-1+TIM3+) and non-exhausted (PD-1−TIM3−) CD8+ tumor-infiltrating lymphocytes calculated from confocal images in J. Means ± S.D. of >10 fields of view (at least 720 cells) are shown. p values were calculated by t-test. See also Figures S1, S2.
The adoptively transferred CA-RIT-NFAT1-expressing cells survived in vivo and were able to reach the infection site, although at lower percentages and total numbers compared to control cells, as judged by their presence in spleens of recipient mice 5 days after infection (Figure S1K and data not shown). Compared to cells transduced with DBDmut-CA-RIT-NFAT1, a higher percentage of CA-RIT-NFAT1-expressing cells expressed PD-1, TIM3 and LAG3, inhibitory surface receptors characteristic of exhausted T cells (Figure 1C–D).
To assess the impaired function of CA-RIT-NFAT1-expressing T cells in a different in vivo system, we utilized a tumor model in which influenza hemagglutinin (HA)-specific CL4 TCR transgenic T cells were transduced with CA-RIT-NFAT1 or DBDmut-CA-RIT-NFAT1 (Bauer et al., 2014; Marangoni et al., 2013). The cells were expanded in vitro, then transferred into congenic mice that had previously received HA-expressing CT26 tumors (CT26HA) subcutaneously in one flank and CT26 tumors that did not express HA in the contralateral flank. Tumor growth was assessed daily for 8 days after T cell transfer, and the expression of exhaustion-associated surface markers on the transferred cells was evaluated 3 days after transfer (Figure 1E). Control cells expressing the mutant DBDmut-CA-RIT-NFAT1 rejected the CT26HA antigen-expressing tumor without rejecting the contralateral tumor that did not express HA (Figure 1F), whereas cells expressing CA-RIT-NFAT1 showed diminished effector activity (Figure 1F) despite being present in the tumor at similar frequencies as cells expressing DBDmut-CA-RIT-NFAT1 (Figure 1G). As in the Listeria model, we observed a higher frequency of expression of the inhibitory markers PD-1, TIM3 and LAG3 in CA-RIT-NFAT1-expressing cells recovered from the tumor, compared to cells expressing DBDmut-CA-RIT-NFAT1 (Figure 1H–I).
Overall, even in the presence of endogenous NFAT proteins, CA-RIT-NFAT1 directly or indirectly upregulated the expression of several markers of T cell exhaustion on the CD8+ T cells, and induced a negative feedback transcriptional program that attenuated CD8+ T cell responses in two different settings in vivo.
Exhausted cells retain the ability to signal through NFAT
We asked whether NFAT could translocate to the nucleus in in vivo exhausted T cells. CT26HA tumors were implanted in Thy1.1+ recipients that were then injected with regulatory T cells that recognize the HA antigen; this regimen induces exhaustion of endogenous CD8+ T cells (Bauer et al., 2014). Ten days after tumor injection, mice were sacrificed, and tumor-infiltrating T cells were restimulated ex vivo with plate-bound anti-CD3s for 15 min. The cells were then fixed and sorted to separate the exhausted PD-1+TIM3+ cell and control PD-1TIM3- cell populations (Figure S2A), and stained for endogenous NFAT1 and DAPI (Figures 1J, S2B). The results show clearly that exhausted CD8+ T cells are permissive for NFAT1 nuclear translocation upon activation through the TCR, although higher amounts of TCR stimulation are required than for non-exhausted cells (Figure 1K). Thus, in vivo-generated exhausted T cells can activate NFAT nuclear import, thus further reinforcing the exhaustion phenotype in the absence of AP-1 cooperation.
The transcriptional program induced by CA-RIT-NFAT1 overlaps with that of exhausted/anergic T cells
To define the transcriptional program induced by CA-RIT-NFAT1, we performed RNA-seq on CD4+ and CD8+ T cells transduced with empty or CA-RIT-NFAT1 retrovirus. Almost 2,000 genes showed altered expression (p<0.05) in CA-RIT-NFAT1-expressing CD8+ T cells compared to mock-transduced cells, with approximately half of the genes showing increased expression and half decreased expression (Figure 2A, top panel). Similar results were obtained for CD4+ T cells (Figure 2A, bottom panel). There was substantial overlap in genes differentially expressed in CD4+ and CD8+ T cells (one-tailed version of Fisher’s exact test; p-value <10−10), indicating that ectopic expression of CA-RIT-NFAT1 has similar transcriptional effects in both cell types (Figure 2B–D). There was also a highly significant overlap between the transcriptional profiles induced in CD8+ and CD4+ T cells by CA-RIT-NFAT1 and those observed in exhausted CD8+ T cells and anergic CD4+ T cells in vivo (22/56 genes and 99/371 genes respectively; p-value <10−10) (Doering et al., 2012; Okamura et al., 2009; Wherry et al., 2007)(Figure 2E–F, Table 1, Figure S3E; Tables S1, S2).
Figure 2. The transcriptional program induced by CA-RIT-NFAT1 in T cells overlaps with that of in vivo exhausted/anergic T cells.
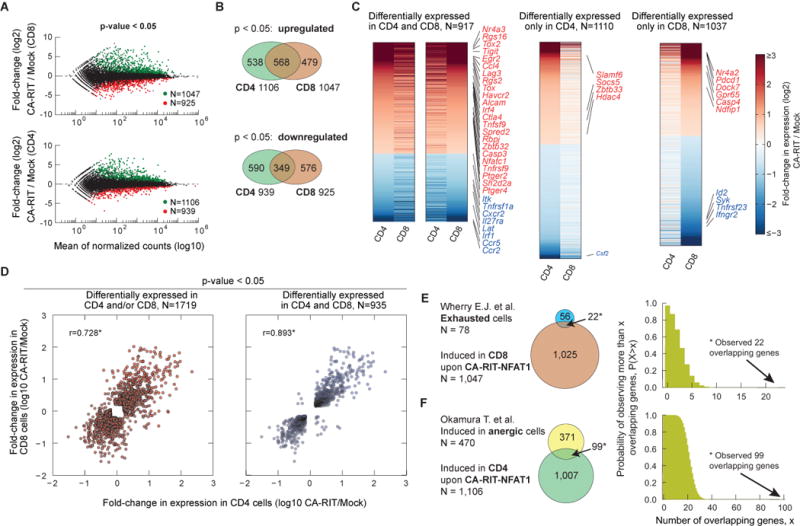
(A) Genes whose expression is altered upon CA-RIT-NFAT1 expression in CD8+ (top panel) or CD4+ (bottom panel) T cells. Changes in gene expression in CA-RIT-NFAT1-expressing versus mock-transduced cells plotted against overall gene expression. (B) Venn diagram displaying numbers of genes significantly induced (top panel) or downregulated (bottom panel) in CD8+ and/or CD4+ T cells. (C) Heatmap representation of changes in gene expression in CA-RIT-NFAT1-transduced over mock-transduced CD4+ and CD8+ T cells (p<0.05). Each horizontal line represents one gene, ordered by gene expression (highest to lowest fold change). 18 genes which showed opposite trends in CD4+ and CD8+ T cells were omitted (depicted in the upper left and lower right quadrants of D, right panel). (D) Scatter plot representing changes in gene expression in CA-RIT-NFAT1-transduced cells versus mock-transduced cells (RPKM values from RNA-Seq) in CD8+ T cells (y axis) versus CD4+ T cells (x-axis). Each dot represents a gene. Pearson correlation coefficients are indicated. *, p<10−10. (E) Overlap of genes significantly upregulated upon CA-RIT-NFAT1 expression in CD8+ T cells with genes significantly upregulated in either in vivo-generated exhausted CD8+ T cells (top) (cluster #1,(Wherry et al., 2007)), or in anergic CD4+ CD25− CD45RBlo LAG3+ anergic T cells compared to CD4+ CD25− CD45RBlo Lag3− T cells (bottom) (Okamura et al., 2009). (F) Plots show the probability of observing more than N overlapping genes if sets with 1,047 and 78 genes (top) or 1,007 and 480 genes (bottom) are sampled randomly from the set of all genes. * p-value for the overlap < 10−10 (one-tailed Fisher’s exact test). See also Tables S1, S2, S6 and Figure S3.
