

Abstract
The hypoxic response in cells and tissues is mediated by the family of hypoxia-inducible factor (HIF) transcription factors that play an integral role in the metabolic changes that drive cellular adaptation to low oxygen availability. HIF expression and stabilization in immune cells can be triggered by hypoxia, but also by other factors associated with pathological stress: e.g., inflammation, infectious microorganisms, and cancer. HIF induces a number of aspects of host immune function, from boosting phagocyte microbicidal capacity to driving T cell differentiation and cytotoxic activity. Cellular metabolism is emerging as a key regulator of immunity, and it constitutes another layer of fine-tuned immune control by HIF that can dictate myeloid cell and lymphocyte development, fate, and function. Here we discuss how oxygen sensing in the immune microenvironment shapes immunological response and examine how HIF and the hypoxia pathway control innate and adaptive immunity.
Introduction
Hypoxia-inducible factor (HIF) transcription factors are master regulators of the cellular response to hypoxia and coordinate a transcriptional program that ensures optimal functional, metabolic, and vascular adaptation to O2 shortages (Semenza, 2011). HIF-1α is widely expressed and is detected in virtually all innate and adaptive immune populations including macrophages (Cramer et al., 2003), neutrophils (Walmsley et al., 2005), dendritic cells (Jantsch et al., 2008), and lymphocytes (McNamee et al., 2013). HIF-2α expression is also expressed in a range of cell types, including endothelial cells (Hu et al., 2003) and certain immune cells. For example, HIF-2α is expressed in tumor-associated macrophages (Imtiyaz et al., 2010; Talks et al., 2000) as well as CD8+ T cells in response to hypoxia (Doedens et al., 2013), where its expression is influenced by cytokine exposure. HIF-2α stabilization and function in other immune cell types like neutrophils (Imtiyaz et al., 2010; Thompson et al., 2014) and dendritic cells remain largely unexplored. As has been shown in cancer cells (Holmquist-Mengelbier et al., 2006; Keith et al., 2012; Warnecke et al., 2008), differing expression patterns of the HIF-1α and HIF-2α isoforms in immune cells depend on both intrinsic and extrinsic factors, and their resulting balance specifically contributes to the regulation of overlapping or distinct sets of target genes.
Recent work has shown that the HIF transcription factors are key elements in the control of immune cell metabolism and function. The aim of this review is to explore how hypoxia-signaling pathways can trigger HIF expression in the immune system, including unique mechanisms by which immune cells stabilize HIF, and to discuss the functional consequences for immune cell function. The intent is to show how these pathways act on immune cells in pathological states, including infection and cancer.
The Hypoxia Pathway and Stabilization of Hypoxia-Inducible Factor
HIF is a basic loop-helix-loop protein that forms a heterodimeric complex, which acts as a transcriptional regulator of genes whose promoters contain hypoxia response consensus sequences (HREs) (Wang et al., 1995; Wenger et al., 2005). The regulatory complex is comprised of HIF-1β, which is constitutively expressed, and either one of the HIF-α isoforms: HIF-1α or HIF-2α. Additional proteins bind the complex as coactivators and further modulate the transcription of target genes (Arany et al., 1996). Among these direct target genes, enzymes that control the metabolic switch for optimal cellular adaptation to hypoxia, vascular endothelial growth factor (VEGF), and other secreted factors that promote new vessel formation integrate the most well-known HIF downstream network that supports organism development and adaptable physiological responses (Semenza, 2014).
HIF-a subunit stability is posttranscriptionally regulated by oxygen availability through the iron-dependent enzymes prolylhydroxylases (PHDs). When oxygen is available, PHDs are active and hydroxylate HIF-a, marking it for proteasomal degradation in a process mediated by von Hippel-Lindau tumor suppressor protein (VHL)-dependent ubiquitination. If oxygen concentration drops, PHDs become inactive, resulting in HIF-a accumulation. Factor inhibiting HIF (FIH) provides another layer of regulation by hydroxylating asparaginyl residues in HIF1-α and HIF-2α, blocking protein interactions between the HIF-α transactivation domain (CAD) and coactivators like P300 that form an effective transcriptional complex. Apart from O2 as a cofactor, both PHDs and FIH require a-ketoglutarate (2-oxoglutarate) as a limiting electron donor cosubstrate, which is oxidized and decarboxylated to succinate. Ferrous iron and ascorbate serve as cofactors for these hydroxylation reactions (Semenza, 2014). Inflammation, vascular injury, and compromised oxygen availability are all hallmarks of immunological reaction to tissue damage and infection. Limited O2 availability results in a decrease of PHD- and FIH-dependent HIF-α hydroxylation, leading to its stabilization and nuclear translocation (Figure 1A; Semenza, 2014).
Figure 1. Mechanisms of HIF Stabilization by Immune Cells.

(A) O2 dependent. When oxygen is available, HIF-1α is hydroxylated by PHD, enzymes that depend on oxygen and iron as cofactors. When prolylhydroxylated, HIF-1a is polyubiquitinated by VHL, marking it for proteasomal degradation. FIH hydroxylates HIF-1α at asparagine 803, which does not lead to polyubiquitination, but instead blocks interactions between HIF-1α and p300/CBP, a member of the HIF complex that acts as a coactivator of target gene transcription. When oxygen tension drops, PHD and FIH activity is inhibited, leading to HIF-1α accumulation and nuclear translocation, heterodimerization with HIF-1β, and recruitment of p300/CBP. The HIF transcriptional complex binds to hypoxia-response elements (HREs) to control target gene expression. (B) O2 independent. Bacterial products are recognized by TLRs expressed on myeloid cells, signaling through NF-κB to increase Hif1a transcription. In a similar fashion, TCR ligation upon antigen presentation on T lymphocytes leads to increased Hif1a transcription and HIF-1a protein accumulation, even in the presence of oxygen. While the mechanism of Hif1a mRNA induction is not known, activation of PI3K and mTOR appear to be involved in TCR-mediated induction of HIF-1α.
HIF-α stabilization in immune cells can occur in an oxygen-independent manner. Bacterial induction of HIF-1α expression has been documented in macrophages cultured under normoxic conditions in the presence of different pathogens (Figure 1B; Peyssonnaux et al., 2005). Moreover, enterobacterial siderophores, which are secreted high-affinity, iron-chelating agents, can lead to oxygen-independent functional HIF-1α stabilization by limiting iron availability for optimal PHD-mediated hydroxylation (Hartmann et al., 2008). Lipopolysaccharide (LPS), a major cell membrane component of Gram-negative bacteria, induces HIF-1α protein accumulation in macrophages through transcriptional and translational activation, acting independently from hypoxia-induced HIF-1α protein stabilization (Blouin et al., 2004). Nuclear factor-κB (NF-κB), which plays a central role in regulating the immune response to infection, is also required for the bacteria-induced HIF-1α mRNA transcriptional response in macrophages (Rius et al., 2008). Tumor necrosis factor-α (TNF-α), another key host inflammatory mediator, can induce HIF-1α expression in macrophages harvested from wounds and cultured under normoxic conditions (Albina et al., 2001), providing another link between inflammation and HIF stabilization in immune cells. Oxidized low-density lipoprotein, which can induce inflammation and in turn promote atherosclerosis, also leads to HIF-1α protein accumulation in human macrophages through a mechanism involving reactive oxygen species (ROS) (Shatrov et al., 2003). Similar to macrophages, ROS production by activated T cells (Sena et al., 2013) may constitute another mechanism of HIF induction. ROS can drive HIF-1a transcription and function by either activating the HIF-1a promoter (Bonello et al., 2007) or regulating hydroxylase function, especially that of the asparaginyl hydroxylase FIH (Masson et al., 2012).
