

Abstract
Background
PPHN is associated with decreased lung angiogenesis and impaired pulmonary vasodilatation at birth. Prostanoids are important modulators of vascular tone and angiogenesis. We hypothesized that altered levels of prostacyclin (PGI2), a potent vasodilator, and thromboxane (TXA2), a vasoconstrictor, contribute to impaired angiogenesis of pulmonary artery endothelial cells (PAEC) in PPHN.
Methods
PAEC were isolated from fetal lambs with PPHN induced by prenatal ductus arteriosus constriction or sham operated controls. Expression and activity of PGI2 synthase (PGIS) and TXA2 synthase (TXAS), expression of cyclooxygenases 1 and 2 (COX-1 and COX-2) and the role of PGIS/TXAS alterations in angiogenesis were investigated in PAEC from PPHN and control lambs.
Results
PGIS protein and activity were decreased and PGIS protein tyrosine nitration was increased in PPHN PAEC. In contrast, TXAS protein and its stimulated activity were increased in PPHN PAEC. COX-1 and COX-2 proteins were decreased in PPHN PAEC. Addition of PGI2 improved in vitro tube formation by PPHN PAEC, whereas, indomethacin decreased tube formation by control PAEC. PGIS knockdown decreased the in vitro angiogenesis in control PAEC, whereas, TXAS knockdown increased the in vitro angiogenesis in PPHN PAEC.
Conclusion
Reciprocal alterations in PGI2 and TXA2 may contribute to impaired angiogenesis in PPHN.
INTRODUCTION
Persistent pulmonary hypertension of the newborn (PPHN) represents a failure of the normal postnatal adaptation that occurs at birth in pulmonary circulation. It is characterized by decreased blood vessel density in the lungs (1) and impaired pulmonary vasodilatation at birth, both of which lead to postnatal persistence of high pulmonary vascular resistance (PVR). The increased PVR can result from a decrease in vasodilator signals, or increase in vasoconstrictor signals by pulmonary artery endothelial cells (PAEC) in PPHN. Nitric oxide (NO) and prostacyclin (PGI2) are two key mediators involved in pulmonary vasodilatation at birth (2–4). Although alterations in NO-cGMP system have been extensively studied in PPHN, the role of altered prostanoid signaling in PPHN remains unclear. Inhaled NO therapy has improved the outcomes in PPHN; however, some neonates do not respond to this therapy (5). Impaired vascular growth in the lung may contribute to this failure of response to NO (6).
PGI2 is a prostanoid synthesized from arachidonic acid through cyclooxygenase (COX) - PGI2 synthase (PGIS) pathway. PGH2, the catalytic end product of COX activity and a vasoconstrictor itself, is further metabolized by PGIS to PGI2. PGI2 is synthesized primarily in vascular cells, especially in the vascular endothelium (7). PGI2 causes vasodilatation by activating adenylate cyclase in the vascular smooth muscle cells via a G protein coupled receptor, which increases cAMP synthesis. A surge in PGI2 in pulmonary circulation during perinatal transition contributes to pulmonary vasodilatation at birth (3). Regulation of PGIS, which directs the synthesis of PGI2, during fetal life and its alterations in PPHN remain unclear. PGI2 also modulates blood vessel formation (8) and decreases in PGI2 levels may lead to impaired angiogenesis in PPHN. However, the role of PGI2 as a mediator of angiogenesis during perinatal transition remains unexplored. Thromboxane A2 (TXA2), another arachidonic acid metabolite generated from PGH2 by thromboxane synthase (TXAS), is a potent pulmonary vasoconstrictor (9), particularly during hypoxia. TXA2 is believed to promote angiogenesis during inflammation but its effect on angiogenesis in developing lungs is also unknown. An imbalance between PGI2 and TXA2 may be involved in the pathogenesis of PPHN. Previous studies have shown that impaired PGI2 signaling leads to impaired vasodilation in the ductal constriction model of PPHN (10). However, the role of altered prostaglandin signaling in impaired angiogenesis in PPHN has not been previously investigated. Oxidative stress impairs vasodilatation and angiogenesis (11, 12) in PPHN and may modulate the release of prostanoids in PPHN (13, 14).
We hypothesized that PPHN is associated with an altered balance of PGI2 and TXA2, which in turn leads to impaired angiogenesis. We investigated our hypothesis in the fetal lamb model of PPHN induced by intrauterine ductal constriction.
RESULTS
PGI2 and PGIS alterations in PPHN
Basal levels of 6-Keto-PGF1α, a stable metabolite of PGI2 were decreased by 4-fold in pulmonary artery endothelial cells (PAEC) from PPHN lambs when compared to control cells (Figure IA, n= 12, p<0.05). ATP-stimulated 6-Keto-PGF1α (PGI2 metabolite) levels were also significantly lower in PPHN than control cells. PGIS protein levels were decreased by 2.5-fold in PPHN compared to control PAEC (Figure 1B, n=14, p<0.05). PGIS mRNA levels were not different between control and PPHN cells (Figure 1C, n=7, p=0.12). Tyrosine nitration of PGIS was increased in PPHN cells by 2-fold compared to control PAEC (Figure 1D, n=6, p<0.05).
Figure 1.