Table 1. Partial list of genes expressed in exhausted and/or anergic T cells that are also induced in CA-RIT-NFAT1-expressing CD4 and CD8 T cells.
The table shows representative genes (identified by RNA-seq) that are significantly induced in CD4+ and CD8+ T cells upon CA-RIT-NFAT1 expression and also overlap with genes expressed in in vivo-generated anergic and exhausted T cells.
| Cell surface receptors and ligands | ||
| Lag3 | Lymphocyte-activation gene 3 | * |
| Tnfrsf9 | Tumor necrosis factor receptor superfamily member 9; 4-1BB | * |
| Ptger2 | Prostaglandin E2 receptor 2 | * |
| Havcr2 | Hepatitis A virus cellular receptor 2; TIM-3 | * |
| Alcam | Activated Leukocyte cell adhesion molecule | * |
| Tigit | T cell immunoreceptor with Ig and ITIM domains | + |
| Ctla4 | Cytotoxic T-Lymphocyte Antigen 4 | + |
| Ptger4 | Prostaglandin E2 receptor 4 | + |
| Tnfrsf1b | Tumor necrosis factor receptor superfamily member 1b | + |
| Ccl4 | Chemokine (C-C motif) ligand 4 | + |
| CD109 | Cluster of Differentiation 109 | # |
| CD200 | Cluster of Differentiation 200; Ox-2 | # |
| Tnfsf9 | Tumor necrosis factor superfamily member 9; 4-1BBL | # |
| Nrp1 | Neuropilin-1 | # |
| Sema4c | Semaphorin-4C | # |
| Ptprj | Receptor-type tyrosine-protein phosphatase eta | # |
| Il21 | Interleukin 21 | # |
| Tspan2 | Tetraspanin-2 | # |
| Transcription factors | ||
| Ikzf2 | IKAROS family zinc finger 2; Helios | * |
| Egr2 | Early growth response protein 2 | * |
| Tox | Thymocyte selection-associated high mobility group box protein | * |
| Zeb2 | Zinc finger E-box-binding homeobox 2 | * |
| Irf4 | Interferon Regulatory Factor | * |
| Nfatc1 | Nuclear Factor of Activated T cells c1; NFAT2 | * |
| Zbtb32 | Zinc finger and BTB domain-containing protein 32; ROG | # |
| Rbpj | Recombinant binding protein suppressor of hairless | # |
| Hif1a | Hipoxia-inducible factor 1-alpha | # |
| Signaling | ||
| Rgs16 | Regulator of G-protein signaling 16 | * |
| Sh2d2a | SH2 domain-containing protein 2A | + |
| Nucb1 | Nucleobindin-1 | + |
| Plscr1 | Phospholipid scramblase 1 | + |
| Ptpn11 | Tyrosine-protein phosphatase non-receptor type 11 | # |
| Prkca | Protein kinase C alpha (PKCa) | # |
| Plscr4 | Phospholipid scramblase 4 | # |
| Others | ||
| Casp3 | Caspase 3 | * |
| Gpd2 | Glycerol-3-phosphate dehydrogenase | + |
| Gas2 | Growth arrest-specific protein 2 | + |
| Sh3rf1 | SH3 domain containing ring finger 1 | # |
| Nhedc2 | Na+/H+ exchanger domain containing 2 | # |
| Plek | Pleckstrin | # |
| Tnfaip2 | Tumor necrosis factor, alpha-induced protein 2 | # |
| Ctsb | Cathepsin B | # |
genes that are upregulated in both anergic and exhausted T cells;
genes whose expression is significantly upregulated in exhausted T cells but is not statistically significant in anergic T cells;
genes whose expression is significantly upregulated in anergic T cells but is not statistically significant in exhausted T cells. In vivo data obtained from (Doering et al., 2012; Okamura et al., 2009; Wherry et al., 2007).
CA-RIT-NFAT1-expressing T cells showed increased protein and mRNA expression of the inhibitory receptors LAG3, TIM3, PD-1 and GITR, based on flow cytometry, RNA-seq and quantitative PCR (Figure 2, S3A–D). Upregulation of the inhibitory receptors depended on NFAT1 DNA-binding, since it was not observed in cells expressing DBDmut-CA-RIT-NFAT1 (Figure S3B and data not shown), despite higher expression of this DNA-binding mutant compared to CA-RIT-NFAT1 (Figure S1E). Several genes coding for transcription factors (Prdm1, Bhlhe40, Irf4, Ikzf2, Zeb2, Lass6, Tox, Eomes) were recently identified in a network analysis (Doering et al., 2012) as potentially contributing to induction of the exhausted state; except for Eomes, all these transcription factors are also upregulated in CA-RIT-NFAT1-expressing cells (Figure S3E). Moreover, genes encoding other categories of negative regulatory proteins (e.g. diacylglycerol kinase, several phosphatases) were also expressed at higher amounts in cells expressing CA-RIT-NFAT1 compared to mock-transduced cells (Figure S3E). Thus even in the absence of interaction with AP-1, NFAT1 can induce exhaustion/anergy-associated genes in both CD8+ and CD4+ T cells.
CA-RIT-NFAT1 directly upregulates gene expression by binding to gene promoter regions
To determine which genes were likely to be direct targets of NFAT1 and CA-RIT-NFAT1, we analysed the genomic distribution of both proteins in CD8+ T cells by chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq). Because only very small numbers of exhausted T cells can be obtained from mice, it was not possible to perform ChIP-seq experiments to define NFAT binding sites in exhausted CD8+ T cells in vivo. Instead, we compared the genome-wide distribution of endogenous NFAT1 with that of ectopically expressed CA-RIT-NFAT1 in CD8+ T cells left resting or stimulated in vitro.
Wild type (WT) or NFAT1-deficient naïve P14+ Tcra−/− CD8+ T cells were stimulated with anti-CD3 and anti-CD28, retrovirally transduced with empty vector or CA-RIT-NFAT1, and expanded in IL-2-containing media to yield effector CTL (Pipkin et al., 2010). 6 days later, cells were either left untreated (resting) or restimulated with PMA and ionomycin for 1 h, and chromatin was prepared and immunoprecipitated with anti-mouse NFAT1. Immunoprecipitation was specific, as evidenced by the low number of background peaks in NFAT1-deficient compared to control cells (Figure 3A). For subsequent analysis, we removed peaks also present in NFAT1-deficient cells and therefore considered background. As expected from the constitutive nuclear localization of CA-RIT-NFAT1 (Figure S1A), similar numbers of CA-RIT-NFAT1 peaks were observed in resting and restimulated NFAT1-deficient cells transduced with CA-RIT-NFAT1 (Figure 3B, middle panels). Comparison with the genome reference (mm9; Figure 3B, right panel) showed that both NFAT1 and CA-RIT-NFAT1 peaks were enriched at promoters/transcription start sites (TSS), exons and the first introns of genes (Figure 3B, 3C).
