

Abstract
To identify potential selective and resistance-breaking mosquitocides against the African malaria vector Anopheles gambiae, we investigated the acetylcholinesterase (AChE) inhibitory and mosquitocidal properties of isoxazol-3-yl dimethylcarbamates (15), and the corresponding 3-oxoisoxazole-2(3H)-dimethylcarboxamide isomers (14). In both series, compounds were found with excellent contact toxicity to wild-type susceptible (G3) strain and multiply resistant (Akron) strain mosquitoes that carry the G119S resistance mutation of AChE. Compounds possessing good to excellent toxicity to Akron strain mosquitoes inhibit the G119S mutant of An. gambiae AChE (AgAChE) with ki values at least 10- to 600-fold higher than that of propoxur, a compound that does not kill Akron mosquitoes at the highest concentration tested. On average, inactivation of WT AgAChE by dimethylcarboxamides 14 was 10-20 fold faster than that of the corresponding isoxazol-3-yl dimethylcarbamates 15. X-ray crystallography of dimethylcarboxamide 14d provided insight into that reactivity, a finding that may explain the inhibitory power of structurally-related inhibitors of hormone-sensitive lipase. Finally, human/An. gambiae AChE inhibition selectivities of these compounds were low, suggesting the need for additional structural modification.
Keywords: Malaria, Anopheles gambiae, acetylcholinesterase, carboxamide, carbamate
1. Introduction
Malaria is one of the deadliest diseases known to mankind. Recent estimates suggest that approximately 207 million malaria cases and 625,000 deaths were attributed to this disease alone in 2012.1 This problem is particularly severe in Sub-Saharan Africa where 90% of the cases occur. The much awaited malaria vaccine RTS,S/AS01 may help reduce the burden in malaria endemic regions. However, at present it has shown only partial efficacy in reducing malaria episodes in vaccinated children.2 Until an effective vaccine is in place, other effective and economical interventions such as vector control are still needed. Insecticide treated nets (ITNs) have shown efficacy in reducing malaria transmission in the past in many malaria endemic regions.3-5 At present, this method of intervention relies exclusively on pyrethroids, owing to their low mammalian toxicity and high insecticidal and repellant activity against the malaria vector, Anopheles gambiae. However, due to their widespread use, there has been an upsurge in mosquito populations resistant to this class of insecticides.6 This development threatens to compromise the efficacy of ITNs, and has motivated the development of other classes of insecticides with different modes of action. Acetylcholinesterase (AChE) inhibitors appear promising in this regard, in view of their efficacy as an indoor wall application (“indoor residual spraying”, IRS) against adult mosquitoes. However, none of the AChE inhibitors approved by the World Health Organization (WHO) for IRS (e.g. 1, 2) have been approved for ITNs, perhaps due to concerns of human toxicity. Thus, new safe and effective insecticides against the susceptible and resistant strains of Anopheles gambiae are needed for deployment on ITNs.
Our recently disclosed series of aryl methylcarbamates (e.g. 5-7, Figure 1) has shown high selectivity for An. gambiae AChE (AgAChE) vs human AChE (hAChE).7,8 These compounds bearing a γ-branched substituent exhibit up to 500-fold selectivity for AgAChE over hAChE, and are toxic to wild-type (WT) An. gambiae (G3 strain, MR4, CDC). This high selectivity must arise from amino acid substitutions near the active sites of AgAChE and hAChE.9 Although we cannot yet point to specific residue substitutions that 5-7 exploit to achieve selective inhibition, we have developed a ligand-based selectivity model.8 However, these compounds, like aryl methylcarbamates 1-4, are not appreciably toxic to Akron strain An. gambiae (MR4, CDC). Carbamate resistance of the Akron strain is known to arise from a G119S mutation in the oxyanion hole of AChE.10,11 To compensate for reduced active site volume arising from this mutation, we prepared and assayed pyrazol-4-yl carbamates (e.g. 8e, 8g, 8h Figure 1) against susceptible (G3) and resistant (Akron) strain An. gambiae. Their excellent contact toxicity towards both strains and inhibition of wild-type (WT) and G119S AgAChE suggests that a small core structure might be the key to combat carbamate resistance stemming from the G119S mutation.12 The excellent Akron toxicity of commercial insecticide aldicarb (9) can also be explained by this steric argument.12
Figure 1.