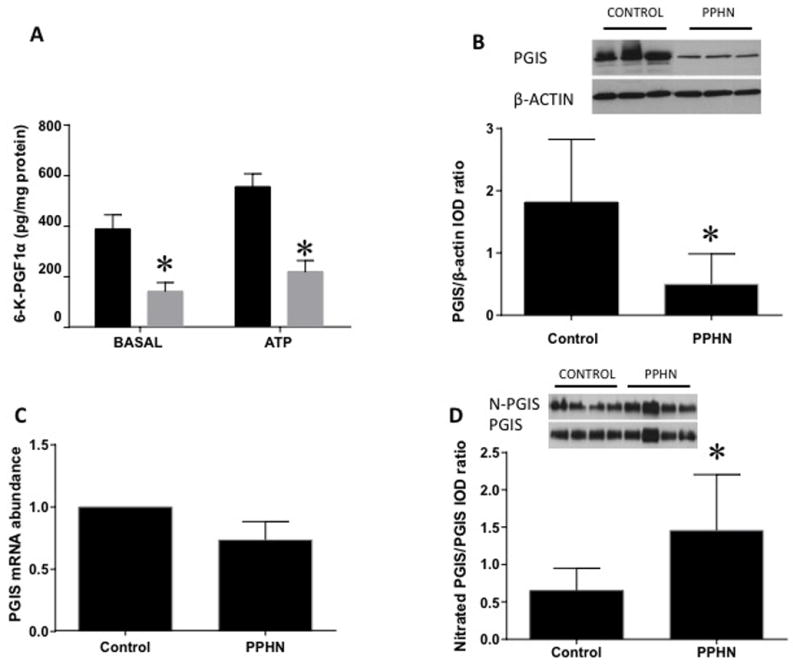
Alterations in prostacyclin synthase (PGIS) protein levels and activity in pulmonary artery endothelial cells (PAEC) in persistent pulmonary hypertension of the newborn (PPHN). A: Levels of 6-Keto-PGF1α, a stable metabolite of PGI2, were decreased in PPHN (gray bar) at basal level and after ATP stimulation (n=12, p<0.05 from control PAEC) compared to control cells (filled bar). B:PGIS protein levels were decreased in PPHN (n=14, p<0.05). Representative western blots were shown with β-actin used as loading control. C:PGIS mRNA levels were not different between PPHN and control PAEC (n=7, p=0.12). D: PGIS nitration (N-PGIS) was higher in PPHN than control PAEC. (n=6, p<0.05).*Indicates p<0.05 compared to control PAEC.
TXA2 levels and TXAS expression in PPHN
ATP-stimulated levels of TXB2, a stable metabolite of TXA2 were increased by two-fold in PAEC from PPHN lambs compared to control PAEC (Figure 2A, n=11, p<0.05), although basal levels of TXB2 were not different (data not shown). TXAS protein levels were increased in PPHN cells compared to controls (Figure 2B, n=14, p<0.05). TXAS mRNA abundance was significantly decreased in PAEC from PPHN lambs compared to control lambs (Figure 2C, n=9, p<0.05).
Figure 2.
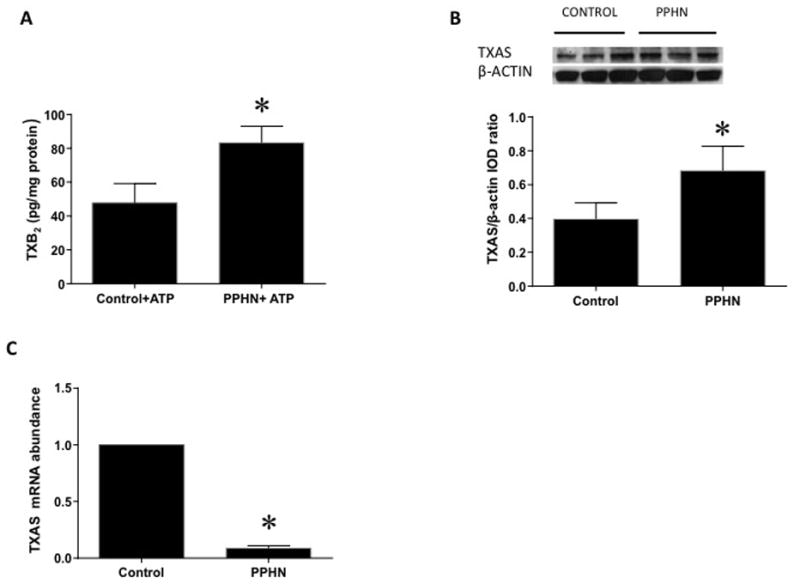
Thromboxane B2 (TXB2, stable metabolite of TXA2) levels were increased with ATP stimulation in PPHN cells (A, n=11, p<0.05 from control PAEC). Thromboxane synthase (TXAS) protein levels were increased in PPHN cells (B, n=14, p<0.05). In contrast, TXAS mRNA levels were decreased in PPHN cells (C, n=11, p<0.05).*p<0.05 compared to control.
COX-1 and COX-2 expression in PPHN
COX-1 protein levels were decreased in PPHN compared to control PAEC (Figure 3A, n=11, p<0.05). COX-1 mRNA levels were not different between PPHN and control PAEC (Figure 3C, n=8, p=0.26). COX-2 protein levels were decreased in PPHN compared to control PAEC (Figure 3B, n=11, p<0.05). COX-2 mRNA levels were also not different between PPHN and control PAEC (Figure 3D, n=8, p=0.67).
Figure 3.