Figure 3. CA-RIT-NFAT1 directly upregulates gene expression by binding to promoter regions.
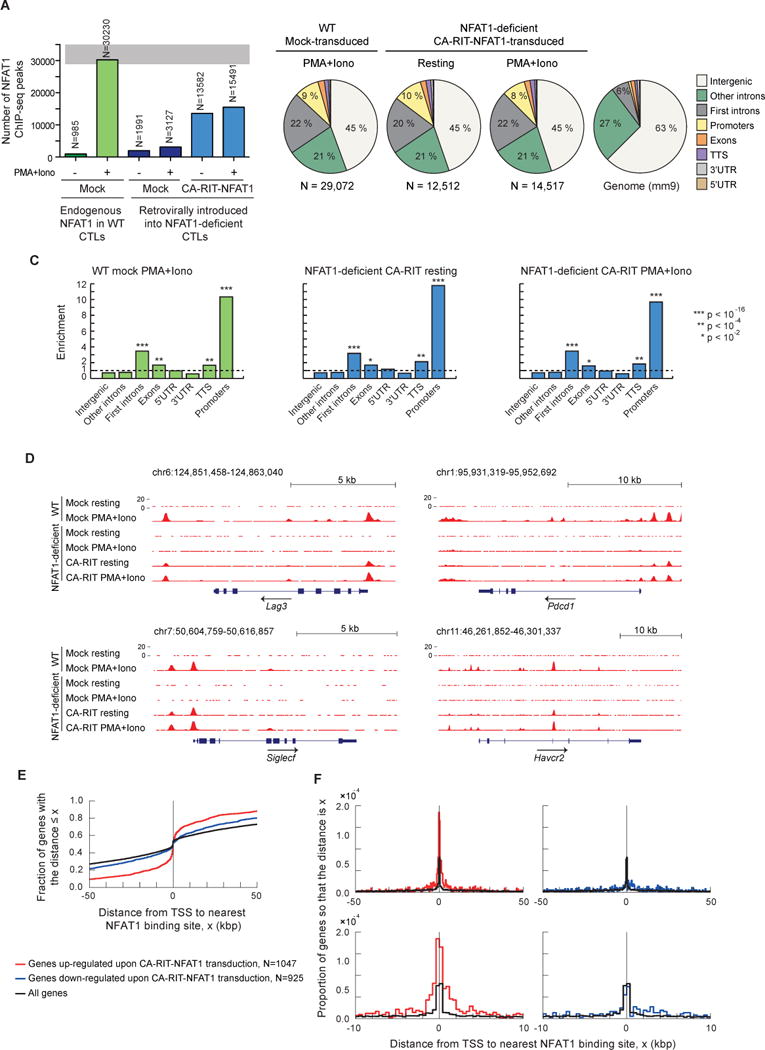
ChIP-seq results for endogenous NFAT1 in WT cells and for CA-RIT-NFAT1 in transduced NFAT1-deficient cells, either untreated or restimulated with PMA and ionomycin for 1h. (A) Number of NFAT1 and CA-RIT-NFAT1 ChIP-seq peaks identified by HOMER. (B) Pie charts show the genomic distribution of ChIP-seq peaks for endogenous NFAT1 in WT cells transduced with empty vector (left pie chart), or CA-RIT-NFAT1 ChIP-seq peaks in NFAT1-deficient cells transduced with CA-RIT-NFAT1 (middle two pie charts). Representation of the annotated regions in the mouse mm9 genome is shown for comparison (right pie chart). (C) Enrichment of NFAT1 binding sites in annotated genomic regions based on their relative abundance in the mouse genome (mm9). Statistical analysis was performed using Fisher’s exact test. ***, p<2e-16; ** p<1e-10; * p<1e-2. (D) Genome browser views of Lag3, Pdcd1 (encoding PD-1), Siglec5 and Havcr2 (encoding Tim3) loci showing the distribution of NFAT1 and CA-RIT-NFAT1 ChIP-seq peaks. (E–F) Cumulative plots (E) or probability per base pair (F) of the distance between the closest NFAT1 binding site relative to the transcription start site of all genes (black curves), genes significantly (p < 0.05) upregulated (red curves) or downregulated (blue curves) in NFAT1-deficient cells transduced with CA-RIT-NFAT1 in resting conditions. See also Figure S4.
Genome browser views of genes encoding the inhibitory receptors PD-1, LAG3, TIM3, and Siglec-F illustrate that endogenous NFAT1 binds only after PMA and ionomycin stimulation (Figures 3D, S4A, compare lines 1 and 2), whereas CA-RIT-NFAT1 ChIP-seq peaks were observed in both resting and stimulated cells (compare lines 5 and 6). These ChIP-seq peaks were not observed in NFAT1-deficient T cells (lines 3 and 4), attesting to the specificity of our NFAT1 antibody. There was substantial overlap in ChIP-seq peaks for endogenous NFAT1 and CA-RIT-NFAT1, as well as ChIP-seq peaks of CA-RIT-NFAT1 binding under resting and stimulated conditions (Figures 3D, S4A). Regions of NFAT1 but not CA-RIT-NFAT1 binding and vice versa are discussed below.
To understand the genome-wide binding patterns of NFAT1 and CA-RIT-NFAT1, we examined the ChIP-seq data in more detail. The distribution of CA-RIT-NFAT1-bound regions with respect to the nearest TSS differed depending on whether the genes transcribed from that TSS were up- or down-regulated by CA-RIT-NFAT1. The majority of upregulated genes had a CA-RIT-NFAT1 binding site within 2.5 kb of the TSS, but the binding sites were widely distributed with respect to the TSS of down-regulated genes (Figure 3E–F). These results suggest that CA-RIT-NFAT1 directly regulates the transcription of up-regulated genes by binding to sites in the vicinity of their promoter/TSS regions. Figure S4E shows the distribution of ChIP-seq peaks for other transcription factors with respect to the TSS (see Supplemental Discussion).
Sequence motif analyses of NFAT1 and CA-RIT-NFAT1 binding sites in CTLs
We used a motif-search algorithm (Homer) to compare sequence motifs enriched above background in NFAT1 and CA-RIT-NFAT1 ChIP-seq peaks (Figure 4). We considered ChIP-seq peaks that bound endogenous NFAT1 in stimulated cells (n ~29,000), and CA-RIT-NFAT1 under resting or stimulated conditions (n ~12,500 and ~14,500 respectively; Figure 4A). Notably, most NFAT1 ChIP-seq peaks did not overlap with CA-RIT-NFAT1 ChIP-seq peaks (Figure 4A, left and middle panels), suggesting that at these sites, NFAT binding is strongly stabilized by AP-1 (likely by ~20-fold or more; (Rao et al., 1997)). Among these is the well-known composite NFAT:AP-1 element in the Il2 promoter (Chen et al., 1998; Jain et al., 1993; Jain et al., 1992) (Figure 4B, left panel). Similar composite elements are known to occur in the promoter and enhancer regions of several other calcium-responsive, cyclosporin A (CsA)-sensitive genes (Hogan et al., 2003; Rao et al., 1997).
Figure 4. ChIP-seq peaks for endogenous NFAT1 are enriched for composite NFAT:AP-1 sites.