Structure of commercial carbamates (1-4, 9), and previously reported aryl (5-7) and pyrazol-4-yl (8e, 8g, 8h) carbamates.
In this paper we disclose the synthesis, mosquitocidal and AChE inhibitory properties of carboxamides and carbamates based on an isoxazole core. The insecticidal properties of 3-oxoisoxazole-2(3H)-carboxamides have been documented in the patent literature,13 but their contact toxicity to An. gambiae was not described. We will show that compounds from both structural classes can have high contact toxicity towards both WT and Akron resistant strain An. gambiae. The high insecticidal activity against resistant strain An. gambiae is particularly noteworthy.
2. Synthesis
Acylated Meldrum's acids 11a-p were synthesized in moderate to high yield from 10 and the requisite acid chloride (or acid), using the published literature procedure (Scheme 1).14 The R substituents were chosen to investigate the role of branching on inhibition selectivity, since they will reside at C5 of the isoxazole 13 (cf. 5-7 Figure 1). Compounds 11b-i feature α-branched alkyl groups, 11j-l feature β-branched alkyl groups, and 11m-p feature γ-branched alkyl groups. The subsequent thermolysis of 11 with N, O-bis-t-Boc hydroxylamine using the previously reported procedure14 did not proceed smoothly in every case. In some instances (e.g. 12b, 12c, 12j, 12k), the product was obtained in low yield, whereas in other cases (12m-o), it could not be purified due to contamination with residual BocNHOBoc. To optimize the yield for these substrates, we took 1 equiv. of the BocNHOBoc and 1.8 equiv. of the compound 12l and heated the reaction to 90 °C for 1.5-2 h. The reaction time for this step is crucial to avoid loss in yield due to incomplete conversion and competing side reactions. Using this modification, we could improve the yield of compound 12 for some substrates (e.g. 12a,12f-i, 12l) and the product could be isolated by column chromatography except in the case of 12d, where we proceeded to the next step with the crude product. The cyclization of β-keto hydroxamic acid 12 proceeded in high yields even when the acid equivalents were reduced to half of that recommended in the literature.14 The final carboxamide and carbamate products were obtained regioselectively through judicious use of two different protocols. The reaction of isoxazol-3-ol 13a-p with dimethylcarbamoyl chloride under neutral conditions gave 3-oxoisoxazole-2(3H)-carboxamides 14a-h, j-p as the major products and isoxazol-3-yl dimethylcarbamate 15a-p as the minor products (Scheme 1). Under neutral conditions, the more nucleophilic ring nitrogen attacks the carbonyl carbon of carbamoyl chloride forming dimethylcarboxamides as the major product. However, under basic conditions, 13a-p gave isoxazol-3-yl dimethylcarbamate 15a-p as the major product, with dimethylcarboxamides 14a-p formed in 5-17% yield (Scheme 1).
Scheme 1.

Synthesis of 5-substituted 3-oxoisoxazole-2(3H)-carboxamides14, 16 and isoxazol-3-yl carbamates 15. Reagents and conditions: (i) Pyridine, DCM, 0 °C, 15 min; RC(O)Cl, 0 °C, 1.5 h; RT, 1.5 h; (ii) RCOOH, Et3N, DECP, DMF, 0 °C, 30 min; RT, 16 h; (iii) NH(Boc)(OBoc), toluene, 90 °C, 1.5-2 h; (iv) NH(Boc)(OBoc), toluene, 65 °C, 16 h; (v) conc. HCl, MeOH, 50 °C, 2.5 h; (vi) ClC(O)NMe2, toluene, reflux, 16 h; (vii) KOt-Bu, THF, 0 °C, 15 min; ClC(O)NMe2, RT, 16 h; (viii) KOt-Bu, THF, 0 °C, 15 min; ClC(O)NHMe, RT, 16 h.