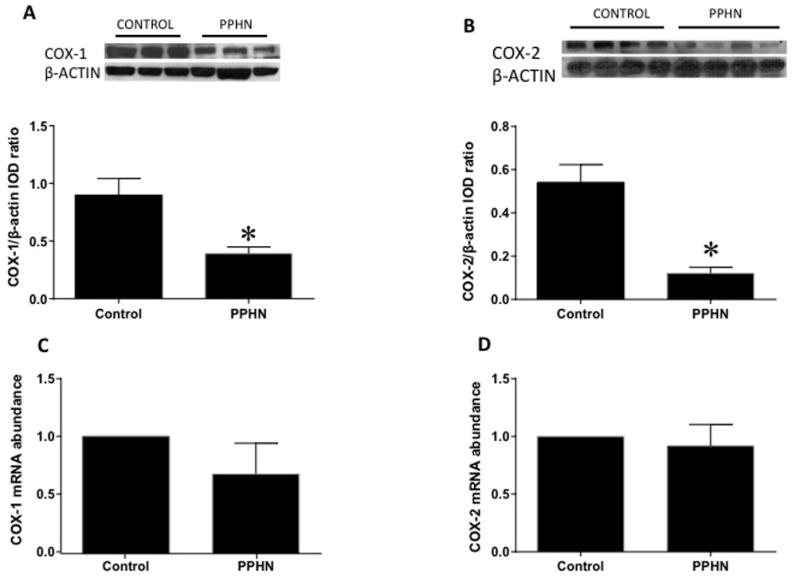
Cyclooxygenase-1 (COX-1, panel A) and Cyclooxygenase-2 (COX-2, panel B) protein levels were decreased in PAEC from PPHN lambs (A, n=11, p<0.05). COX-1 (panel C) and COX-2 (panel D) mRNA levels were not different between PPHN and control PAEC (n=8, p>0.05).*p<0.05 compared to control.
In vitro angiogenesis in PPHN
Total tube length was decreased in PPHN compared to control PAEC (Figure 4A, B), as we reported previously (12). When indomethacin was added to control PAEC, the tube length and branch point number were decreased (Figure 4B&C). Exogenous PGI2 improved tube formation in indomethacin treated cells (Figure 4A–C). PGI2 also restored tube length and branch point number in PAEC from PPHN lambs to the same level as control PAEC (Figure 4B&C, n=4, p< 0.05).
Figure 4.

A: Representative photomicrograph of indomethacin (IND) and PGI2 effects on in vitro tube formation by PAEC from control and PPHN lambs. Tube length (B) and branch point number (C) were decreased in PPHN cells compared to control PAEC. Indomethacin decreases tube length (A and B) and branchpoint number in control PAEC (A and C). Decreased tube length (A&B) and branch point number (A&C) in PPHN was improved by adding PGI2 (100 pg/ml) to the medium.* p< 0.05 from untreated control PAEC and † from untreated PPHN cells. Open bar, untreated cells; filled bar, indomethacin treated and crosshatch, PGI2 treated cells. Scale bar=200 μ.
Effect of PGIS Knockdown on in vitro angiogenesis
Tube length decreased by four-fold and branch point number decreased by eight-fold when PGIS expression was knocked down by siRNA in control PAEC when compared to non-silencing RNA (NS-siRNA) treated PAEC (Figure 5A, n=6, p<0.05,). The gap in PAEC with PGIS knockdown,6 h after a scratch was 562.5±17.3 μm compared to 347.5±9.5 μm for NS-siRNA cells, indicating slower recovery from injury (Figure 5B, n=10, p<0.05). On cell invasion assay, 74.8±6.4 of PGIS knockdown cells invaded the membrane as compared to 134±6.4 in NS-siRNA treated PAEC under high power field (5C, n=4, p<0.05).
Figure 5.
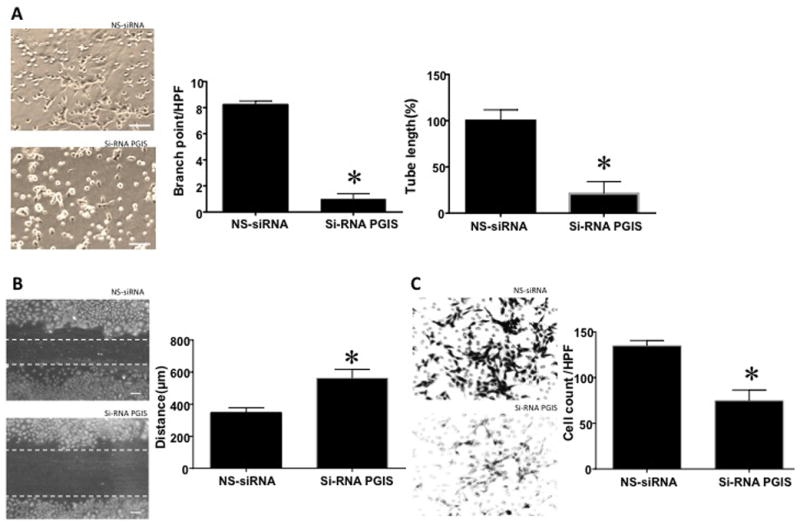
A: PGIS knockdown with si-RNA decreases in vitro tube formation by control PAEC. Branch point number (n=4, p<0.05) and tube length (n=6, p<0.05) were decreased in control PAEC after application of PGIS-siRNA, compared to non-silencing (NS)-siRNA. Scale bar = 200 μ. B: PGIS si-RNA increased the gap in monolayer of control PAEC after a scratch, indicating longer recovery from injury compared to NS-RNA (n=10, p<0.05). Scale bar=100 μ. C: PGIS si-RNA also decreased the number of control cells able to invade through the Boyden chamber as compared to NS-siRNA treated control cells on cell invasion assay (n=4, p<0.05). For all studies, * indicates p<0.05 compared to NS-siRNA treated PAEC.
Effect of TXAS Knockdown on angiogenesis
Tube length was increased when TXAS was knocked down in control and PPHN cells as compared to NS- siRNA treated control and PPHN cells (6A, C, D, n=10, p<0.05). Branch point number was increased in PPHN cells after TXAS knockdown (6D, n=10, p<0.05). The gap in TXAS knockdown PPHN cells (6B and D) and control cells (6D) after 6 h was significantly less on scratch recovery test, indicating that TXAS knockdown has led to faster recovery from injury (n=20, p<0.05).