Adaptive immune cells can also stabilize HIF-1a through oxygen-independent mechanisms (Figure 1B). T cell receptor (TCR) ligation induces substantial accumulation of HIF-1α mRNA and protein, especially in the proinflammatory T helper 17 (Th17) CD4+ T cell lineage, by a mechanism dependent on STAT3 (Dang et al., 2011; Ikejiri et al., 2012; Nakamura et al., 2005; Shi et al., 2011; Wang et al., 2014), where IL-6 and transforming growth factor-β (TGF-β) enhance HIF-1α mRNA expression via a signal transduction and activator of transcription (STAT3)-dependent mechanism (Dang et al., 2011; Kojima et al., 2002; Shi et al., 2011). In contrast, stimulation of CD4+ T cells differentiated with cytokines promoting other helper subsets showed minimal increases in HIF-1a mRNA or protein upon activation (Dang et al., 2011; Shi et al., 2011). CD8+ T cells also rapidly stabilize HIF-1α and HIF-2α proteins upon in vitro TCR crosslinking in normoxic conditions (Doedens et al., 2013; Finlay et al., 2012; Wang et al., 2011). Further, interleukin-4 (IL-4), which induces HIF-2α stabilization in macrophages (Takeda et al., 2010), also contributes to HIF-2α accumulation in activated CD8+ T cells, while IL-2 conditions favor HIF-1 can induce inflammation and in turn promote atherosclerosis, also leads to HIF-1α protein accumulation in human macrophages through a mechanism involving reactive oxygen species (ROS) (Shatrov et al., 2003). Similar to macrophages, ROS production by activated T cells (Sena et al., 2013) may constitute another mechanism of HIF induction. ROS can drive HIF-1α transcription and function by either activating the HIF-1α promoter (Bonello et al., 2007) or regulating hydroxylase function, especially that of the asparaginyl hydroxylase FIH (Masson et al., 2012).
Adaptive immune cells can also stabilize HIF-1α through oxygen-independent mechanisms (Figure 1B). T cell receptor (TCR) ligation induces substantial accumulation of HIF-1a mRNA and protein, especially in the proinflammatory T helper 17 (Th17) CD4+ T cell lineage, by a mechanism dependent on STAT3 (Dang et al., 2011; Ikejiri et al., 2012; Nakamura et al., 2005; Shi et al., 2011; Wang et al., 2014), where IL-6 and transforming growth factor-β (TGF-b) enhance HIF-1a mRNA expression via a signal transduction and activator of transcription (STAT3)-dependent mechanism (Dang et al., 2011; Kojima et al., 2002; Shi et al., 2011). In contrast, stimulation of CD4+ T cells differentiated with cytokines promoting other helper subsets showed minimal increases in HIF-1α mRNA or protein upon activation (Dang et al., 2011; Shi et al., 2011). CD8+ T cells also rapidly stabilize HIF-1α and HIF-2αproteins upon in vitro TCR crosslinking in normoxic conditions (Doedens et al., 2013; Finlay et al., 2012; Wang et al., 2011). Further, interleukin-4 (IL-4), which induces HIF-2α stabilization in macrophages (Takeda et al., 2010), also contributes to HIF-2α accumulation in activated CD8+ T cells, while IL-2 conditions favor HIF-1α accumulation (Doedens et al., 2013). Thus, HIF is positioned to be a key player in the integration of TCR- and cytokine receptor-mediated signals involved in CD4+ helper lineage commitment and CD8+ effector differentiation in a fashion independent of oxygen availability. This suggests a potential role for HIF-1α in promoting T-cell-mediated inflammation and indicates that microenvironmental cues and cytokines that drive T cell fate and differentiation are also crucial in shaping the hypoxia response in T cell accumulation (Doedens et al., 2013). Thus, HIF is positioned to be a key player in the integration of TCR- and cytokine receptor-mediated signals involved in CD4+ helper lineage commitment and CD8+ effector differentiation in a fashion independent of oxygen availability. This suggests a potential role for HIF-1α in promoting T-cell-mediated inflammation and indicates that microenvironmental cues and cytokines that drive T cell fate and differentiation are also crucial in shaping the hypoxia response in T cells.
HIF and innate immunity
Inflammation
Monocyte and neutrophil development and differentiation are not affected by specific deletion of HIF-1α in the myeloid lineage, as shown by conditional gene targeting (Cramer et al., 2003). In contrast, mice with HIF-1α myeloid-specific deletion showed impaired inflammatory responses, which are linked to a metabolic defect characterized by lower glycolytic rates and energy generation when HIF-1a is absent. As a result, macrophages show impaired aggregation, invasion, and motility. Notably, myeloid HIF-1α contributes to inflammation in rheumatoid arthritis synovial tissue (Cramer et al., 2003), which is hypoxic (Konisti et al., 2012).
Indeed, the relationship between the hypoxic and inflammatory responses is tightly controlled (Figure 2A). As noted above, hypoxia and bacterial infections result in increased nuclear factor-kappa B (NF-κB) activity in phagocytes, which in turn induces HIF-1α mRNA transcription (Cummins et al., 2006; Fitzpatrick et al., 2011; Rius et al., 2008). Together with NF-kB, the p42/p44 mitogen-activated protein kinase (MAPK) (Erk) pathway is involved in LPS-induced HIF-1α induction in human monocytes (Frede et al., 2006).
Figure 2. Hypoxia Pathway and Innate Immunity.

(A) Inflammation. HIF-1α and NF-κB crosstalk regulates essential inflammatory functions in myeloid cells. HIF-1α increases macrophage aggregation, invasion, and motility and drives the expression of proinflammatory cytokines. HIF-1α also increases neutrophil survival by inhibiting apoptosis and triggers NF-κB-dependent neutrophilic inflammation. (B) Infection. HIF-1α increases intracellular bacterial killing by macrophages and also promotes granule protease production and release of nitric oxide (NO) and TNF-α, which in turn further contribute in antimicrobial control. (C) Cancer. As a result of hypoxia, HIF-1α is stabilized in cancer cells, increasing the production of chemotactic factors like CCL5, CXCL12, VEGF, endothelins ET-1 and ET-2, and semaphorin3A (Sema3A), which result in myeloid cell recruitment to the tumor. Once located in 31 hypoxic regions, tumor-associated macrophages stabilize HIF-1α, which contributes to tumor-promoting inflammation, angiogenesis, and impaired lymphocyte function. (D) Metabolism. HIF-1α in myeloid cells increases the transcription of key glycolytic enzymes, resulting on increased glucose uptake and glycolytic rate. Importantly, pyruvate entry in the citric acid cycle (TCA) is inhibited and converted to lactate, which is released to the extracellular compartment. This metabolic adaptation results in a decreased O2 consumption by limiting the rate of oxidative phosphorylation (OXPHOS). M1 and M2 polarized macrophages show different metabolic pathway preferences: whereas M1 rely on glycolysis as energy source, M2 have steady OXPHOS metabolism.
Neutrophils are short-lived innate immune cells that contribute to the inflammatory response. Hypoxia delays neutrophil apoptosis in a reversible manner, through a process that can be mimicked by iron chelator agents (Mecklenburgh et al., 2002). In fact, hypoxia induces neutrophil survival through HIF-1a-dependent NF-kB activity that results in sustained inflammation (Walmsley et al., 2005). FIH, PHD1, and PHD2 protein expression in neutrophils are not affected by hypoxia (Walmsley et al., 2005). However, PHD3 expression is increased in neutrophils subjected to hypoxia or isolated from rheumatoid arthritis patients (Walmsley et al., 2011). Selective ablation of PHD3 in neutrophils abrogates this hypoxic prosurvival effect and reduces neutrophilic inflammation in a colitis model, revealing a tight control by the hypoxia pathway (Walmsley et al., 2011). Thus, HIF-1α-induced NF-κB controls neutrophil proinflammatory functions and survival under hypoxia, which requires PHD3 expression. In similar fashion, neutrophil accumulation in a mouse model of colitis induced an inflammatory hypoxic microenvironment that favored resolution, which was abrogated when neutrophils were depleted or lacked respiratory burst, but restored upon pharmacological HIF-1α stabilization (Campbell et al., 2014).