Unexpectedly, reaction of 13a-c, e-g, and i-m with methylcarbamoyl chloride under basic conditions gave N-methyl-3-oxoisoxazole-2(3H)-carboxamide 16 with no trace of the corresponding methylcarbamates (Scheme 1). It is possible that hydrogen bonding between the carbamate NH and the isoxazol-3(2H)-one carbonyl in 16 renders the methylcarboxamides considerably more stable than the methylcarbamate (see Figure 4A below). In that case, methylcarbamates initially formed under basic conditions could undergo O- to N-carbamoyl transfer to yield the observed methylcarboxamides. In fact, we noted that methylcarboxamides such as 16 were themselves unstable in aqueous buffer (pH 7.7) over 30 minutes; it is therefore possible that any traces of methylcarbamate present in the reaction mixture would be even more unstable and decomposed during aqueous workup.
Figure 4.

A) Anisotropic displacement ellipsoid drawings (50%) of X-ray structure of compound 16j. Hydrogen atoms were omitted for clarity, except at the exocyclic N. A predicted hydrogen bond is shown with a dotted black line (H1-O2 distance 2.11 Å). B) Anisotropic displacement ellipsoid drawings (50%) of X-ray structure of compound 14d. These structures were deposited at the Cambridge Structural Database (CCDC-1034990 and CCDC-1034991) and were visualized using OLEX2.17
To probe the effect of the N-substituent on mosquitocidal and enzyme inhibition activities, various N-substituted derivatives of 13g were synthesized as shown in Scheme 2. Compounds 17g, 18g, and 23g were synthesized by reaction of 13g with triphosgene and corresponding amine hydrogen chloride salt (17g) or amine (18g, 23g). Compound 22g was obtained in low yield from the same reaction mixture but appeared to be unstable, and hence was not assayed. Compounds 19g, 20g, and 21g were synthesized by refluxing 13g with corresponding carbamoyl chloride in toluene; the corresponding dimethylcarbamates 24g, 25g, and 26g, were obtained in the same reactions. As expected, carboxamides predominated under neutral conditions, except in the case of the carbamoyl chloride derived from pyrrolidine, where the carbamate 25g was obtained as the major product (Scheme 2).
Scheme 2.

Derivatization of isoxazol-3-ol 13g. Reagents and conditions: (i) Triphosgene, DCM, RT, 2h; DIPEA, HX or HX•HCl, RT, 30 min; Or (ii) ClC(O)X, toluene, reflux, 16 h.
2.1. Characterization of the 3-oxoisoxazole-2(3H)-carboxamides and isoxazol-3-yl carbamates
The final products were characterized and identified using 1H and 13C NMR, mass spectroscopy, and in two cases (14d, 16j) X-ray diffraction. Assignments of the carboxamide and carbamate structures were made in the following way. As illustrated in Figure 2, dimethylcarboxamides (e.g. 14a) show a broad singlet for the N-Me protons in the 1H NMR spectrum, whereas dimethylcarbamates (e.g. 15a) show two well-resolved singlets for these protons. The barrier to rotation of the C(O)-N bond in analogous urea-type dimethylcarboxamide analogues ranges from 8-13 kcal/mol,15 whereas in dimethylcarbamates it is higher, typically ~16 kcal/mol.16 These consistent differences in barriers to rotation presumably correspond to differences in the C(O)-N double bond character of ureas and carbamates. Similar behavior is seen in the 13C NMR spectra of these compounds (Figure 2B), where the N-Me carbons of 14a appear as very low intensity, broadened signals, and the N-Me carbons of 15a appear as two sharp resonances.
Figure 2.

A) The N-Me regions of the 1H NMR spectra of 3-oxoisoxazole-2(3H)-carboxamide 14a and isoxazol-3-yl carbamate 15a. B) The N-Me regions of the 13C NMR spectra of these compounds.
Another characteristic difference between 3-oxoisoxazole-2(3H)-carboxamides (both methyl- and dimethyl), and isoxazol-3-yl dimethylcarbamates was seen in 1H NMR chemical shifts of H-4 (Figure 3). This proton appeared between 5.27 ppm and 5.58 ppm for carboxamides 14 and 16, whereas for dimethylcarbamates 15 it appeared in isoxazol-3-yl the range 6.07 - 6.17 ppm (Figure 3). To correlate this chemical shift difference to structure, we noted that the H-4 chemical shift of dimethylcarboxamide 14j was similar to that of methylcarboxamide 16j. The identity of 16j as a methylcarboxamide was confirmed by X-ray crystallography (Figure 4A).