DISCUSSION
We observed that PPHN is associated with decreased PGIS expression and activity in the PAEC. We also observed a reciprocal increase in the TXAS expression and activity, which suggests a shift of prostanoid metabolism in pulmonary vascular endothelium towards the release of vasoconstrictors over vasodilators in PPHN. Impaired angiogenesis in PPHN may be a downstream effect of decreased PGIS function; exogenous PGI2 restored angiogenesis in PPHN and knockdown of PGIS in normal cells impaired angiogenesis. Similarly, knockdown of TXAS improved angiogenesis in PPHN.
Several previous studies have mechanistically linked high PVR in PPHN to impaired vasodilation and to vascular remodeling (15, 16). However, PPHN is also associated with reduced blood vessel density in the lungs (17). Chronic intrauterine pulmonary hypertension impairs lung angiogenesis and alveolar development leading to lung hypoplasia (17). Impairment of angiogenesis can also contribute to increased PVR and potentially account for failure of response to vasodilators, such as inhaled NO (6). Therefore, investigation of the mechanisms involved in the impaired angiogenesis in PPHN is critical to improve our current understanding and management of this condition.
Studies of PAEC isolated from fetal lambs with prenatal ductal constriction demonstrated that a decrease in NO and VEGF signaling causes impaired in vitro angiogenesis function (1). The role of altered prostaglandin system in the impaired angiogenesis function of PAEC in PPHN, however, remains unclear and motivated our current studies. We observed that the endothelial dysfunction in PPHN includes an alteration in PGI2-TXA2 signaling in the PAEC and contributes to impaired angiogenesis. PGI2 induces angiogenesis in vivo, via activation of the PPAR-δ signaling pathway and up regulation of local VEGF formation (7, 18, 19). Our results support a role for PGI2 in the induction of angiogenesis in fetal PAEC. The specific role of TXA2, a vasoconstrictor, in the regulation of angiogenesis function remains unclear with conflicting reports based on the specific vascular bed and condition studied. TXA2 has been reported to mediate angiogenesis function in the development of tumor metastasis (20). However, TXA2 receptor activation can also inhibit endothelial cell migration (21). Our study suggests that TXA2 decreases angiogenesis in fetal PAEC in vitro and may contribute to impaired angiogenesis in PAEC from PPHN lambs.
PGI2 is primarily produced by the vascular endothelium (22, 23) and contributes to both vasodilation and angiogenesis (8). PGI2 has been identified as a potent pulmonary vasodilator in the fetus and the newborn and its synthesis is developmentally regulated (24, 25). Fike et al have previously shown that PGI2 levels and PGIS abundance were decreased in piglets with hypoxia induced pulmonary hypertension (26), consistent with our observations in the fetal lamb model of PPHN induced by prenatal ductal constriction. We selected this fetal lamb model since previous studies demonstrated that it closely simulates PPHN observed in newborn infants (15, 16). Our studies suggest that alterations in prostaglandin system are an important component of the impaired angiogenesis observed in this model. Although the decrease in PGI2 levels in PPHN PAEC was consistent with the decrease in PGIS protein levels, PGIS mRNA did not decrease, suggesting post-translational regulation of PGIS protein in PPHN.
PPHN is characterized by the presence of increased oxidative stress in PAEC, which can potentially impair angiogenesis (1, 12, 27). Reaction between superoxide radical and NO generates peroxynitrite, which can cause tyrosine nitration of proteins (28, 29). We investigated tyrosine nitration of PGIS, a post-translational modification that can impair PGIS function, as a potential explanation for the decreased PGI2 levels. PGIS contains tyrosine residue 430, which when nitrated, decreases the catalytic reaction of the heme –thiolate center of PGIS. Tyrosine nitration of PGIS has been previously shown to reduce its activity (13, 14). Increased tyrosine nitration of PGIS in our PPHN cells may also contribute to a decrease in PGI2 levels, leading to endothelial dysfunction and impaired angiogenesis in PPHN. Previous studies have also shown that PGIS nitration in vitro leads to an increase in the levels of PGH2, an intermediate product in the COX pathway, which can be a vasoconstrictor by itself or be a substrate for TXA2 (30, 31). Thus, decreased utilization of PGH2 by PGIS can lead to increased synthesis of TXA2, consistent with our observation in this study. We did not measure the levels of PGH2 since it is an unstable transient compound.
Levels of TXA2, a potent vasoconstrictor prostanoid, were elevated in our PPHN cells. In the pulmonary arteries of newborn pigs, chronic hypoxia did not alter the TXA2 formation (26, 30, 31). These data point to differences in the mechanism of pulmonary hypertension in the 2 models. Although we did not observe differences in the basal TXA2 levels, the ATP-stimulated levels were significantly increased in PPHN PAEC. This alteration may be relevant during birth related transition, which is associated with a surge in ATP levels in pulmonary circulation (32). Although TXAS protein levels were significantly increased in PPHN cells, we observed a decrease in TXAS mRNA levels in PPHN cells. This may be an adaptive response to increased TXAS protein levels in PPHN and suggest post-translational modifications of TXAS in PPHN.