HIF-2α also regulates essential inflammatory functions of immune cells. Mice lacking HIF-2α in the myeloid compartment fail to mount an inflammatory response upon LPS challenge, due to impaired tumor necrosis factor-α (TNF-α), interferon-g (IFN-γ), IL-12, and IL-1β production. In contrast to proinflammatory cytokine expression, HIF-2a was not found to control nitric oxide (NO) production or alter macrophage activation marker expression (Imtiyaz et al., 2010). Although this study and others (Mecklenburgh et al., 2002; Walmsley et al., 2005) fail to detect HIF-2α mRNA or protein in bone marrow-derived neutrophils, a recent publication highlights that hypoxia, hydroxylase inhibition, and bacteria can induce HIF-2α expression in human and murine peripheral blood neutrophils (Thompson et al., 2014). Mice lacking myeloid HIF-2αa have reduced neutrophilic inflammation and increased neutrophil apoptosis. Neutrophils isolated from arthritis patients also express increased amounts of HIF-2α protein (Thompson et al., 2014).
The identification of the involvement of the hypoxia pathway in proinflammatory responses has supported efforts to develop novel therapeutic approaches to treat inflammatory disorders. In this regard, HIF-1α inhibition could be a potential strategy for limiting exacerbated responses, such as arthritis (Giaccia et al., 2003). Another approach consists of PHD inhibition, which has shown therapeutic potential in preclinical models of colitis (Cummins et al., 2008; Fraisl et al., 2009; Robinson et al., 2008).
Bacterial Infection
Since bacterial infection typically results in a hypoxic tissue microenvironment, in vivo models of infection have made an important contribution to our understanding of the role of the hypoxia pathway in innate immune cells called upon to fight pathogens.
HIF-1α-deficient macrophages have an impaired metabolic adaptation to hypoxia, which results in reduced motility and migration. Moreover, HIF-1α-deficient macrophages show decreased capacity to kill bacteria. Although the rate of phagocytic uptake of group B Streptococcus was similar, intracellular killing and lysis is severely impaired when HIF-1α is genetically ablated (Cramer et al., 2003). Other studies using different pathogens have pointed to a potential role of hypoxia and HIF-1α in the control of bacterial phagocytosis (Figure 2B; Anand et al., 2007).
A subsequent study using HIF-1α conditional gene targeting in myeloid cells confirmed that both hypoxia and HIF-1α regulate the intracellular killing by macrophages infected with group A Streptococcus (GAS) and Pseudomonas aeruginosa. Conversely, VHL-deficient macrophages showed enhanced bactericidal capacity (Peyssonnaux et al., 2005). In vivo experiments indicated that mice were more susceptible to GAS infection when lacking myeloid HIF-1α, resulting in more severe necrotic skin ulcers and greater weight loss. In these studies HIF-1a was correlated with iNOS expression, driving NO and TNF-α production; these in turn have an important role in regulating the antimicrobial action of myeloid cells (Peyssonnaux et al., 2005). In similar fashion, neutrophil bactericidal activity is controlled by HIF-1α. Although HIF-1α-deficient neutrophils migrate to GAS-induced skin lesions at similar rates compared to their HIF-1α-competent counterparts, they present markedly decreased production of granule proteases and cathelicidin antimicrobial peptide (Peyssonnaux et al., 2005). Moreover, neutrophils isolated from patients with heterozygous germline mutations in VHL exhibited increased phagocytosis of streptococci (Walmsley et al., 2006). Exploiting these relationships, pharmacological stabilization of HIF-1α enhanced phagocyte-mediated clearance of methicillin-resistant Staphylococcus aureus in vitro and in vivo (Okumura et al., 2012).
Macrophage metabolism and polarization
An emerging topic is the control of myeloid cell function by metabolic activity. As we have discussed, the environmental milieu promotes HIF-1a expression in myeloid cells fighting against pathogens or cancer, driving a metabolic shift to modulate oxygen consumption and energy choices. Macrophages undergoing LPS-induced M1 polarization have a robust metabolic shift toward glycolysis, pentose-phosphate pathway activity, and basal oxidative phosphorylation, whereas immunomodulatory IL-4-induced M2-like macrophages have moderate glycolytic activity but higher oxidative activity (Figure 2C; Haschemi et al., 2012). This work underlines the importance of metabolic reprogramming in the macrophage response to microbial stimuli. Determining the extent to which the hypoxia pathway links myeloid metabolic adaptation with inflammatory response is an exciting field of new investigation.
HIF-2α is known to have a more discrete role in the glycolytic adaptation to hypoxia in cancer cells (Hu et al., 2003) and macrophages (Imtiyaz et al., 2010). Although arginase has an important role in the HIF-2α contribution to IL-4-induced M2 polarization, the fact that HIF-2α is selectively induced in M2-like macrophages (Takeda et al., 2010) could indicate a potential metabolic mechanism by which increased expression of HIF-2α outcompete HIF-1α in its transcriptional activity, limiting the glycolytic shift associated to M1 polarization. However, it is still not clear whether metabolic adaptation is cause or consequence of the myeloid cellular differentiation program in inflammatory responses. Moreover, HIF-1α can also drive immunosuppressive functions by myeloid cells, i.e., inducible NO synthase (iNOS) production (Peyssonnaux et al., 2005), and macrophages lacking HIF-2α fail to mount an inflammatory response upon LPS challenge (Imtiyaz et al., 2010), indicating that the different functions of the HIF isoforms on macrophage polarization are not completely exclusive, but sometimes overlapping and dependent on the pathophysiological context.
Viral Infection
HIF-1α activity is induced in response to viral infection, but accumulating evidence suggests that the net consequence can favor the pathogen rather than the host. In particular, certain viruses have evolved mechanisms to stabilize HIF-1α to exert an antiapoptotic effect that promotes survival of the infected cell. Hepatitis B HBx protein increases HIF-1α transactivation by enhancing its association with cAMP-response element binding protein (CREB)-binding protein (BP) (Yoo et al., 2003). Kaposi’s sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen (LANA) targets VHL for degradation via ubiquitination, increasing HIF protein expression (Cai et al., 2006) and Epstein-Barr virus (EBV) oncoprotein LMP1 boosts HIF-1α concentrations by increasing the proteasomal degradation of PHD1 and PHD3 that normally mark HIF-1α for degradation (Kondo et al., 2006). In HIV-1 and JCV polyomavirus infection, HIF-1α directly stimulates the transcription of key viral HIV-1 genes by associating with the HIV-1 long terminal repeat (Deshmane et al., 2009) or JCV early promoter (Pina-Oviedo et al., 2009), respectively.
Dendritic Cells and Antigen Presentation
The function and in vivo relevance of HIF-1α in dendritic cells (DCs) are still not completely clear. HIF-1α stabilization does not affect DC maturation in the absence of proinflammatory stimuli (Elia et al., 2008). LPS-treated human monocyte-derived DCs cultured under hypoxia express lower amounts of costimulatory molecules and maturation markers, resulting in decreased T cell proliferation upon antigen presentation. In contrast, hypoxia induces the production of proinflammatory cytokines (TNF-α, IL-1β) by DCs (Mancino et al., 2008), modulates chemokine receptor expression (Ricciardi et al., 2008), and decreases DC migration in response to C-C chemokine receptor type 7 (CCR7) ligands (Mancino et al., 2008; Qu et al., 2005; Zhao et al., 2005).
Upon toll-like receptor (TLR) ligation, DCs stabilize HIF-1α (Jantsch et al., 2011) and undergo a metabolic shift similar to that observed in other activated and hypoxic immune cells, relying on aerobic glycolysis to support maturation and function (Jantsch et al., 2008; Krawczyk et al., 2010). Pharmacological HIF-1αstabilization with a PHD inhibitor increased major histocompatibility complex (MHC) and costimulatory molecule expression by DCs, which in turn induced greater T cell proliferation, serving to produce higher titers of antibodies on immunization with the model antigen ovalbumin (Bhandari et al., 2013). However, the role of the hypoxia pathway in this process is still to be fully determined, and robust in vivo models and experimentation are needed to ascertain whether HIF-1α could affect antigen presentation, key for the crosstalk between the innate and adaptive immune system.