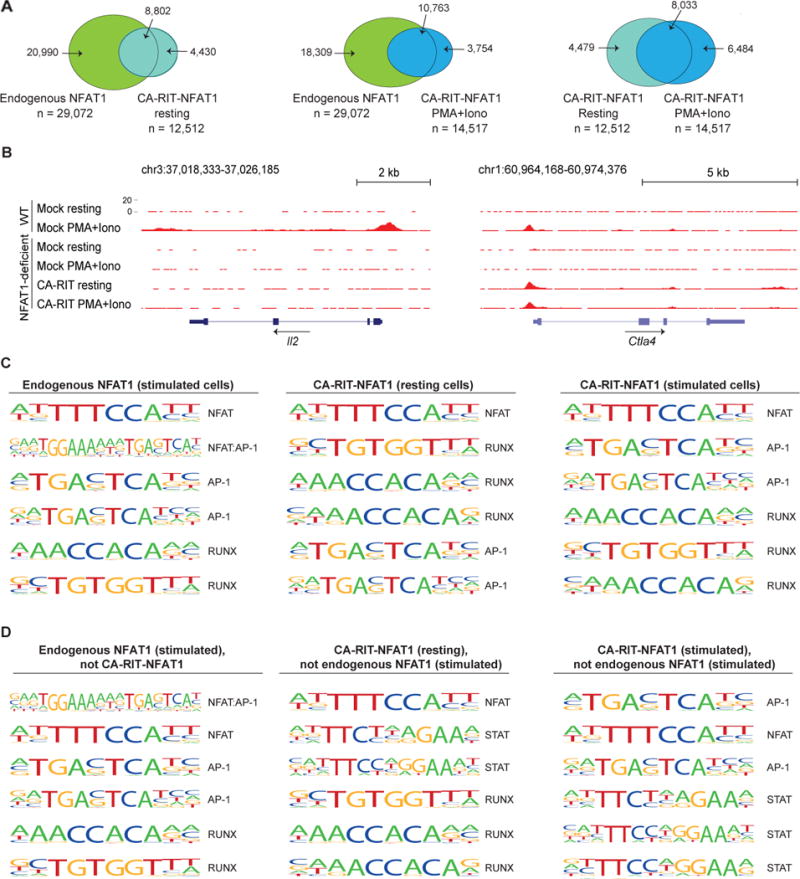
(A) Venn diagrams showing the overlap of ChIP-seq peaks in the different conditions. (B) Genome browser views of Il2 and Ctla4 loci showing the distribution of ChIP-seq peaks. (C) List of the top six most representative motifs ranked based on the p-values. The HOMER program searches for published motifs, and therefore duplicate AP-1 and Runx sequences are listed. (D) Motifs identified as enriched only in peaks from one set of ChIP-seq data as indicated. See also Table S3.
In ChIP-seq peaks for endogenous NFAT1, the top two enriched motifs were the consensus monomer NFAT binding element (TTTCCA, complement TGGAAA) and a composite NFAT:AP-1 element (TGGAAAnnnTGAG/CTCA) (Figure 4C, left panel). Composite NFAT1:AP-1 element was the top enriched motif in the subset of NFAT1 ChIP-seq peaks that did not bind CA-RIT-NFAT1 (Figure 4D, left panel), whereas the monomer NFAT element was the top enriched motif in the subset of NFAT1 ChIP-seq peaks that also bound CA-RIT-NFAT1 (data not shown). As an example of the latter group, the proximal Ctla4 promoter contains the monomer NFAT binding element TGGAAAAT (Figure 4B, right panel). These data confirm that the RIT mutation abolishes NFAT1:AP-1 interaction in cells as it does in vitro, and emphasize that despite its inability to engage in cooperative interactions with AP-1, CA-RIT-NFAT1 binds functionally and with measurable affinity to many NFAT1 binding sites.
Finally, a substantial number of the CA-RIT-NFAT1 ChIP-seq peaks detected in resting and stimulated cells were not observed in stimulated WT cells (Figure 4A, Table S4). These peaks showed low enrichment for NFAT:AP-1 composite sites (Figure 4C), and no enrichment for κB-like sites such as the site in the Rnf128 promoter that binds NFAT1 homodimers [GTAACGTTTCC or GGATTCTTCC] (Soto-Nieves et al., 2009); Rnf128, which encodes the E3 ligase Grail, is upregulated in anergic CD4+ T cells (Fathman and Lineberry, 2007). Rather, the strong enrichment for consensus NFAT binding motifs in CA-RIT-NFAT1 ChIP-seq peaks suggests that monomer binding is dominant under these conditions, perhaps stabilized through protein-protein interactions with other partner proteins (see Supplementary Discussion).
NFAT-deficient CD8+ T cells show diminished expression of exhaustion-associated inhibitory surface receptors
To examine the requirement for endogenous NFAT proteins in the regulation of exhaustion-associated genes, we bred our conventional gene-disrupted NFAT 1-deficient mice (Xanthoudakis et al., 1996) to mice deficient in NFAT2 in the T cell lineage (Figure S5A; see Methods). When differentiated into ‘memory’ CTL (Pipkin et al., 2010) and restimulated with PMA and ionomycin in vitro, NFAT1, 2-double deficient CD8+ T cells showed clearly decreased induction of the effector cytokines IL-2 and IFN-γ compared to WT or singly NFAT1- or NFAT2-deficient CD8+ T cells (Figure 5A); they also showed strongly diminished expression of the inhibitory surface receptors LAG3, TIM3 and PD-1 (Figure 5C and data not shown). The residual expression was due to NFAT4, the third NFAT protein present in immune cells: NFAT1, 2-deficient cells additionally transduced with shRNA against NFAT4 (Figure 5B) produced little or no IL-2 or interferon-γ (IFN-γ) upon restimulation (Figure 5A) and expressed little or no LAG3 and PD-1 either under resting conditions or after restimulation (Figure 5C), indicating that all 3 NFAT proteins contribute to CD8+ T cell activation and exhaustion. Notably, there was no defect in proliferation of the NFAT1, 2-deficient CD8+ T cells under these conditions (Figure S5B).
Figure 5. NFAT family members regulate the expression of inhibitory receptors in vitro.
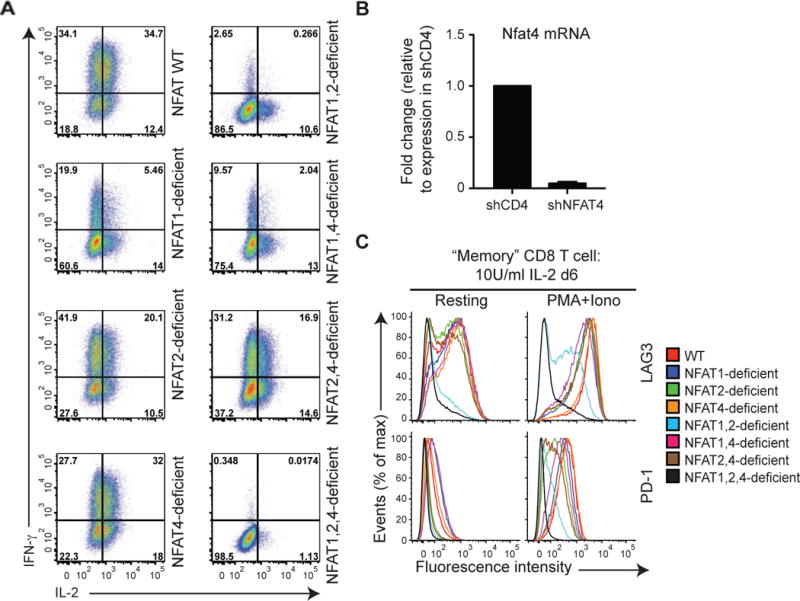
Naïve WT, NFAT 1-deficient, NFAT2-deficient or NFAT1, 2-deficient CD8+ T cells were purified by FACS, and transduced with a retrovirus encoding Ametrine (a variant GFP) and shRNAs targeting either CD4 (negative control) or NFAT4 to yield single, double or triple NFAT-deficient cells. On day 6 after activation, cells were restimulated with PMA and ionomycin for 6 h (A and C right panels), or left untreated (B and C left panels). IFN-y and IL-2 production (A) or expression of cell surface receptors (C) was determined by flow cytometry of Ametrinehi cells. A representative experiment out of two is shown. (B) Expression of mRNA encoding NFAT4 was assessed by quantitative real time PCR and normalized to mRNA encoding L32 ribosomal protein. The amount of expression in control shCD4 cells was set at 1. Combined results from 3 independent experiments are shown (mean ± SEM). See also Figure S5.