Our investigation of upstream enzymes in PG pathway was motivated by previous studies showing increased risk of the babies for PPHN following intrauterine exposure to non-steroidal anti-inflammatory drugs (NSAIDs) during late gestation (33). One of the critical rate-limiting steps in prostanoid production is the conversion of arachidonic acid by COX to prostaglandin G2 (PGG2) and subsequent peroxidation of PGG2 to PGH2 by the same enzyme. COX-1 is a constitutive enzyme whereas COX-2 is inducible and may be present only under inflammatory conditions, induction being under transcription control (34). Fike et al noted increased abundance in COX-1 and no change in COX-2 in hypoxic neonatal pigs (26). We observed decreased expression of both COX-1 and COX-2 in PPHN cells. The difference in our results may also be due to differences in the animal models used or changes specific to PAEC. The limitation of our study is that our observations were made in isolated PAEC and the alterations in PGIS/TXAS signaling may not be directly extrapolated to in vivo lung alterations in infants with PPHN. We also limited our studies to the role of altered PGIS/TXAS signaling in the angiogenesis function of PAEC and did not investigate its role in the vasodilator response of pulmonary arteries.
In conclusion, PPHN is associated with decreased PGIS expression and function and increased TXAS expression and function in PAEC. These reciprocal alterations could potentially explain the decreased angiogenesis previously noted in PPHN. Whether PGI2 restores vasodilatation by improving angiogenesis in neonates with PPHN requires further investigation.
METHODS
Creation of PPHN model
PPHN was induced by prenatal constriction of ductus arteriosus in the fetal lambs from 128 to 136 d gestation, per methods described previously (16, 35). Control lambs had thoracotomy and exposure of DA, which was left undisturbed. The study protocol was approved by the Institutional Animal Care and Use Committee of Medical College of Wisconsin. Fetal lungs were harvested at 136 d (term = 140 d) for the isolation of pulmonary arteries and PAEC. Studies were done in PAEC.
Antibodies and Chemicals
Rabbit anti PGIS (1:500), anti-COX-2 (1:250) and anti-TXAS antibodies (1:250), mouse anti-COX-1 Antibody (1:250), 6-keto prostaglandin F1α EIA and thromboxane B2 EIA Kits were from Cayman Chemical Company (Ann Arbor, MI). PGIS siRNA (bovine), TXAS siRNA (bovine), non-silencing siRNA (NS-siRNA) and Immunocruz IP/WB Optima F System were obtained from Santa Cruz biotechnology, Inc. (Santa Cruz, CA). Growth factor-reduced Matrigel was from BD Biosciences (Bedford, MA). Cell invasion assay kit was obtained from Chemicon International (Temecula, CA). PGIS, TXAS, COX-1 and COX-2 primers were obtained from Invitrogen (Carlsbad, CA). Mouse β-actin (1:1,000) and polyclonal nitrotyrosine antibody (Clone 409) and other chemicals were obtained from Sigma Aldrich (St. Louis, MO). Polyclonal PGI2 synthase antibody for IP was obtained from ProSci Inc. (San Diego, CA). Horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit IgG antibodies (1:10,000) were obtained from Bio-Rad (Hercules, CA).
Isolation and culture of endothelial cells
PAEC were isolated with 0.1% collagenase type A (Roche Molecular Biochemical, Indianapolis, IN) and characterized using techniques described previously (35). Cells for individual experiments were grown to confluence in 100 mm plates for immunoprecipitation, 60 mm plates for western blots, 24-well plates for angiogenesis activity assays, 6 well plates for mRNA extraction or transfection and 24- well plates for PGI2 and TXA2 assays. We studied PAEC from control and PPHN lambs between passages 4–6.
Western blot analysis for proteins
PAEC were grown to confluence and cell lysates were prepared in modified RIPA or MOPS buffer. Protein content was determined by bicinchoninic acid (BCA) method. A 20–45 μg aliquot of protein was resolved by 10% SDS-PAGE. Separated proteins were transferred to nitrocellulose membrane and then blotted with specific antibodies overnight at 4° C after blocking with 5% skim milk in TBS with 0.1% Tween 20 (TBS-T). Membranes were then blotted with HRP – conjugated anti-mouse or anti-rabbit IgG antibodies. Enhanced chemiluminescence agents (ECL) were used to visualize the bands. Relative band densities (IOD) were then quantified using Image J (NIH). Readings obtained were normalized to corresponding β-actin signal, used as the loading control.
Co-immunoprecipitation studies (35) for PGIS nitration
PAEC at 90% confluence in 100 mm round dishes were treated with HBSS for 10 min. The supernatant was then aspirated and cells were lysed in RIPA buffer. Samples were sonicated and removal of cell debris was done by centrifugation. PGIS was immunoprecipitated from a 500 μg protein aliquot using a specific antibody (35). The immunoprecipitated proteins were separated by SDS-PAGE and transferred to nitrocellulose membrane. Membranes were blocked with 5% skim milk in TBS-T and then immunoblotted for PGIS and for nitrotyrosine with specific antibodies. Bands were visualized and quantified as described above. IOD ratio of nitrotyrosine to PGIS was calculated to assess the relative levels of protein nitration.
Measurement of PGIS and TXAS activity
Concentrations of 6-keto-PGF1-α, the stable metabolite of PGI2 and TXB2, the stable metabolite of TXA2, were determined in the supernatants of confluent control and PPHN cells by EIA using a commercially available kit (Cayman Chemical). Basal levels of 6-keto-PGF1-α and TXB2 were assessed from supernatant collected over 2 h from the cells kept in each well of 24-well plates. 6-keto-PGF1-α and TXB2 release in response to the physiologic agonist, ATP (10−5) was studied after 15 min exposure, followed by the collection of supernatant. Levels of 6-keto-PGF1-α and TXB2 were normalized to protein concentration in each well and expressed as picograms/milligram of protein.