Innate immunity and cancer
Myeloid cells are a prominent population infiltrating solid tumors. Macrophages are recruited into tumors, and although they have the ability to inhibit angiogenesis and fight malignant progression, they are commonly associated with a microenvironment that predisposes to tumor immune escape, new vessel formation, and metastasis (Figure 2D).
Hypoxia, as a prominent feature of solid tumors, leads to HIF-1α stabilization in cancer cells and contributes to the recruitment of bone-marrow-derived myeloid cells by at least two key mechanisms. First, HIF-1α drives the production of chemokines and chemokine receptors that could influence myeloid recruitment (Murdoch et al., 2004). For instance, CCL5 and C-X-C motif chemokine 12 (CXCL12, also known as SDF1a) production by hypoxic tumor cells is driven by HIF-1a (Du et al., 2008; Lin et al., 2012), and chemokine receptor C-X-C motif chemokine receptor (CXCR4) expression by myeloid and tumor cells is regulated by the VHL-HIF axis (Schioppa et al., 2003; Staller et al., 2003). Second, HIF-1α in tumor cells increases the secretion of cytokines, mainly VEGF, which can act as a chemoattractant for myeloid cells (Leek et al., 2000), an effect that is dependent on VEGF receptor 2 (VEGFR2) expression on macrophages (Dineen et al., 2008). Endothelins ET-1 and ET-2 are also chemoattractants for monocyte/macrophage recruitment into tumors, and their expression by tumor cells is induced by HIF-1α (Grimshaw, 2007; Grimshaw et al., 2002).
Hypoxia-induced tumor-produced semaphorin3A (Sema3A) also contributes to recruitment of macrophages into hypoxic tumors by binding neuropilin-1 (Nrp-1) and PlexinA1/A4 coreceptors and signaling through VEGFR1. When macrophages ocalize to hypoxic areas, macrophage Nrp-1 but not PlexinA1/ A4 is repressed by hypoxia, leading to tumor-associated macrophage (TAM) accumulation. Deletion of Nrp-1 on macrophages prevents TAM infiltration in hypoxic regions, resulting in delayed tumor growth, which is in turn characterized by impaired vascularization and improved antitumor immunity (Casazza et al., 2013).
Once recruited, the influence of different factors present in the tumor microenvironment results in different TAM phenotypes, which can be classified into different subsets. For instance, TAMs infiltrating hypoxic tumor regions have been associated with an increased and proangiogenic M2-like phenotype, characterized by lower MHC-II expression compared to TAMs located in normoxic areas (Laoui et al., 2014; Movahedi et al., 2010).
TAMs contribute to tumor-promoting chronic inflammation in part by directly dampening adaptive immunity, as well as by secreting extracellular factors that fuel endothelial and malignant cell proliferation and survival. Targeted deletion of HIF-1α in macrophages resulted in reduced tumor growth in a spontaneous PYMT mammary adenocarcinoma model, which was shown to result from improved lymphocyte proliferative capacity and function when myeloid HIF-1α is absent (Doedens et al., 2010; Lewis and Murdoch, 2005). Myeloid-derived suppressor cells are another stromal component that can dampen antitumor immunity, and HIF-1α can modulate their function and differentiation into tumor-associated macrophages (Corzo et al., 2010) and their expression of the immune checkpoint ligand PD-L1 (Noman et al., 2014).
Macrophages can promote angiogenesis (Lin et al., 2006). Tie2-expressing monocytes are considered an important lineage that contributes to tumor vascularization (De Palma et al., 2005) and Tie2 expression in this population is regulated by hypoxia, which could contribute to its recruitment and function (Lewis et al., 2007).
VEGF is the most studied endothelial-mitogen among macrophage-secreted factors modulated by hypoxia (Shweiki et al., 1992), and its induction in myeloid cells is driven by HIF-1α (Cramer et al., 2003; Eubank et al., 2011; Roda et al., 2012). The contribution of macrophage-derived VEGF to angiogenesis and malignancy has been studied in a variety of tumor models (Stockmann et al., 2008). Upon macrophage depletion, delayed mammary carcinoma progression can be restored by VEGF (Lin et al., 2007).
Although it is accepted that VEGF contributes to tumorigenesis (Goel and Mercurio, 2013), when VEGF is specifically deleted in the myeloid lineage, spontaneous mammary PYMT tumors developed faster, as a result of blockade of the angiogenic switch and vascular normalization (Stockmann et al., 2008). This study highlighted the multifaceted aspects of TAMs and TAM-derived VEGF on tumor progression, and suggested that separate from total VEGF expression in tumors, myeloid-derived VEGF could fine-tune angiogenesis upon endothelial-macrophage interactions.
Hypoxic TAMs can also contribute to increased VEGF bioavailability by releasing MMP-9, a matrix metalloproteinase induced by HIF-1α, resulting in increased neovascularization and tumor cell invasiveness (Du et al., 2008). On the other hand, primary tumor-released VEGF can induce MMP-9 on macrophages present in the premetastatic niche, promoting metastasis (Hiratsuka et al., 2002). HIF-1α signaling in primary mammary tumors leads to the induction of members of the lysyl oxidase (LOX) family, preconditioning the lungs for bone-marrow-derived cell recruitment prior to metastatic colonization (Wong et al., 2011).
HIF-2α is also expressed in TAMs. Absence of HIF-2α impairs infiltration, migration, and chemotactic receptor expression by TAMs, resulting in reduced inflammatory murine-associated cancer (Imtiyaz et al., 2010). In contrast to HIF-1α, HIF-2α does not control the hypoxic induction of VEGF in macrophages (Ahn et al., 2014; Eubank et al., 2011), but it can induce soluble VEGF receptor 1 (sVEGFR-1) secretion by hypoxic macrophages, which neutralizes VEGF (Roda et al., 2011) and raises another potential opposing role for different HIF isoforms.
HIF and adaptive immunity
In the process of T cell development, lymphoid progenitors migrate from the bone marrow to the thymus, an organ known to be hypoxic under physiological conditions (Hale et al., 2002). T cell precursors that lack CD4 or CD8 coreceptor surface expression initiate TCR rearrangements and navigate multiple developmental checkpoints and selection events that coordinate MHC restriction and functional activity with lineage before becoming mature gd or ab CD4+ and CD8+ T cells that migrate to peripheral lymphoid tissues. In contrast to myeloid VHL deletion, which does not affect neutrophil and monocyte development (Cramer et al., 2003), specific deletion of VHL in thymocytes early in development by a cre recombinase expressed under the control of the proximal Lck promoter resulted in a dramatic reduction in thymic cellularity. Additional deletion of HIF-1α with VHL deficiency restored thymocyte development and prevented excessive apoptosis associated with VHL deletion (Biju et al., 2004), suggesting a tightly regulated role of the hypoxia pathway in lymphocyte development. On the other hand, loss of HIF-1α does not obviously impact thymic subsets or CD4+ and CD8+ T cells in secondary lymphoid organs (Kojima et al., 2002). However, HIF-1a deficiency in the lymphoid compartment does result in abnormal B cell development and autoimmunity (Kojima et al., 2002); interestingly, CD19-cre-mediated B cell-specific deletion prevents hypoxic cell cycle arrest in B cells, an indication of HIF-1α’s importance in lymphoid cell homeostasis (Goda et al., 2003).
Given the substantial induction of HIF-1α mRNA and protein in response to T cell activation and the well-known role of HIF in driving glycolytic metabolism, several groups have recently explored potential HIF function in adaptive immune responses. How HIF activity impacts T cell activation and differentiation is a timely topic because it is now also thought that changes in metabolic activity not only accompany the dramatic expansion and acquisition of effector function driven by recognition of antigen, but may also be intimately involved in fate decisions of differentiating effector cells. Proinflammatory Th1, Th2, and Th17 cell populations generated in vitro display increased glycolysis and less OXPHOS than in-vitro-induced T regulatory cells, which show a greater reliance on lipid oxidation and OXPHOS (Michalek et al., 2011; Shi et al., 2011). Consistent with HIF’s role in promoting glycolysis, HIF-1a is highly expressed by Th17 cells. Deletion of HIF-1α in T cells impairs differentiation of CD4+ Th17 producing cells in vitro and in MOG/CFA-induced experimental autoimmune encephalomyelitis (EAE) (Dang et al., 2011; Shi et al., 2011). Inversely, hypoxia and deletion of VHL or miR-210, which also promote stabilization of HIF-1α, can enhance generation of Th17 CD4+ cells (Dang et al., 2011; Ikejiri et al., 2012; Wang et al., 2014). The inhibition of glycolysis, in turn, reduces Th17 cell induction and protects from EAE, suggesting that HIF regulation of metabolic activity underlies its role in T helper cell subset differentiation (Shi et al., 2011). However, HIF-1α also may favor the Th17 cell lineage by directly promoting expression of RORgt (RAR-related orphan receptor gt) and its target IL-17 (Dang et al., 2011).