To ask whether NFAT family members were required for induction of the inhibitory receptors in vivo, we utilized a mouse model of infection with LCMV. WT, NFAT1-deficient, NFAT2-deficient or NFAT1 and NFAT2 double-deficient naïve CD8+ P14+ Tcra−/− T cells (CD45.2+) were transferred into CD45.1+ congenic mice. The mice were then infected with LCMV Armstrong 5 strain, which induces an acute infection (Wherry et al., 2007); eight days after infection, expression of the inhibitory receptors was determined in the transferred cells ex vivo (Figure 6A). Under these conditions in vivo, there was only a minor upregulation of LAG3; however, consistent with our in vitro studies, expression of all tested inhibitory receptors was strongly decreased in cells lacking both NFAT1 and NFAT2 (Figure 6A). Based on our in vitro experiments (Figure 5), the residual expression is most likely due to NFAT4.
Figure 6. NFAT family members regulate the expression of inhibitory receptors in vivo.
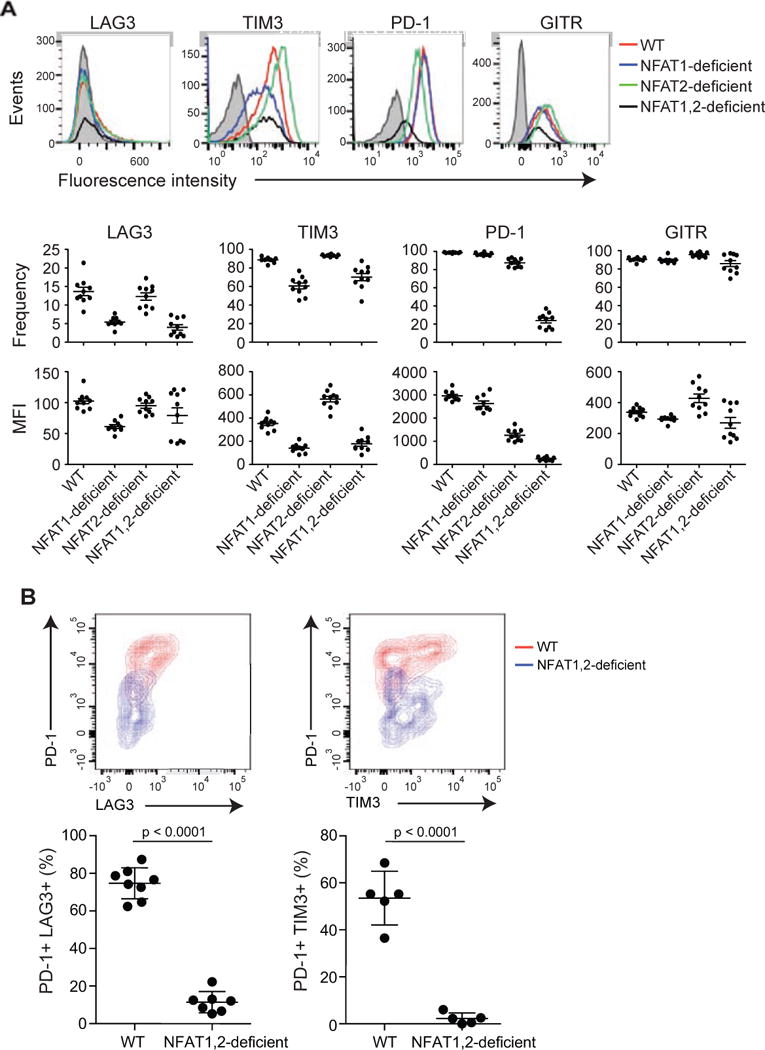
(A) Acute infection. Naïve P14+ Tcra−/− WT, NFAT1-deficient, NFAT2-deficient or NFAT1, 2-deficient CD8+ T cells (CD45.2+) were transferred intravenously into CD45.1+ congenic mice, after which the mice were injected with LCMV (2 × 105 PFU) intraperitoneally. Eight days after infection, splenocytes were harvested and the expression of inhibitory receptors on the adoptively transferred CD8+ CD45.2+ cells was evaluated. Top panel, a representative example for each genotype is shown; bottom panels, data for 8–10 mice in each group are summarized as mean ± SEM, with each dot representing a mouse. A representative experiment out of two is shown. The gray histogram shows unstained control. (B) Chronic infection. WT mice and mice lacking both NFAT1 and NFAT2 in T cells were infected with LCMV clone 13. Eighteen days after infection, splenocytes were harvested and expression of inhibitory receptors on CD8+ CD44+ H2Db-gp33-41+ cells was determined by flow cytometry. Top panel, representative contour plots. Bottom panel, results from individual mice (2–3 independent experiments; each mouse is represented by a dot). p values were calculated using t-test.
We then evaluated the role of NFAT proteins using LCMV clone 13, which induces a chronic infection that leads to CD8+ T cell exhaustion (Wherry et al., 2007). Mice lacking both NFAT1 and NFAT2 showed a dramatic decrease in the frequency of cells co-expressing PD-1 and TIM3 or LAG3 (Figure 6B). Together, our results support a direct role for NFAT family members in controlling the expression of inhibitory receptors in CD8+ T cells, both in cell culture and in mice. Not all surface receptors are affected, however: for instance, the expression of CD44 was unimpaired in the absence of NFAT (data not shown).
DISCUSSION
In this study, we demonstrate that NFAT proteins, established regulators of T cell activation, also participate in the transcriptional program of CD8+ T cell exhaustion. To dissect these two aspects of NFAT function, an essential tool was CA-RIT-NFAT1, an engineered form of NFAT1 that is both constitutively active and unable to interact with AP-1. Using this engineered protein, we have defined, for the first time, the genome-wide localization of NFAT1 under conditions where it does not cooperate with AP-1, as well as the NFAT-dependent transcriptional profile induced under these conditions. Moreover, we have used mice and T cells deficient for multiple NFAT family members to show that NFAT proteins directly and redundantly control cytokine expression as well as expression of exhaustion-related genes in CD8+ T cells. We note that some of our detailed findings may be specific to the experimental systems used here, and may or may not apply to CD8+ T cell exhaustion observed in other model systems or in human diseases.