Quantification of mRNA abundance
PAEC were plated in 6-well plates and grown to confluence. RNA extraction was done using RNAeasy mini kit from Qiagen (Germantown, MD). Complementary DNA was synthesized from the extracted RNA using the iScript cDNA synthesis kit (Bio-Rad). The PCR primers were designed using Primer3 and primer sequences are provided online through protocol exchange. Real time RT-PCR was performed using the iQ5 multicolor real-time PCR detection system (Bio-Rad). The PCR cycle started at 95°C for 3 min followed by 40 cycles of 95°C for 15 s and 58°C for 30s. Melting temperatures were monitored for each pair of primers, and single-peak melting temperature was observed for all of the primer pairs. Number of the threshold cycle (Ct) for each target mRNA was corrected against the corresponding Ct of β-actin mRNA. 2−ΔΔCt was then shown as fold-change in mRNA abundance with each treatment.
PGIS/TXAS knockdown
Knockdown of PGIS and TXAS in control PAEC was accomplished with specific siRNAs (bovine siRNA, sc-270412 and sc-270480, Santa Cruz Biotechnology) and Lipofectamine RNAiMAX Transfection reagent (Invitrogen) using a protocol from the manufacturer. Transfection efficiency was more than 60% with use of FITC-conjugated NS-RNA in previous experiments in our laboratory. After treatment with PGIS siRNA (30 pmoles), TXAS siRNA (30 pmoles), or NS-siRNA and Lipofectamine RNAiMAX (5 μl) in six-well plates for 20 min, PAEC (1.5 × 105) were incubated for 48 h in a humidified CO2 incubator and were then used for performing immunoblots or angiogenesis studies.
Angiogenesis assays
In vitro tube formation assay
Tube formation assay was performed in vitro using Matrigel (BD Biosciences, San Jose, CA) in 24-well plates (12). After thawing overnight at 4°C, 300 μl of Matrigel was added to designated wells and was incubated for 30 min at 37°C for solidification. PAEC (1.5 × 105) were added on top of the solidified Matrigel. Tube formation by PAEC was monitored over the next 4–6 h (12). One representative picture was taken per well using an inverted microscope at 20 × (objective) magnification (Nikon Eclipse TE2000, Nikon Instruments Inc., Melville, NY). Total tube length was measured in the presence/absence of PGI2 (100 pg/ml) at concentrations that simulate physiologic levels. Indomethacin (10−5 M) was then added to some wells, to inhibit PGI2 release. Data from control and PPHN PAEC were compared. Tube formation was also studied in control PAEC with/without PGIS knockdown and in PPHN cells with/without TXAS knockdown to determine their role in angiogenesis.
Monolayer scratch recovery assay
PAEC with/without PGIS or TXAS knockdown were grown to confluence in 6-well plates. Scratch lines were created by a 1-ml pipette tip in each well, and the wells were gently rinsed with HBSS twice to remove the detached cells. The cells were kept in serum free media for one hour, followed by addition of Dulbecco’s modified eagle medium (DMEM) with 20% FCS. The distances between the frontlines of recovery were measured for comparison 4–6 h later (12).
Cell invasion assay
Matrigel – coated Transwells with 8-μm pores (Chemicon International) were used for the cell invasion assay. 300 μl of serum free DMEM was placed in each insert and incubated at 37°C for 1 h to soak inserts. The serum-free DMEM was replaced with DMEM containing 1 × 105 PAEC with/without PGIS knockdown while DMEM with 20% FCS was applied to the outer chamber. The plate was then incubated for 4–6 h. The outer surface of the insert was stained by the staining solution provided in the kit. PAEC that invaded through the 8-μm pores were counted under the microscope.
STATISTICAL ANALYSES
Data are shown as mean ± SEM. Student’s t-test was used for normally distributed data, and Mann-Whitney U-test was used for data that did not pass the normality test for comparison between two groups. Two-way ANOVA with Student-Newman-Keuls post-hoc test was used when more than two groups were compared. Data were analyzed with Graph Pad PRISM software (Graph pad Inc., La Jolla, CA). A p value <0.05 was considered statistically significant.
Figure 6.
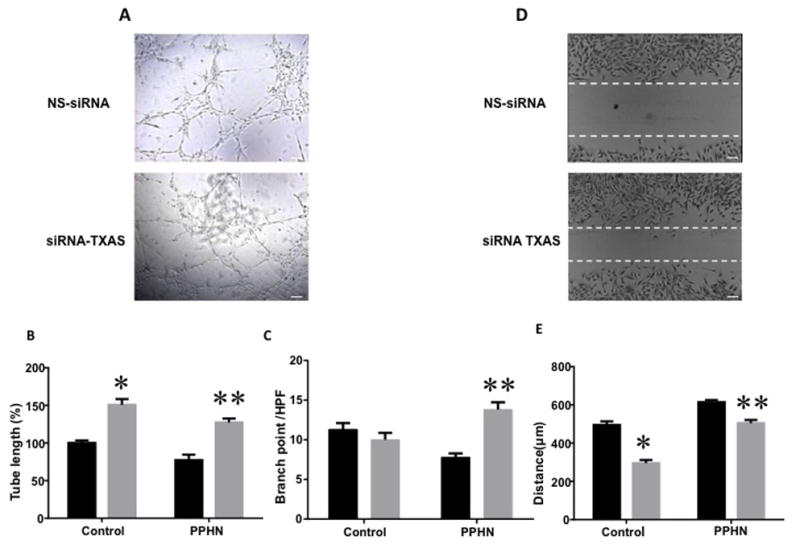
A: TXAS knockdown with si-RNA improves tube formation in PPHN cells. Scale bar = 100 μM. Tube length (B) and branch point number (C) increased significantly after application of TXAS-siRNA, compared to NS-siRNA in both control and PPHN cells (n=10, p<0.05). D, TXAS si-RNA also decreased the gap after a scratch in PAEC from PPHN lambs compared to non-silencing (NS)-siRNA treatment, indicating shorter recovery from injury. Scale bar = 100 μ. TXAS knockdown decreased gap distance significantly in both control and PPHN cells (E, n=20, p<0.05). For all studies, * indicates p<0.05 compared to NS-siRNA treated control cells and ** from NS-siRNA treated PPHN cells. Black bar, NS-siRNA and gray bar, TXAS siRNA.