The generation of regulatory T (Treg) cells in the peripheral immune compartment is entwined with induction of Th17 CD4+ cells, as both rely on TGF-b, which can in turn promote expression of both RORgt and FOXP3; these latter are key transcription factors regulating the development of each respective cell type (Littman and Rudensky, 2010). Forkhead binding protein 3 (FOXP3) antagonizes RORgt, unless IL-6 is present to drive Th17 cell development; thus, the generation of these two lineages is linked to the inflammatory milieu and is generally reciprocal. Consistent with HIF-1α promoting Th17 cell formation, these groups also observed enhanced Treg cell differentiation with deletion of HIF-1a in conditions favoring Th17 cells (Clambey et al., 2012; Dang et al., 2011; Shi et al., 2011); inhibition of glycolysis had the same effect (Shi et al., 2011). One proposed mechanism for HIF attenuation of Treg cell development is direct binding and targeting of FOXP3 for degradation (Dang et al., 2011; Darce et al., 2012). However, in conditions promoting Treg cell induction, hypoxia can enhance FOXP3 expression and Treg cell accumulation, and in vivo HIF-1a-deficient Treg cells display impaired suppressive function in T-cell-mediated colitis (Clambey et al., 2012). Thus, hypoxia and HIF activity may regulate CD4+ T cell differentiation differently in divergent tissue microenvironments (Figure 3A).
Figure 3. Hypoxia Pathway and Adaptive Immunity.
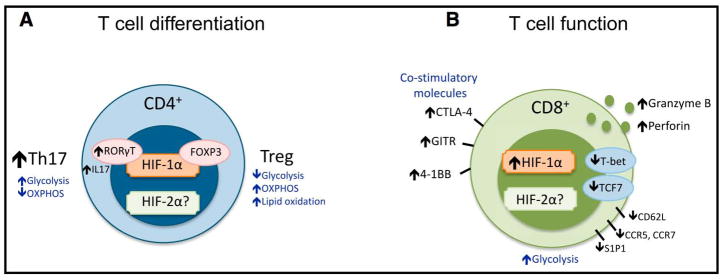
(A) T cell differentiation. Upon activation, HIF-1α is strongly induced in conditions that favor differentiation of CD4+ Th17 cells. HIF-1α can increase the expression of RORgt, which in turn promotes IL-17 production and Th17 cell development. Th17 and Treg cells also rely on different metabolic pathways. HIF-1α can bind FOXP3 protein and impact its function and degradation, and in some cases, promotes Foxp3 mRNA induction and supports Treg cell function. HIF-1α-driven glycolytic shift supports Th17 cell differentiation, while lack of HIF-1α can support Treg cell differentiation under Th17 cell differentiation conditions. (B) HIF-1α stabilization by activated CD8+ T cells results in increased expression and release of important cytolytic molecules (granzyme B, perforin), increased expression of costimulatory/inhibitory molecules (CTLA-4, GITR, 4-1BB), and altered migration and chemokine receptor expression.
HIF-1a can also drive metabolic adaptations in CD8+ T cells and control expression of many molecules associated with effector function and migration (Doedens et al., 2013; Finlay et al., 2012). The earliest phases of activation-induced metabolic reprogramming appear to be Myc dependent, as HIF-deficient T cells induce glycolysis (Wang et al., 2011). However, as CD8+ T cells differentiate to effector cells, loss of HIF-1β impairs expression of genes controlling glycolysis and glycolytic capacity, albeit not completely (Finlay et al., 2012). Interestingly, HIF-1β-deficient T cells show diminished expression of effector molecules, such as perforin and granzymes, and altered expression of molecules involved in T cell migratory capacity. HIF-1β-deficient CD8+ T cells maintained CD62L expression in conditions where wild-type cells lost CD62L, yet displayed increased T cell homing to secondary lymphoid organs, perhaps due to aberrant upregulation of CXCR5, CCR7, and S1P1R (Finlay et al., 2012). This study and others establish HIF and glycolysis downstream of the mechanistic target of rapamycin (mTOR), coupling induction of HIF-1α with metabolic changes and regulation of CD8+ effector cell function and trafficking (Figure 3B; Finlay et al., 2012; Nakamura et al., 2005; Shi et al., 2011).
Enhanced HIF activity resulting from deletion of VHL promotes glycolytic metabolism and effector function by CD8+ T cells (Doedens et al., 2013), further highlighting a role for HIF in host protection. Hypoxia, via a HIF-dependent mechanism, induces expression of many key T cell activation receptors (4-1BB, glucocorticoid-induced TNFR family related gene [GITR], OX40), inhibitory receptors (lymphocyte activation gene-3 [LAG-3], cytotoxic T-lymphocyte-associated protein 4 [CTLA-4]), and effector molecules (Granzyme B), while suppressing expression of key transcription factors (T-bet, Eomes, TCF-1) in CD8+ effector cells. VHL-deficient T cells are potent effectors, clearing virus and slowing tumor growth when wild-type CD8+ T cells lose effector function and undergo progressive “exhaustion” due to chronic antigen exposure. Stabilization of HIF in CD8+ T cells apparently can bypass inhibitory receptor signaling typically associated with the dampening of effector capacity when antigen cannot be cleared. However, the exhaustion-induced loss of function by CD8+ T cells serves to limit immunopathology in persistent infections, and the exuberant response of VHL-deficient T cells also mediates host death when mice are infected with a strain of virus that cannot be efficiently cleared by the immune system. Thus, manipulation of HIF activity in CD8+ T cells may serve to exacerbate or avert immunopathology, promote pathogen clearance, or diminish tumor burden, depending on the context.
Adaptive immunity and cancer
In the context of cancer immunology, tumor-infiltrating lymphocytes (TILs) comprise a heterogeneous population. CD4+ Th1 cells and CD8+ cytotoxic lymphocytes have been generally linked to a better prognosis in cancer patients (Tosolini et al., 2011). On the other hand, T regulatory cells that suppress self-reactive T cells, protecting the host from autoimmunity, can be deleterious in mounting an effective antitumor immune response; indeed, high numbers of Treg cells and low CD8+ T cell:Treg cell ratios have been associated with a worse prognosis (Sato et al., 2005). Although solid malignancies are oxygen deprived due to an imbalance between vascularization and cellular demands, it is not known how hypoxia signaling in TILs affects migration, survival, crosstalk, and immune function of both effector and suppressive cells.
The effects of HIF signaling in cancer cells are better established and lead to invasion, angiogenesis, metastasis, and secretion of tumor-derived factors like cytokines and growth factors that recruit immunosuppressive cells. As previously discussed, intratumoral hypoxia and HIF-1α stabilization in cancer cells and stromal components such as endothelial cells and TAMs induce VEGF secretion. VEGF acts as a Treg cell chemotactic factor by binding to Neuropilin-1 (NP-1). Mice deficient in T-cell-specific NP-1 have impaired infiltration of Treg cell into tumors, which results in impaired melanoma tumor growth (Hansen et al., 2012). HIF-1α can also promote TGF-b secretion by cancer cells (Deng et al., 2013). TGF-β, itself a critical modulator of inflammation, acts as an important chemo-attractant for Treg cell recruitment into tumors and can also bind NP-1 (Glinka and Prud’homme, 2008). CCL28 is another chemokine that is secreted by hypoxic tumor cells and attracts CXCR10+ Treg cells to the tumor site, promoting tumor tolerance and angiogenesis (Facciabene et al., 2011). In addition, an increase in the intratumoral pool of adenosine as a result of hypoxia has been proposed as an immune-tolerance mechanism (Sitkovsky and Lukashev, 2005).