Figure 3.
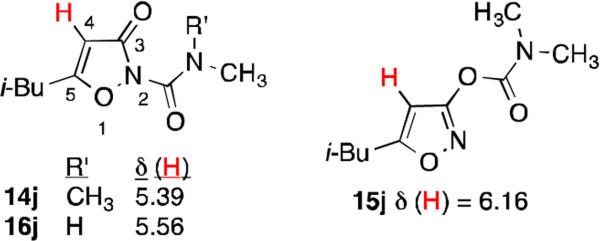
Characteristic chemical shifts of H-4 in 3-oxoisoxazole-2(3H)-carboxamides and isoxazol-3-yl dimethylcarbamates (ppm), as illustrated by 14j, 16j, and 15j respectively.
The identity of 14d (H-4 δ = 5.42 ppm) as a dimethylcarboxamide was also confirmed by X-ray crystallography (Figure 4B). The presence of the second methyl group (C9) eliminates the hydrogen bond seen in 16j, and steric interaction of this group with O2 causes the exocyclic amide moiety to rotate out of the plane of the isoxazole ring. We will return to this point below in our discussion of inactivation rate constants ki.
3. Tarsal contact toxicity of 3-oxoisoxazole-2(3H)-carboxamides and isoxazol-3-yl carbamates
Contact toxicity is a critical property for any insecticide that might be deployed on ITNs. Tarsal contact toxicity was determined towards G3 (WT, susceptible) and Akron (G119S, carbamate-resistant) strain An. gambiae, using a modification of the standard WHO tarsal contact toxicity protocol.18 In the discussion below, contact toxicities will be classified as excellent toxicity (LC50 < 100 μg/mL), good (LC50 = 100-199 μg/mL), moderate (LC50 = 200-399 μg/mL), and poor (LC50 ≥ 400 μg/mL). As mentioned previously, commercial carbamates 1-4 bearing a phenyl core displayed excellent toxicities to the susceptible G3 strain of An. gambiae (LC50 = 16-39 μg/mL), but no measurable toxicity to resistant Akron strain (Table 1). In contrast pyrazol-4-yl methylcarbamate 8e and aldicarb 9 had excellent toxicities to both strains12 (Table 1). We attributed the low resistance ratios of these two compounds in part to their smaller core structures, and were thus interested to learn whether the isoxazol-3-yl core could also confer high toxicity to Akron strain An. gambiae. As noted above, isoxazol-3-yl methylcarbamates could not be isolated, and methylcarboxamides 16 proved unstable in aqueous buffer. Therefore our investigations focused on dimethylcarboxamides 14, and dimethylcarbamates 15. The carboxamide structure represents a significant departure from the structure of aryl and pyrazole carbamates that we previously studied. The possible effects of disubstitution on the carbamate nitrogen on contact toxicity were similarly unknown. We were pleasantly surprised to find that a number of dimethylcarboxamides were appreciably toxic to G3 and Akron strain An. gambiae (Table 1). Compound 14a did not show any contact toxicity at the highest concentration tested (1000 μg/mL, Table 1), perhaps as a consequence of low lipophilicity. However, α-branched dimethylcarboxamides (14b-h) showed appreciable toxicities towards G3 and Akron strain (Table 2). Compound 14b (R = c-Pr) showed moderate toxicity towards G3 strain, but was considerably less toxic against Akron strain. The open-chain analog carboxamides 14c (R = i-Pr), and 14g (R = 2-pentyl) showed moderate and good G3 toxicity respectively, and were even more toxic to the Akron strain. Compound 14d (R = c-Bu) exhibited excellent toxicity towards both strains of An. gambiae. The compounds with highest G3 toxicity in the α-branched series were 14e (R = s-Bu, LC50 = 63 μg/mL) and 14h (R = 3-pentyl, LC50 = 38 μg/mL), and they showed good and excellent Akron toxicity respectively. Amongst the β-branched dimethylcarboxamides explored (14j-l), only 14j (R = i-Bu) showed moderate or better G3 toxicity, but Akron toxicity was lower. Among the γ-branched compounds explored (14m-p), the carboxamide 14m (R = i-pentyl) was the most toxic and showed equivalent LC50 values towards G3 and Akron strain. A sharp decrease in G3 and Akron toxicity was observed from carboxamides 14n (R = CH2CH2-c-Pr) to 14p (R = CH2CH2-c-C5H9), concomitant with the increase in the size of the pendant carbocyclic ring. Finally, a range of compounds (17g-21g) were examined to assess the effect of varying the substitution on the exocyclic nitrogen. Only 18g (ethylmethylcarboxamide) showed poor or better toxicity to G3 strain, and it was 2-3 fold less toxic than the dimethylcarboxamide 14g. These data may suggest that the N-substituents of 17g-21g are too big to inhibit AgAChE well, and we address this point below. The low toxicity of azetidine carboxamide 17g relative to dimethylcarboxamide 14g may be due to instability of the azetidine ring, as discussed below.