Acknowledgments
Financial statement:
Our study was supported by National Institutes of Health (Bethesda, MD) grants RO3HD-073274 (RJT), RO3HD-065841 (GGK) and RO1HL-057268 (GGK) and CTSI UL1RR031973 from Advancing Healthier Wisconsin Foundation (Milwaukee, WI) grant (RJT and GGK). GGK was supported by Muma Endowed Chair in Neonatology (Milwaukee, WI).
Footnotes
DISCLOSURE STATEMENT:
Authors do not have any financial ties to products in the study or potential/perceived conflicts of interest.
Contributor Information
Chaitali N. Mahajan, Department of Pediatrics, University of Kansas Medical Center, Kansas City, Kansas, USA
Adeleye J. Afolayan, Department of Pediatrics and Children’s Research Institute, Medical College of Wisconsin, Milwaukee, Wisconsin, USA
Annie Eis, Department of Pediatrics and Children’s Research Institute, Medical College of Wisconsin, Milwaukee, Wisconsin, USA.
Ru-Jeng Teng, Department of Pediatrics and Children’s Research Institute, Medical College of Wisconsin, Milwaukee, Wisconsin, USA.
Girija G. Konduri, Department of Pediatrics and Children’s Research Institute, Medical College of Wisconsin, Milwaukee, Wisconsin, USA.
References
- 1.Gien J, Seedorf GJ, Balasubramaniam V, Markham N, Abman SH. Intrauterine pulmonary hypertension impairs angiogenesis in vitro: role of vascular endothelial growth factor nitric oxide signaling. Am J Respir Crit Care Med. 2007;176:1146–1153. doi: 10.1164/rccm.200705-750OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abman SH, Chatfield BA, Hall SL, McMurtry IF. Role of endothelium-derived relaxing factor during transition of pulmonary circulation at birth. Am J Physiol. 1990;259:H1921–1927. doi: 10.1152/ajpheart.1990.259.6.H1921. [DOI] [PubMed] [Google Scholar]
- 3.Leffler CW, Hessler JR. Perinatal pulmonary prostaglandin production. Am J Physiol. 1981;241:H756–759. doi: 10.1152/ajpheart.1981.241.5.H756. [DOI] [PubMed] [Google Scholar]
- 4.Velvis H, Moore P, Heymann MA. Prostaglandin inhibition prevents the fall in pulmonary vascular resistance as a result of rhythmic distension of the lungs in fetal lambs. Pediatr Res. 1991;30:62–68. doi: 10.1203/00006450-199107000-00013. [DOI] [PubMed] [Google Scholar]
- 5.Neonatal Inhaled Nitric Oxide Study G. Inhaled nitric oxide in full-term and nearly full-term infants with hypoxic respiratory failure. N Engl J Med. 1997;336:597–604. doi: 10.1056/NEJM199702273360901. [DOI] [PubMed] [Google Scholar]
- 6.Goldman AP, Tasker RC, Haworth SG, Sigston PE, Macrae DJ. Four patterns of response to inhaled nitric oxide for persistent pulmonary hypertension of the newborn. Pediatrics. 1996;98:706–713. [PubMed] [Google Scholar]
- 7.Gryglewski RJ, Korbut R, Ocetkiewicz A. Generation of prostacyclin by lungs in vivo and its release into the arterial circulation. Nature. 1978;273:765–767. doi: 10.1038/273765a0. [DOI] [PubMed] [Google Scholar]
- 8.Pola R, Gaetani E, Flex A, et al. Comparative analysis of the in vivo angiogenic properties of stable prostacyclin analogs: a possible role for peroxisome proliferator-activated receptors. J Mol Cell Cardiol. 2004;36:363–370. doi: 10.1016/j.yjmcc.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 9.Saito M, Tatsumi K, Kasahara Y, et al. Effects of thromboxane A2 on chronic hypoxic pulmonary hypertension in the rat. Nihon Kyobu Shikkan Gakkai Zasshi. 1996;34:37–44. [PubMed] [Google Scholar]
- 10.Lakshminrusimha S, Porta NF, Farrow KN, et al. Milrinone enhances relaxation to prostacyclin and iloprost in pulmonary arteries isolated from lambs with persistent pulmonary hypertension of the newborn. Pediatr Crit Care Med. 2009;10:106–112. doi: 10.1097/PCC.0b013e3181936aee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konduri GG, Bakhutashvili I, Eis A, Pritchard K., Jr Oxidant stress from uncoupled nitric oxide synthase impairs vasodilation in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2007;292:H1812–1820. doi: 10.1152/ajpheart.00425.2006. [DOI] [PubMed] [Google Scholar]
- 12.Teng RJ, Eis A, Bakhutashvili I, Arul N, Konduri GG. Increased superoxide production contributes to the impaired angiogenesis of fetal pulmonary arteries with in utero pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2009;297:L184–195. doi: 10.1152/ajplung.90455.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zou MH, Ullrich V. Peroxynitrite formed by simultaneous generation of nitric oxide and superoxide selectively inhibits bovine aortic prostacyclin synthase. FEBS Lett. 1996;382:101–104. doi: 10.1016/0014-5793(96)00160-3. [DOI] [PubMed] [Google Scholar]
- 14.Zou M, Martin C, Ullrich V. Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. Biol Chem. 1997;378:707–713. doi: 10.1515/bchm.1997.378.7.707. [DOI] [PubMed] [Google Scholar]