Another contribution of hypoxia to tumor immune escape that is mediated by HIF-1a is a decrease in cancer cell susceptibility to T-cell-mediated killing (Noman et al., 2009). Hypoxia also induces tumor cell autophagy, a process that confers resistance to immune-mediated lysis by cytotoxic lymphocytes (Noman et al., 2011) and natural killer (NK) cells (Baginska et al., 2013). Programmed cell death 1 (PD-1) and CTLA-4 act as coinhibitory receptors on activated T cells and are important targets for cancer immunotherapy strategies based on the administration of blocking antibodies. HIF-1α drives CTLA-4 expression on CD8+ T cells (Doedens et al., 2013) and HIF-1α also increases PD-L1 expression on hypoxic cancer cells to promote immune escape (Barsoum et al., 2014). HIF-1α has also been shown to upregulate other costimulatory receptors that are potential targets for cancer immunotherapy: OX40, GITR, and 4-1BB (Doedens et al., 2013; Palazón et al., 2011, 2012).
An increasing number of agents aiming to inhibit HIF-1α are under development for cancer treatment (Onnis et al., 2009). Given the cellular heterogeneity in the tumor microenvironment, and the fact that the contribution of HIF to malignant and stromal cell populations could differentially drive tumor progression, ongoing early clinical trials will help to understand the impact of HIF inhibition and other novel agents targeting the hypoxia pathway in terms of immune control and cancer progression. As a result, new rational therapeutic combinations targeting hypoxia and angiogenesis (Rapisarda and Melillo, 2012) or novel immunotherapies (Mellman et al., 2011) could help to overcome the multiple mechanisms of tumor resistance.
Concluding remarks
Recent research has highlighted the importance of hypoxic response and HIF signaling in controlling immune responses. Although HIF-1α is considered central in the control of the response, emerging data support the notion that HIF-2α can exert different and sometimes opposing roles. All of these results are in part a demonstration of the critical nature of the tissue microenvironment in immunity.
All immune cells are present in a physiologically complex and fluctuating tissue environment, whereby pH, oxygen, nutrients, and metabolites are both in and out of homeostatic balance relative to each other and to the “normal” environment of that immune cell. Add to the equation the physiological chaos of a virulent infection or a necrotic tumor, and the need to understand the relationship between immunological function and immune cell response to this flux becomes central to understanding the immune response. As the analysis of immunophysiology becomes more sophisticated, the opportunities for development of relevant and effective immunotherapies can only continue to expand.
Acknowledgments
Funding has been provided by NIH for A.G. (A1096852, A1072117), V.N., and R.S.J. (A1096852), Welcome Trust for R.S.J., and Marie Curie IEF fellowship for A.P. 21
References
- Ohn GO, Seita J, Hong BJ, Kim YE, Bok S, Lee CJ, Kim KS, Lee JC, Leeper NJ, Cooke JP, et al. Transcriptional activation of hypoxia-inducible factor-1 (HIF-1) in myeloid cells promotes angiogenesis through VEGF and S100A8. Proc Natl Acad Sci USA. 2014;111:2698–2703. doi: 10.1073/pnas.1320243111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albina JE, Mastrofrancesco B, Vessella JA, Louis CA, Henry WL, Jr, Reichner JS. HIF-1 expression in healing wounds: HIF-1alpha induction in primary inflammatory cells by TNF-alpha. Am J Physiol Cell Physiol. 2001;281:C1971–C1977. doi: 10.1152/ajpcell.2001.281.6.C1971. [DOI] [PubMed] [Google Scholar]
- Anand RJ, Gribar SC, Li J, Kohler JW, Branca MF, Dubowski T, Sodhi CP, Hackam DJ. Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1alpha-dependent manner. J Leukoc Biol. 2007;82:1257–1265. doi: 10.1189/jlb.0307195. [DOI] [PubMed] [Google Scholar]
- Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingston DM. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA. 1996;93:12969–12973. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, Medves S, Zimmer J, Oudin A, Niclou SP, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci USA. 2013;110:17450–17455. doi: 10.1073/pnas.1304790110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74:665–674. doi: 10.1158/0008-5472.CAN-13-0992. [DOI] [PubMed] [Google Scholar]
- Bhandari T, Olson J, Johnson RS, Nizet V. HIF-1a influences myeloid cell antigen presentation and response to subcutaneous OVA vaccination. J Mol Med. 2013;91:1199–1205. doi: 10.1007/s00109-013-1052-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biju MP, Neumann AK, Bensinger SJ, Johnson RS, Turka LA, Haase VH. Vhlh gene deletion induces Hif-1-mediated cell death in thymocytes. Mol Cell Biol. 2004;24:9038–9047. doi: 10.1128/MCB.24.20.9038-9047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin CC, Page EL, Soucy GM, Richard DE. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1alpha. Blood. 2004;103:1124–1130. doi: 10.1182/blood-2003-07-2427. [DOI] [PubMed] [Google Scholar]
- Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, Kietzmann T, Gorlach A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol. 2007;27:755–761. doi: 10.1161/01.ATV.0000258979.92828.bc. [DOI] [PubMed] [Google Scholar]
- Cai QL, Knight JS, Verma SC, Zald P, Robertson ES. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2006;2:e116. doi: 10.1371/journal.ppat.0020116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, Bayless AJ, Scully M, Saeedi BJ, Golden-Mason L, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity. 2014;40:66–77. doi: 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, Deschoemaeker S, Van Ginderachter JA, Tamagnone L, Mazzone M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/ Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24:695–709. doi: 10.1016/j.ccr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC, Stenmark KR, Colgan SP, Eltzschig HK. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA. 2012;109:E2784–E2793. doi: 10.1073/pnas.1202366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, et al. HIF-1a regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. HIF-1alpha is essential for myeloid cellmediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA. 2006;103:18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, Taylor CT. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134:156–165. doi: 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darce J, Rudra D, Li L, Nishio J, Cipolletta D, Rudensky AY, Mathis D, Benoist C. An N-terminal mutation of the Foxp3 transcription factor alleviates arthritis but exacerbates diabetes. Immunity. 2012;36:731–741. doi: 10.1016/j.immuni.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, Naldini L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Deng B, Zhu JM, Wang Y, Liu TT, Ding YB, Xiao WM, Lu GT, Bo P, Shen XZ. Intratumor hypoxia promotes immune tolerance by inducing regulatory T cells via TGF-b1 in gastric cancer. PLoS ONE. 2013;8:e63777. doi: 10.1371/journal.pone.0063777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmane SL, Mukerjee R, Fan S, Del Valle L, Michiels C, Sweet T, Rom I, Khalili K, Rappaport J, Amini S, Sawaya BE. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxia-inducible factor 1alpha expression. J Biol Chem. 2009;284:11364–11373. doi: 10.1074/jbc.M809266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS, Beck AW, Barnett CC, Fleming JB, Brekken RA. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008;68:4340–4346. doi: 10.1158/0008-5472.CAN-07-6705. [DOI] [PubMed] [Google Scholar]
- Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW, Johnson RS. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70:7465–7475. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, Johnson RS, Goldrath AW. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. 2013;14:1173–1182. doi: 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegué E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia AR, Cappello P, Puppo M, Fraone T, Vanni C, Eva A, Musso T, Novelli F, Varesio L, Giovarelli M. Human dendritic cells differentiated in hypoxia down-modulate antigen uptake and change their chemokine expression profile. J Leukoc Biol. 2008;84:1472–1482. doi: 10.1189/jlb.0208082. [DOI] [PubMed] [Google Scholar]