Table 1.
Tarsal contact toxicity of control compounds (1-4, 8e, 9) and N,N-dialkyl-3-oxoisoxazole-2(3H)-carboxamides 14, 17-21 to susceptible (G3) and resistant (Akron) strains of An. gambiae.
| Compound | Ra | An. gambiae G3 bLC50 μg/mL (95% CI) | An. gambiae Akron bLC50 μg/mL (95% CI) | cRR |
|---|---|---|---|---|
| 1d | NA | 39 (32-45) | >5,000d | >130 |
| 2d | NA | 16 (14-17) | >5,000 | >310 |
| 3d | NA | 16 (11-25) | >5,000 | >310 |
| 4d | NA | 37 (14-16) | >5,000 | >130 |
| 8ed | NA | 96 (89-104) | 81 (78-89) | 0.8 |
| 9d | NA | 70 (66-74) | 32 (30-35) | 0.5 |
| 14a | Me | 0 % @ 1,000 μg/mL | ND | - |
| 14b | c-Pr | 278 (225-345) | 10 % @ 1,000 μg/mL | - |
| 14c | i-Pr | 316 (22-428) | 182 (138-253) | 0.6 |
| 14d | c-Bu | 79 (57-116) | 74 (55-105) | 0.9 |
| 14e | s-Bu | 63 (49-81) | 129 (91-196) | 2.0 |
| 14f | c-C5H11 | 105 (78-157) | 118 (89-171) | 1.1 |
| 14g | 2-pentyl | 153 (109-211) | 75 (53-111) | 0.5 |
| 14h | 3-pentyl | 38 (28-53) | 40 (31-54) | 1.1 |
| 14j | i-Bu | 201 (145-284) | 25 % @ 200 μg/mL | - |
| 14k | 2-methyl-butyl | 0 % @ 1,000 μg/mL | ND | |
| 14l | neo-pentyl | 80 % @ 1,000 μg/mL | ND | - |
| 14m | 3-methyl-butyl | 73 (58-93) | 68 (54-87) | 0.9 |
| 14n | CH2CH2-c-Pr | 133 (71-226) | 144 (82-229) | 1.1 |
| 14o | CH2CH2-c-Bu | 30 % @ 1,000 μg/mL | ND | - |
| 14p | CH2CH2-c-C5H9 | 0 % @ 1,000 μg/mL | ND | - |
| 17g | 2-pentyl | 78 % @ 1,000 μg/mL | ND | - |
| 18g | 2-pentyl | 400 (320-483) | 614 (537-684) | 1.5 |
| 19g | 2-pentyl | 15 % @ 1,000 μg/mL | ND | - |
| 20g | 2-pentyl | 77 % @ 1,000 μg/mL | ND | - |
| 21g | 2-pentyl | 45 % @ 1,000 μg/mL | ND | - |
NA signifies not applicable.