- 15.Morin FC., 3rd Ligating the ductus arteriosus before birth causes persistent pulmonary hypertension in the newborn lamb. Pediatr Res. 1989;25:245–250. doi: 10.1203/00006450-198903000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Abman SH, Shanley PF, Accurso FJ. Failure of postnatal adaptation of the pulmonary circulation after chronic intrauterine pulmonary hypertension in fetal lambs. J Clin Invest. 1989;83:1849–1858. doi: 10.1172/JCI114091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grover TR, Parker TA, Balasubramaniam V, Markham NE, Abman SH. Pulmonary hypertension impairs alveolarization and reduces lung growth in the ovine fetus. Am J Physiol Lung Cell Mol Physiol. 2005;288:L648–654. doi: 10.1152/ajplung.00288.2004. [DOI] [PubMed] [Google Scholar]
- 18.Katusic ZS, Santhanam AV, He T. Vascular effects of prostacyclin: does activation of PPARdelta play a role? Trends Pharmacol Sci. 2012;33:559–564. doi: 10.1016/j.tips.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He T, Lu T, d’Uscio LV, Lam CF, Lee HC, Katusic ZS. Angiogenic function of prostacyclin biosynthesis in human endothelial progenitor cells. Circ Res. 2008;103:80–88. doi: 10.1161/CIRCRESAHA.108.176057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nie D, Lamberti M, Zacharek A, et al. Thromboxane A(2) regulation of endothelial cell migration, angiogenesis, and tumor metastasis. Biochem Biophys Res Commun. 2000;267:245–251. doi: 10.1006/bbrc.1999.1840. [DOI] [PubMed] [Google Scholar]
- 21.Ashton AW, Ware JA. Thromboxane A2 receptor signaling inhibits vascular endothelial growth factor-induced endothelial cell differentiation and migration. Circ Res. 2004;95:372–379. doi: 10.1161/01.RES.0000138300.41642.15. [DOI] [PubMed] [Google Scholar]
- 22.Shaul PW, Campbell WB, Farrar MA, Magness RR. Oxygen modulates prostacyclin synthesis in ovine fetal pulmonary arteries by an effect on cyclooxygenase. J Clin Invest. 1992;90:2147–2155. doi: 10.1172/JCI116100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaul PW, Pace MC, Chen Z, Brannon TS. Developmental changes in prostacyclin synthesis are conserved in cultured pulmonary endothelium and vascular smooth muscle. Am J Respir Cell Mol Biol. 1999;20:113–121. doi: 10.1165/ajrcmb.20.1.3135. [DOI] [PubMed] [Google Scholar]
- 24.Cassin S, Winikor I, Tod M, et al. Effects of prostacyclin on the fetal pulmonary circulation. Pediatr Pharmacol (New York) 1981;1:197–207. [PubMed] [Google Scholar]
- 25.Brannon TS, North AJ, Wells LB, Shaul PW. Prostacyclin synthesis in ovine pulmonary artery is developmentally regulated by changes in cyclooxygenase-1 gene expression. J Clin Invest. 1994;93:2230–2235. doi: 10.1172/JCI117220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fike CD, Kaplowitz MR, Pfister SL. Arachidonic acid metabolites and an early stage of pulmonary hypertension in chronically hypoxic newborn pigs. Am J Physiol Lung Cell Mol Physiol. 2003;284:L316–323. doi: 10.1152/ajplung.00228.2002. [DOI] [PubMed] [Google Scholar]
- 27.Wedgwood S, Steinhorn RH, Bunderson M, et al. Increased hydrogen peroxide downregulates soluble guanylate cyclase in the lungs of lambs with persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol. 2005;289:L660–666. doi: 10.1152/ajplung.00369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou MH. Peroxynitrite and protein tyrosine nitration of prostacyclin synthase. Prostaglandins Other Lipid Mediat. 2007;82:119–127. doi: 10.1016/j.prostaglandins.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Hink U, Oelze M, Kolb P, et al. Role for peroxynitrite in the inhibition of prostacyclin synthase in nitrate tolerance. J Am Coll Cardiol. 2003;42:1826–1834. doi: 10.1016/j.jacc.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 30.Fike CD, Kaplowitz MR, Zhang Y, Pfister SL. Cyclooxygenase-2 and an early stage of chronic hypoxia-induced pulmonary hypertension in newborn pigs. J Appl Physiol (1985) 2005;98:1111–1118. doi: 10.1152/japplphysiol.00810.2004. discussion 1091. [DOI] [PubMed] [Google Scholar]
- 31.Fike CD, Pfister SL, Slaughter JC, et al. Protein complex formation with heat shock protein 90 in chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Heart Circ Physiol. 2010;299:H1190–1204. doi: 10.1152/ajpheart.01207.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konduri GG, Mattei J. Role of oxidative phosphorylation and ATP release in mediating birth-related pulmonary vasodilation in fetal lambs. Am J Physiol Heart Circ Physiol. 2002;283:H1600–1608. doi: 10.1152/ajpheart.00245.2002. [DOI] [PubMed] [Google Scholar]
- 33.Alano MA, Ngougmna E, Ostrea EM, Jr, Konduri GG. Analysis of nonsteroidal antiinflammatory drugs in meconium and its relation to persistent pulmonary hypertension of the newborn. Pediatrics. 2001;107:519–523. doi: 10.1542/peds.107.3.519. [DOI] [PubMed] [Google Scholar]
- 34.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 35.Konduri GG, Ou J, Shi Y, Pritchard KA., Jr Decreased association of HSP90 impairs endothelial nitric oxide synthase in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2003;285:H204–211. doi: 10.1152/ajpheart.00837.2002. [DOI] [PubMed] [Google Scholar]