- Eubank TD, Roda JM, Liu H, O’Neil T, Marsh CB. Opposing roles for HIF-1a and HIF-2a in the regulation of angiogenesis by mononuclear phagocytes. Blood. 2011;117:323–332. doi: 10.1182/blood-2010-01-261792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P, Zhang L, Coukos G. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475:226–230. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, Panteleyev AA, Okkenhaug K, Cantrell DA. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209:2441–2453. doi: 10.1084/jem.20112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick SF, Tambuwala MM, Bruning U, Schaible B, Scholz CC, Byrne A, O’Connor A, Gallagher WM, Lenihan CR, Garvey JF, et al. An intact canonical NF-kB pathway is required for inflammatory gene expression in response to hypoxia. J Immunol. 2011;186:1091–1096. doi: 10.4049/jimmunol.1002256. [DOI] [PubMed] [Google Scholar]
- Fraisl P, Aragonés J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov. 2009;8:139–152. doi: 10.1038/nrd2761. [DOI] [PubMed] [Google Scholar]
- Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem J. 2006;396:517–527. doi: 10.1042/BJ20051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccia A, Siim BG, Johnson RS. HIF-1 as a target for drug development. Nat Rev Drug Discov. 2003;2:803–811. doi: 10.1038/nrd1199. [DOI] [PubMed] [Google Scholar]
- Glinka Y, Prud’homme GJ. Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J Leukoc Biol. 2008;84:302–310. doi: 10.1189/jlb.0208090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol. 2003;23:359–369. doi: 10.1128/MCB.23.1.359-369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013;13:871–882. doi: 10.1038/nrc3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimshaw MJ. Endothelins and hypoxia-inducible factor in cancer. Endocr Relat Cancer. 2007;14:233–244. doi: 10.1677/ERC-07-0057. [DOI] [PubMed] [Google Scholar]
- Grimshaw MJ, Wilson JL, Balkwill FR. Endothelin-2 is a macrophage chemoattractant: implications for macrophage distribution in tumors. Eur J Immunol. 2002;32:2393–2400. doi: 10.1002/1521-4141(200209)32:9<2393::AID-IMMU2393>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Hale LP, Braun RD, Gwinn WM, Greer PK, Dewhirst MW. Hypoxia in the thymus: role of oxygen tension in thymocyte survival. Am J Physiol Heart Circ Physiol. 2002;282:H1467–H1477. doi: 10.1152/ajpheart.00682.2001. [DOI] [PubMed] [Google Scholar]
- Hansen W, Hutzler M, Abel S, Alter C, Stockmann C, Kliche S, Albert J, Sparwasser T, Sakaguchi S, Westendorf AM, et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J Exp Med. 2012;209:2001–2016. doi: 10.1084/jem.20111497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann H, Eltzschig HK, Wurz H, Hantke K, Rakin A, Yazdi AS, Matteoli G, Bohn E, Autenrieth IB, Karhausen J, et al. Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology. 2008;134:756–767. doi: 10.1053/j.gastro.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012;15:813–826. doi: 10.1016/j.cmet.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, Shipley JM, Senior RM, Shibuya M. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. 2002;2:289–300. doi: 10.1016/s1535-6108(02)00153-8. [DOI] [PubMed] [Google Scholar]
- Holmquist-Mengelbier L, Fredlund E, Löfstedt T, Noguera R, Navarro S, Nilsson H, Pietras A, Vallon-Christersson J, Borg A, Gradin K, et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell. 2006;10:413–423. doi: 10.1016/j.ccr.2006.08.026. [DOI] [PubMed] [Google Scholar]
- Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikejiri A, Nagai S, Goda N, Kurebayashi Y, Osada-Oka M, Takubo K, Suda T, Koyasu S. Dynamic regulation of Th17 differentiation by oxygen concentrations. Int Immunol. 2012;24:137–146. doi: 10.1093/intimm/dxr111. [DOI] [PubMed] [Google Scholar]
- Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, Hammond R, Gimotty PA, Keith B, Simon MC. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest. 2010;120:2699–2714. doi: 10.1172/JCI39506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, Volke M, Gläsner J, Warnecke C, Wiesener MS, et al. Hypoxia and hypoxiainducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. 2008;180:4697–4705. doi: 10.4049/jimmunol.180.7.4697. [DOI] [PubMed] [Google Scholar]
- Jantsch J, Wiese M, Schödel J, Castiglione K, Gläsner J, Kolbe S, Mole D, Schleicher U, Eckardt KU, Hensel M, et al. Toll-like receptor activation and hypoxia use distinct signaling pathways to stabilize hypoxia-inducible factor 1a (HIF1A) and result in differential HIF1A-dependent gene expression. J Leukoc Biol. 2011;90:551–562. doi: 10.1189/jlb.1210683. [DOI] [PubMed] [Google Scholar]
- Keith B, Johnson RS, Simon MC. HIF1a and HIF2a: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, Gu H, Nomura S, Caldwell CC, Kobata T, Carmeliet P, Semenza GL, Sitkovsky MV. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1alpha-deficient chimeric mice. Proc Natl Acad Sci USA. 2002;99:2170–2174. doi: 10.1073/pnas.052706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Seo SY, Yoshizaki T, Wakisaka N, Furukawa M, Joab I, Jang KL, Pagano JS. EBV latent membrane protein 1 up-regulates hypoxia-inducible factor 1alpha through Siah1-mediated down-regulation of prolyl hydroxylases 1 and 3 in nasopharyngeal epithelial cells. Cancer Res. 2006;66:9870–9877. doi: 10.1158/0008-5472.CAN-06-1679. [DOI] [PubMed] [Google Scholar]
- Konisti S, Kiriakidis S, Paleolog EM. Hypoxia—a key regulator of angiogenesis and inflammation in rheumatoid arthritis. Nat Rev Rheumatol. 2012;8:153–162. doi: 10.1038/nrrheum.2011.205. [DOI] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, Movahedi K, Houbracken I, Schouppe E, Elkrim Y, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014;74:24–30. doi: 10.1158/0008-5472.CAN-13-1196. [DOI] [PubMed] [Google Scholar]
- Leek RD, Hunt NC, Landers RJ, Lewis CE, Royds JA, Harris AL. Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. J Pathol. 2000;190:430–436. doi: 10.1002/(SICI)1096-9896(200003)190:4<430::AID-PATH538>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Lewis C, Murdoch C. Macrophage responses to hypoxia: implications for tumor progression and anti-cancer therapies. Am J Pathol. 2005;167:627–635. doi: 10.1016/S0002-9440(10)62038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CE, De Palma M, Naldini L. Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res. 2007;67:8429–8432. doi: 10.1158/0008-5472.CAN-07-1684. [DOI] [PubMed] [Google Scholar]
- Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- Lin EY, Li JF, Bricard G, Wang W, Deng Y, Sellers R, Porcelli SA, Pollard JW. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol Oncol. 2007;1:288–302. doi: 10.1016/j.molonc.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Wan S, Sun L, Hu J, Fang D, Zhao R, Yuan S, Zhang L. Chemokine C-C motif receptor 5 and C-C motif ligand 5 promote cancer cell migration under hypoxia. Cancer Sci. 2012;103:904–912. doi: 10.1111/j.1349-7006.2012.02259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140:845–858. doi: 10.1016/j.cell.2010.02.021. [DOI] [PubMed] [Google Scholar]
- Mancino A, Schioppa T, Larghi P, Pasqualini F, Nebuloni M, Chen IH, Sozzani S, Austyn JM, Mantovani A, Sica A. Divergent effects of hypoxia on dendritic cell functions. Blood. 2008;112:3723–3734. doi: 10.1182/blood-2008-02-142091. [DOI] [PubMed] [Google Scholar]
- Masson N, Singleton RS, Sekirnik R, Trudgian DC, Ambrose LJ, Miranda MX, Tian YM, Kessler BM, Schofield CJ, Ratcliffe PJ. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 2012;13:251–257. doi: 10.1038/embor.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamee EN, Korns Johnson D, Homann D, Clambey ET. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol Res. 2013;55:58–70. doi: 10.1007/s12026-012-8349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecklenburgh KI, Walmsley SR, Cowburn AS, Wiesener M, Reed BJ, Upton PD, Deighton J, Greening AP, Chilvers ER. Involvement of a ferroprotein sensor in hypoxia-mediated inhibition of neutrophil apoptosis. Blood. 2002;100:3008–3016. doi: 10.1182/blood-2002-02-0454. [DOI] [PubMed] [Google Scholar]