Our data demonstrate that NFAT controls CD8+ T cell exhaustion by binding directly to regulatory regions of many exhaustion-associated genes, including the Pdcd1 (PD-1) and Havcr2 (TIM3) promoters and cis-regulatory regions. Although NFAT1 and CA-RIT-NFAT1 occupy many overlapping sites, endogenous NFAT1 demonstrates a strong preference for canonical NFAT:AP-1 composite sites under stimulation conditions that also activate AP-1, whereas CA-RIT-NFAT1 shows a strong preference for monomeric NFAT sites. In some cases a two-step mechanism may apply: for instance, LAG3 is induced by Egr2 (Okamura et al., 2009), but NFAT1 binds near both the Egr2 and Lag3 TSSs (Figures 3D, S4A), suggesting that NFAT induces Egr2, after which NFAT and Egr2 cooperate to induce the expression of downstream target genes. Other pathways may also be involved: Notch (Mathieu et al., 2013) and type-I IFN (Terawaki et al., 2011) signaling regulate Pdcd1 expression, and MEK kinase (Yoon et al., 2011) and T-bet (Anderson et al., 2010) regulate TIM3 expression.
The transcriptional program evoked by CA-RIT-NFAT1 in CD8+ T cells overlaps with that observed in anergic CD4+ T cells. mRNAs encoding E3 ligases (Fathman and Lineberry, 2007; Heissmeyer et al., 2004; Nurieva et al., 2011); phosphatases (Macian et al., 2002); and other signaling proteins including caspase 3, RGS proteins and diacylglycerol kinases (Joshi and Koretzky, 2013; Macian et al., 2002) are upregulated in both cases. The combined action of these negative regulators is likely to underlie the diminished activation of ZAP-70 and PLCγ1 and the consequent decrease in Ca2+ influx that we observe in CA-RIT-NFAT1-expressing T cells. We propose that this negative feedback program becomes dominant in CD4+ T cells under conditions of ineffective costimulation (Mueller, 2010; Nurieva et al., 2011), and in CD8+ T cells in the context of prolonged low-grade antigenic stimulation and/or inflammation, as encountered in chronic viral infections and cancer (Schietinger and Greenberg, 2014; Wherry, 2011).
Our data are consistent with previous studies on the role of NFAT in CD8+ T cell exhaustion. Transcriptional profiling revealed higher expression of mRNA encoding NFAT2 in exhausted murine CD8+ T cells isolated directly ex vivo, compared with all other T cell subsets (naïve, effector, memory) examined (Wherry et al., 2007); NFAT2 was shown to be nuclear in tolerant CD8+ T cells (Srinivasan and Frauwirth, 2007); and Pdcd1 expression was shown to be regulated by NFAT2 (Oestreich et al., 2008). Although Agnellini et al. (2007) reported that NFAT2 was not present in the nucleus of exhausted CD8+ T cells generated during chronic, high-dose LCMV infection in mice after 16 h of stimulation (they did not examine other NFAT proteins), an alternative interpretation of their data is that the short inducible isoform of NFAT2 (Chuvpilo et al., 2002) is poorly induced after restimulation of exhausted CD8+ T cells. Indeed, the higher basal Ca2+ concentrations observed by Agnellini et al. in exhausted CD8+ T cells and in CA-RIT-expressing cells in this report would increase NFAT-dependent induction of NFAT2 under basal conditions, as also observed in ex vivo exhausted cells (Wherry et al., 2007). Moreover, the blunted but not completely blocked Ca2+ influx in restimulated CA-RIT-NFAT1-expressing T cells is consistent with the observation that exhausted CD8+ T cells support Ca2+ influx upon restimulation (Agnellini et al., 2007).
Although in these studies we engineered NFAT1 to eliminate NFAT:AP-1 cooperation, we propose that the NFAT-driven transcriptional program of feedback attenuation of T cell responses is a normal late feature of the immune response. Fos and Jun are elevated only transiently after stimulation (Jain et al., 1992), whereas NFAT can remain in the nucleus for long times even in response to very low elevations of intracellular Ca2+ (Dolmetsch et al., 1997; Marangoni et al., 2013; Oh-hora et al., 2008). Fos expression also declines during chronic infection despite antigen persistence, whereas NFAT2 expression is increased as discussed above (Wherry et al., 2007). Finally, exhausted CD8+ T cells, defined in a tumor setting by PD-1 and TIM3 expression, can support the nuclear translocation of endogenous NFAT1 in response to high levels of TCR stimulation (Figure 1J). The combination of increased basal Ca2+ and a residual low concentration of Ca2+ signaling in anergized/exhausted cells would be sufficient for NFAT proteins to enter and remain in the nucleus to maintain the exhausted state. This would result in the transcriptional induction of exhaustion-related genes, whose expression requires NFAT but not NFAT:AP-1 cooperation as shown here.
Especially in the presence of antigen-specific T regulatory cells, tumor-infiltrating CD8+ T cells display brief and unstable T cell-APC contacts which nevertheless are sufficient to induce prolonged NFAT-dependent transcription and establish a feedback mechanism that results in decreased T cell responses (Marangoni et al., 2013). Under these conditions, delayed export of NFAT from the nucleus results in a “memory” of previous signaling by NFAT (Marangoni et al., 2013). In contrast, signaling pathways regulating Ras, ERK and AP-1 activation exhibit a shorter signal memory (Faroudi et al., 2003), suggesting that tolerance/exhaustion programs are evoked in tumor-infiltrating T cells as a result of an altered balance between NFAT and other transcription factors including AP-1. Consistent with the likelihood that AP-1 does not participate in the exhaustion program, exhausted T cells show not only a higher expression of NFAT2 transcripts but also a concomitant reduction in the AP-1 family member Fos (Wherry, 2011). These findings complement our own observations that the strongest exhaustion phenotype is induced in vitro by CA-RIT-NFAT1, which cannot cooperate with AP-1.
The NFAT-dependent program of anergy/exhaustion has considerable relevance for the clinic. For instance, the PD-1-PD-1 ligand pathway is well established as a major inhibitory receptor pathway involved in T cell exhaustion (Barber et al., 2006), and blocking this pathway during chronic LCMV infection reinvigorates virus-specific CD8+ T cell responses, resulting in lower viral loads (Barber et al., 2006). Moreover, antibodies to PD-1 and CTLA-4 have emerged as surprisingly effective agents for cancer immunotherapy (Schietinger and Greenberg, 2014), and individual or combined blockade of TIM3, PD-1 and LAG3 with antagonist antibodies has been shown to reverse exhaustion efficiently in CD8+ T cells (Blackburn et al., 2009; Jin et al., 2010; Sakuishi et al., 2010). These therapies all act downstream of the NFAT-dependent program of anergy/exhaustion that we have described here.
Our data imply that rescuing AP-1 signaling in exhausted T cells would reverse the exhausted state, and so offer therapeutic value in chronic viral infections and cancer. Conversely, disrupting NFAT:AP-1 interaction would impose a hyporesponsive state, thus offering a potential therapeutic avenue for interfering with autoimmune responses that depend on NFAT:AP-1 cooperation. Since the RIT mutation does not interfere with NFAT:FOXP3 cooperation (data not shown), confirming predictions from the crystal structures that NFAT proteins make partially distinct contacts with Fos-Jun and FOXP proteins (Bandukwala et al., 2011; Chen et al., 1998; Wu et al., 2006), it might be possible to design reagents that disrupt the NFAT:AP-1 interaction but leave T regulatory function intact. Our future studies will address these possibilities.