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J, Mack M, Pipeleers D, In’t Veld P, De Baetselier P, Van Ginderachter JA. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–2234. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Makino Y, Okamoto K, Poellinger L, Ohnuma K, Morimoto C, Tanaka H. TCR engagement increases hypoxia-inducible factor-1 alpha protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J Immunol. 2005;174:7592–7599. doi: 10.4049/jimmunol.174.12.7592. [DOI] [PubMed] [Google Scholar]
- Noman MZ, Buart S, Van Pelt J, Richon C, Hasmim M, Leleu N, Suchorska WM, Jalil A, Lecluse Y, El Hage F, et al. The cooperative induction of hypoxiainducible factor-1 alpha and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. J Immunol. 2009;182:3510–3521. doi: 10.4049/jimmunol.0800854. [DOI] [PubMed] [Google Scholar]
- Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, Buart S, Berchem G, Romero P, Mami-Chouaib F, Chouaib S. Blocking hypoxiainduced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011;71:5976–5986. doi: 10.1158/0008-5472.CAN-11-1094. [DOI] [PubMed] [Google Scholar]
- Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1a, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura CYM, Hollands A, Tran DN, Olson J, Dahesh S, von Kockritz-Blickwede M, Thienphrapa W, Corle C, Jeung SN, Kotsakis A, et al. A new pharmacological agent (AKB-4924) stabilizes hypoxia inducible factor-1 (HIF-1) and increases skin innate defenses against bacterial infection. J Mol Med. 2012;90:1079–1089. doi: 10.1007/s00109-012-0882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med. 2009;13(9A):2780–2786. doi: 10.1111/j.1582-4934.2009.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazon A, Teijeira A, Martınez-Forero I, Hervas-Stubbs S, Roncal C, Penuelas I, Dubrot J, Morales-Kastresana A, Pérez-Gracia JL, Ochoa MC, et al. Agonist anti-CD137 mAb act on tumor endothelial cells to enhance recruitment of activated T lymphocytes. Cancer Res. 2011;71:801–811. doi: 10.1158/0008-5472.CAN-10-1733. [DOI] [PubMed] [Google Scholar]
- Palazon A, Martınez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF, Perez-Gracia JL, Penuelas I, Hervas-Stubbs S, Rouzaut A, et al. The HIF-1a hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discov. 2012;2:608–623. doi: 10.1158/2159-8290.CD-11-0314. [DOI] [PubMed] [Google Scholar]
- Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piña-Oviedo S, Khalili K, Del Valle L. Hypoxia inducible factor-1 alpha activation of the JCV promoter: role in the pathogenesis of progressive multifocal leukoencephalopathy. Acta Neuropathol. 2009;118:235–247. doi: 10.1007/s00401-009-0533-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Yang MX, Kong BH, Qi L, Lam QL, Yan S, Li P, Zhang M, Lu L. Hypoxia inhibits the migratory capacity of human monocyte-derived dendritic cells. Immunol Cell Biol. 2005;83:668–673. doi: 10.1111/j.1440-1711.2005.01383.x. [DOI] [PubMed] [Google Scholar]
- Rapisarda A, Melillo G. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nat Rev Clin Oncol. 2012;9:378–390. doi: 10.1038/nrclinonc.2012.64. [DOI] [PubMed] [Google Scholar]
- Ricciardi A, Elia AR, Cappello P, Puppo M, Vanni C, Fardin P, Eva A, Munroe D, Wu X, Giovarelli M, Varesio L. Transcriptome of hypoxic immature dendritic cells: modulation of chemokine/receptor expression. Mol Cancer Res. 2008;6:175–185. doi: 10.1158/1541-7786.MCR-07-0391. [DOI] [PubMed] [Google Scholar]
- Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134:145–155. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roda JM, Sumner LA, Evans R, Phillips GS, Marsh CB, Eubank TD. Hypoxia-inducible factor-2a regulates GM-CSF-derived soluble vascular endothelial growth factor receptor 1 production from macrophages and inhibits tumor growth and angiogenesis. J Immunol. 2011;187:1970–1976. doi: 10.4049/jimmunol.1100841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roda JM, Wang Y, Sumner LA, Phillips GS, Marsh CB, Eubank TD. Stabilization of HIF-2a induces sVEGFR-1 production from tumor-associated macrophages and decreases tumor growth in a murine melanoma model. J Immunol. 2012;189:3168–3177. doi: 10.4049/jimmunol.1103817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198:1391–1402. doi: 10.1084/jem.20030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. 2011;365:537–547. doi: 10.1056/NEJMra1011165. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014;9:47–71. doi: 10.1146/annurev-pathol-012513-104720. [DOI] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38:225–236. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatrov VA, Sumbayev VV, Zhou J, Brüne B. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1alpha (HIF-1alpha) accumulation via redox-dependent mechanisms. Blood. 2003;101:4847–4849. doi: 10.1182/blood-2002-09-2711. [DOI] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol. 2005;5:712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, Cheresh DA, Johnson RS. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456:814–818. doi: 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A, Johnson RS. Differential activation and antagonistic function of HIF-alpha isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24:491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157:411–421. doi: 10.1016/s0002-9440(10)64554-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AA, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, Williams L, Parmar S, Shaw G, McGrath EE, et al. Hypoxia-inducible factor 2a regulates key neutrophil functions in humans, mice, and zebrafish. Blood. 2014;123:366–376. doi: 10.1182/blood-2013-05-500207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH, Pagés F, Galon J. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011;71:1263–1271. doi: 10.1158/0008-5472.CAN-10-2907. [DOI] [PubMed] [Google Scholar]
- Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201:105–115. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley SR, Cowburn AS, Clatworthy MR, Morrell NW, Roper EC, Singleton V, Maxwell P, Whyte MK, Chilvers ER. Neutrophils from patients with heterozygous germline mutations in the von Hippel Lindau protein (pVHL) display delayed apoptosis and enhanced bacterial phagocytosis. Blood. 2006;108:3176–3178. doi: 10.1182/blood-2006-04-018796. [DOI] [PubMed] [Google Scholar]
- Walmsley SR, Chilvers ER, Thompson AA, Vaughan K, Marriott HM, Parker LC, Shaw G, Parmar S, Schneider M, Sabroe I, et al. Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J Clin Invest. 2011;121:1053–1063. doi: 10.1172/JCI43273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Flach H, Onizawa M, Wei L, McManus MT, Weiss A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat Immunol. 2014;15:393–401. doi: 10.1038/ni.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke C, Weidemann A, Volke M, Schietke R, Wu X, Knaup KX, Hackenbeck T, Bernhardt W, Willam C, Eckardt KU, Wiesener MS. The specific contribution of hypoxia-inducible factor-2alpha to hypoxic gene expression in vitro is limited and modulated by cell type-specific and exogenous factors. Exp Cell Res. 2008;314:2016–2027. doi: 10.1016/j.yexcr.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- Wong CC, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, Fraley SI, Wong CM, Khoo US, Ng IO, et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci USA. 2011;108:16369–16374. doi: 10.1073/pnas.1113483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo YG, Oh SH, Park ES, Cho H, Lee N, Park H, Kim DK, Yu DY, Seong JK, Lee MO. Hepatitis B virus X protein enhances transcriptional activity of hypoxia-inducible factor-1alpha through activation of mitogen-activated protein kinase pathway. J Biol Chem. 2003;278:39076–39084. doi: 10.1074/jbc.M305101200. [DOI] [PubMed] [Google Scholar]
- Zhao W, Darmanin S, Fu Q, Chen J, Cui H, Wang J, Okada F, Hamada J, Hattori Y, Kondo T, et al. Hypoxia suppresses the production of matrix metalloproteinases and the migration of human monocyte-derived dendritic cells. Eur J Immunol. 2005;35:3468–3477. doi: 10.1002/eji.200526262. [DOI] [PubMed] [Google Scholar]