EXPERIMENTAL PROCEDURES
Mice
P14+ TCR transgenic (P14+, Taconic) or OT-II mice were crossed with Tcra−/− mice (the P14 TCR transgene recognizes a peptide from the gp33 protein of mouse lymphocytic choriomeningitis virus (LCMV) presented on H-2Db; the OT-II TCR transgene recognizes a peptide from ovalbumin presented on I-Ab). P14+Tcra−/− mice were further crossed with NFAT1-deficient mice (Xanthoudakis et al., 1996) and with mice deficient in NFAT2 in the T cell lineage, obtained by breeding CD4Cre mice with mice in which exon 3 of the gene encoding NFAT2 was “floxed” (flanked with LoxP sites) (Figure S5A). The resulting mice lack both NFAT1 and NFAT2 in T cells (NFAT1, 2-deficient mice). CL4 mice express a transgenic TCR specific for H-2Kd/HA515–523. All mice were maintained in specific pathogen-free barrier facilities and used according to protocols approved by the La Jolla Institute for Allergy and Immunology animal care and use committees.
Isolation, culture and Retroviral transduction of T Cells
Naïve CD8+ or CD4+ T cells were purified from spleen and lymph nodes harvested from 6- to 8-week-old mice by negative selection or by fluorescence-activated cell sorting. Cells were activated with anti-CD3 and anti-CD28 and maintained in the presence of IL-2. For cytokine production analyses, cells were restimulated with PMA and ionomycin. For retroviral transduction, viral supernatants were generated by transfection of PlatE cells. GFP+ or Ametrine+ transduced cells were purified by fluorescence-activated cell sorting. For more details, see Supplemental Methods.
RNA-seq
For RNA-seq analysis, total RNA was used to isolate poly(A) RNA using the micropoly(A)purist kit (Ambion). The whole transcriptome library kit (Life Technologies) was used to prepare paired-end sequencing libraries. Sequencing was performed using a SOLID4 sequencer (Applied Biosystems) and the sequencing reads in color-space were mapped to the mm9 genome using Tophat (Trapnell et al., 2009). RNA-seq data is available under GEO accession number GSE64409. For more details, see RNA-seq data analyses and Microarray data analysis in Supplemental Methods.
ChIP-seq
Fixed Chromatin was sheared to yield 100–300 bp DNA fragments and immunoprecipitated with protein G/anti-NFAT1 antibody complexes. Immunoprecipitated and de-crosslinked DNA was end-repaired and ligated with barcoded SOLID adaptors. Ligated products were size selected by gel and PCR amplified. Barcoded libraries were sequenced in a SOLID4 sequencer (Applied Biosystems) and the reads were mapped using Bowtie (Langmead et al., 2009) to the mm9 genome in color-space. ChIP-seq data is available under GEO accession number GSE64409. For more details, see ChIP-seq and motif analyses in Supplemental Methods.
Lymphocytic choriomeningitis virus (LCMV) models
Six week-old WT mice or mice lacking NFAT1 and NFAT2 in T cells were infected i.v. with 3×106 PFU of LCMV clone 13. Eighteen days after infection, splenocytes were harvested and cells were stained with antibodies against CD8, CD44, PD-1, LAG3, TIM3 and tetramer (H2Db-gp33-41). Expression of these markers was assessed by flow cytometry.
For adoptive transfer experiments, naïve P14+Tcra−/− CD8+ T cells were isolated from 6 week-old WT or NFAT1-deficient mice, or mice lacking NFAT2 or both NFAT1 and NFAT2 in T cells (all mice are CD45.2+). 5×104 cells were adoptively transferred (i.v.) into B6.SJL (CD45.1+) congenic mice, and then mice were infected with 2×105 PFU of LCMV Arm5 intraperitoneally. Eight days after infection, spleens were harvested, and cells stained for cell surface markers.
Listeria monocytogenes-gp33 in vivo protection assay
Naïve P14+Tcr−/−CD8+T cells were activated with anti-CD3 and anti-CD28, transduced with CA-RIT-NFAT1, DBD-mut-CA-RIT-NFAT1 or empty vector (mock) and expanded with 10 U/ml IL-2 for 4 d. Then, GFPhi cells were purified by FACS, and 5×104 cells were transferred into C56BL/6 recipient mice. One day later, mice were infected with 1×105 CFU Listeria-gp33, kindly provided by Dr. Rafi Ahmed (Kaech et al., 2003). On day 3–5, spleens were harvested and plated on brain heart infusion agar plates for colony counts and cells phenotypically characterized by FACS.
In vivo tumor model
Single-cell suspensions from spleens and LNs of CL4 mice were pulsed with 10 μM HA515–523 peptide for 1 hour at 37°C, then cultured with 10 ng/ml murine rIL-12 for the first 2 days and 20 ng/ml murine rIL-2 for the following 5 days as previously described (Bauer et al., 2014). Cells were then transferred to mice that had been inoculated s.c. in both shaved flanks with 106 viable CT26HA tumour cells suspended in 50 μl HBSS 7 days before. Tumor size was determined by caliper measurements of tumor length and width, and tumor volume was calculated as (l × w2)/2 (Bauer et al., 2014).
To induce exhausted T cells, CT26HA tumours were implanted in Thy1.1 recipients that were then injected with T regulatory cells that recognize the HA antigen (Bauer et al., 2014). Ten days after tumor injection, mice were sacrificed, and tumour-infiltrating T cells were restimulated ex vivo with plate-bound anti-CD3ɛ for 15 min, after which cells were fixed. Then, PD-1+TIM3+ and PD-1TIM3− populations were sorted and stained for NFAT1 and DAPI, and then analyzed for confocal microscopy.
Supplementary Material
HIGHLIGHTS.
-
*
NFAT proteins induce a transcriptional program of CD8+ T cell exhaustion
-
*
CD8+ T cells lacking NFAT fail to express inhibitory surface receptors
-
*
An engineered NFAT that cannot cooperate with AP-1 strongly induces exhaustion
-
*
The engineered NFAT1 blunts TCR signaling and impairs CD8+ function in vivo
Acknowledgments
We thank Drs. Stephan Feske and Ariel Quintana for help with calcium influx experiments, Drs. Camille Fos and Sara Trifari for help with signaling experiments, Ryan Hastie for help in maintaining the colony, and Drs. Runqiang Chen and James Scott-Browne for providing retroviral constructs containing shCD4 and shNFAT4. We thank C. Kim, K. Gunst and L. Nosworthy at the La Jolla Institute Flow Cytometry Facility for help with cell sorting experiments, and Dr. G. Seumois and J. Day of the La Jolla Institute Sequencing facility, and the Genomics Core at The Scripps Research Institute in Florida, for help with next-generation sequencing. This work was funded by NIH grants R01 CA42471 (to AR), R01 AI40127 and AI84167 (to P.G.H. and A.R.), R01 AI095634 (to M.E.P.), EU FP7 grant EC-FP7-SYBILLA-201106 and the Academy of Finland Centre of Excellence in Molecular Systems Immunology and Physiology Research (to T.Ä. and H.L.) and German Research foundation SFB 1054 TP A03 to V.H. G.J.M. was supported by a postdoctoral fellowship from the Jane Coffin Childs Memorial Fund, R.M.P. by a postdoctoral fellowship from the Pew Latin American Fellows Program in the Biomedical Sciences, and T.Ä. by a graduate student fellowship from by the Finnish Doctoral Programme in Computational Sciences FICS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
G.J.M., R.M.P. and A.R. designed the experiments and analyzed data; G.J.M. and R.M.P. performed the experiments; T.A. and H.L. did the computational and statistical analysis of the data; A.R., G.J.M. and R.M.P. wrote the manuscript; E.Y.K., F.M. and T.R.M. designed and performed the tumor model and NFAT nuclear translocation (imaging) experiments; E.D.L. and K.M.A. constructed mice with floxed exon 3 of the gene encoding NFAT2; S.C. provided reagents; M.E.P., H.L. and P.G.H. provided input for experimental design and interpretation and critically evaluated the manuscript. V.H. and Y.C.Z. performed initial experiments relevant to the study.
The authors declare no conflict of interest.
References
- Agnellini P, Wolint P, Rehr M, Cahenzli J, Karrer U, Oxenius A. Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. Proc Natl Acad Sci U S A. 2007;104:4565–4570. doi: 10.1073/pnas.0610335104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AC, Lord GM, Dardalhon V, Lee DH, Sabatos-Peyton CA, Glimcher LH, Kuchroo VK. T-bet, a Th1 transcription factor regulates the expression of Tim-3. Eur J Immunol. 2010;40:859–866. doi: 10.1002/eji.200939842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandukwala HS, Wu Y, Feuerer M, Chen Y, Barboza B, Ghosh S, Stroud JC, Benoist C, Mathis D, Rao A, Chen L. Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity. 2011;34:479–491. doi: 10.1016/j.immuni.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, Mempel TR. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Invest. 2014;124:2425–2440. doi: 10.1172/JCI66375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Glover JN, Hogan PG, Rao A, Harrison SC. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998;392:42–48. doi: 10.1038/32100. [DOI] [PubMed] [Google Scholar]
- Chuvpilo S, Jankevics E, Tyrsin D, Akimzhanov A, Moroz D, Jha MK, Schulze-Luehrmann J, Santner-Nanan B, Feoktistova E, Konig T, et al. Autoregulation of NFATc1/A expression facilitates effector T cells to escape from rapid apoptosis. Immunity. 2002;16:881–895. doi: 10.1016/s1074-7613(02)00329-1. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–1144. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- Faroudi M, Zaru R, Paulet P, Muller S, Valitutti S. Cutting edge: T lymphocyte activation by repeated immunological synapse formation and intermittent signaling. J Immunol. 2003;171:1128–1132. doi: 10.4049/jimmunol.171.3.1128. [DOI] [PubMed] [Google Scholar]
- Fathman CG, Lineberry NB. Molecular mechanisms of CD4+ T-cell anergy. Nat Rev Immunol. 2007;7:599–609. doi: 10.1038/nri2131. [DOI] [PubMed] [Google Scholar]
- Fehr T, Lucas CL, Kurtz J, Onoe T, Zhao G, Hogan T, Vallot C, Rao A, Sykes M. A CD8 T cell-intrinsic role for the calcineurin-NFAT pathway for tolerance induction in vivo. Blood. 2010;115:1280–1287. doi: 10.1182/blood-2009-07-230680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giffin MJ, Stroud JC, Bates DL, von Koenig KD, Hardin J, Chen L. Structure of NFAT1 bound as a dimer to the HIV-1 LTR kappa B element. Nat Struct Biol. 2003;10:800–806. doi: 10.1038/nsb981. [DOI] [PubMed] [Google Scholar]
- Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, Gu H, Liu YC, Dustin ML, Rao A. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5:255–265. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Jain J, Burgeon E, Badalian TM, Hogan PG, Rao A. A similar DNA-binding motif in NFAT family proteins and the Rel homology region. J Biol Chem. 1995;270:4138–4145. [PubMed] [Google Scholar]
- Jain J, McCaffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, Curran T, Rao A. The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature. 1993;365:352–355. doi: 10.1038/365352a0. [DOI] [PubMed] [Google Scholar]
- Jain J, Valge-Archer VE, Rao A. Analysis of the AP-1 sites in the IL-2 promoter. J Immunol. 1992;148:1240–1250. [PubMed] [Google Scholar]
- Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, Freeman GJ, Kuchroo VK, Ahmed R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010;107:14733–14738. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi RP, Koretzky GA. Diacylglycerol kinases: regulated controllers of T cell activation, function, and development. Int J Mol Sci. 2013;14:6649–6673. doi: 10.3390/ijms14046649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–731. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- Macian F, Garcia-Rodriguez C, Rao A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. Embo J. 2000;19:4783–4795. doi: 10.1093/emboj/19.17.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoni F, Murooka TT, Manzo T, Kim EY, Carrizosa E, Elpek NM, Mempel TR. The transcription factor NFAT exhibits signal memory during serial T cell interactions with antigen-presenting cells. Immunity. 2013;38:237–249. doi: 10.1016/j.immuni.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu M, Cotta-Grand N, Daudelin JF, Thebault P, Labrecque N. Notch signaling regulates PD-1 expression during CD8(+) T-cell activation. Immunol Cell Biol. 2013;91:82–88. doi: 10.1038/icb.2012.53. [DOI] [PubMed] [Google Scholar]
- Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol. 2010;11:21–27. doi: 10.1038/ni.1817. [DOI] [PubMed] [Google Scholar]
- Nurieva RI, Liu X, Dong C. Molecular mechanisms of T-cell tolerance. Immunol Rev. 2011;241:133–144. doi: 10.1111/j.1600-065X.2011.01012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol. 2008;181:4832–4839. doi: 10.4049/jimmunol.181.7.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9:432–443. doi: 10.1038/ni1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura H, Aramburu J, Garcia-Rodriguez C, Viola JP, Raghavan A, Tahiliani M, Zhang X, Qin J, Hogan PG, Rao A. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol Cell. 2000;6:539–550. doi: 10.1016/s1097-2765(00)00053-8. [DOI] [PubMed] [Google Scholar]
- Okamura T, Fujio K, Shibuya M, Sumitomo S, Shoda H, Sakaguchi S, Yamamoto K. CD4+CD25-LAG3+ regulatory T cells controlled by the transcription factor Egr-2. Proc Natl Acad Sci U S A. 2009;106:13974–13979. doi: 10.1073/pnas.0906872106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends in immunology. 2014;35:51–60. doi: 10.1016/j.it.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto-Nieves N, Puga I, Abe BT, Bandyopadhyay S, Baine I, Rao A, Macian F. Transcriptional complexes formed by NFAT dimers regulate the induction of T cell tolerance. J Exp Med. 2009;206:867–876. doi: 10.1084/jem.20082731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan M, Frauwirth KA. Reciprocal NFAT1 and NFAT2 nuclear localization in CD8+ anergic T cells is regulated by suboptimal calcium signaling. J Immunol. 2007;179:3734–3741. doi: 10.4049/jimmunol.179.6.3734. [DOI] [PubMed] [Google Scholar]
- Stroud JC, Lopez-Rodriguez C, Rao A, Chen L. Structure of a TonEBP-DNA complex reveals DNA encircled by a transcription factor. Nat Struct Biol. 2002;9:90–94. doi: 10.1038/nsb749. [DOI] [PubMed] [Google Scholar]
- Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, Honjo T. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186:2772–2779. doi: 10.4049/jimmunol.1003208. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Xanthoudakis S, Viola JP, Shaw KT, Luo C, Wallace JD, Bozza PT, Luk DC, Curran T, Rao A. An enhanced immune response in mice lacking the transcription factor NFAT1. Science. 1996;272:892–895. doi: 10.1126/science.272.5263.892. [DOI] [PubMed] [Google Scholar]
- Yoon SJ, Lee MJ, Shin DC, Kim JS, Chwae YJ, Kwon MH, Kim K, Park S. Activation of mitogen activated protein kinase-Erk kinase (MEK) increases T cell immunoglobulin mucin domain-3 (TIM-3) transcription in human T lymphocytes and a human mast cell line. Mol Immunol. 2011;48:1778–1783. doi: 10.1016/j.molimm.2011.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.