Mosquitoes were exposed (1 h) to dried filter papers previously treated with ethanolic solutions of carbamates; mortality was recorded after 24 h. LC50 values derive from the concentrations of inhibitor used to treat the paper. ND designates “not determined”; compounds that did not show significant toxicity to susceptible G3 strain (100% mortality at 1000 μg/mL) were generally not tested on the resistant Akron strain.
Resistance ratio (RR), defined by LC50 (Akron)/LC50 (G3).
Data reported previously.12
Table 2.
Tarsal contact toxicity of isoxazol-3-yl dialkylcarbamates to G3 and Akron strain of An. gambiae.
| Compound | R | An. gambiae G3 aLC50 μg/mL (95% CI) | An. gambiae Akron aLC50 μg/mL (95% CI) | bRR |
|---|---|---|---|---|
| 15a | Me | 0 % @1,000 μg/mL | ND | - |
| 15b | c-Pr | 0 % @1,000 μg/mL | ND | - |
| 15c | i-Pr | 84 (30-124) | 116 (79-151) | 1.4 |
| 15d | c-Bu | 349 (266-456) | 392 (281-593) | 1.1 |
| 15e | s-Bu | 41 (28-58) | 58 (42-92) | 1.4 |
| 15f | c-pentyl | 60 % @ 1,000 μg/mL | ND | - |
| 15g | 2-pentyl | 234 (163-307) | 296 (210-381) | 1.3 |
| 15h | 3-pentyl | 70 % @ 1,000 μg/mL | ND | - |
| 15i | 3-heptyl | 0 % @ 1,000 μg/mL | ND | - |
| 15j | i-Bu | 253 (184-367) | 151 (112-211) | 0.6 |
| 15k | 2-methylbutyl | 30 % @ 1,000 μg/mL | ND | - |
| 15l | neo-pentyl | 0% @ 1,000 μg/mL | ND | - |
| 15m | 3-methylbutyl | 415 (306-582) | 429 (331-563) | 1.0 |
| 15n | CH2CH2-c-Pr | 191 (109-343) | 198 (119-321) | 1.0 |
| 15o | CH2CH2-c-Bu | 40 % @ 1,000 μg/mL | ND | |
| 15p | CH2CH2-c-C5H9 | 674 (575-715) | ND | - |
| 23g | 2-pentyl | 410 (304-590) | 574 (513-633) | 1.4 |
| 25g | 2-pentyl | 0% @ 1,000 μg/mL | ND | - |
| 26g | 2-pentyl | 10% @ 1,000 μg/mL | ND | - |
Mosquitoes were exposed (1 h) to dried filter papers previously treated with ethanolic solutions of carbamates; mortality was recorded after 24 h. LC50 values derive from the concentrations of inhibitor used to treat the paper. ND designates “not determined”; compounds that did not show significant toxicity to susceptible G3 strain (100% mortality at 1000 μg/mL) were generally not tested on the resistant Akron strain.
Resistance ratio (RR), defined by LC50 (Akron)/LC50 (G3).
Turning to the corresponding dimethylcarbamates, 15a bearing a C5-methyl group was not toxic to G3 strain at the highest concentration tested (Table 2). In contrast α-branched analogs 15b-i displayed G3 toxicities ranging from excellent (15c, e) to poor (15b, f, h, i, Table 3). Compounds 15c (R = i-Pr) and 15e (R = s-Bu) also demonstrated good and excellent toxicities towards Akron strain An. gambiae, respectively. One interesting trend is that open-chain analogs typically showed greater G3 toxicity than their cyclic analogs (cf. 15c vs 15b, 15e vs 15d, and 15g vs 15f). Finally 15i (R = 3-heptyl) was not toxic to G3 strain at the highest concentration tested (1000 μg/mL).
Table 3.
Inactivation rate constants (kt) of control compounds (1-4, 8e, 9) and carboxamides 14a-p, 17g-21g for rAgAChE (WT and G119S) and rhAChE (h)
| Compound | R | WT ki (mM−1 min−1)b | G119S ki (mM−1 min−1)b | h ki (mM−1 min−1)b | WT/G119S SRc |
|---|---|---|---|---|---|
| 1d | NA | 266 ± 9 | ±0.037±0.007 | 17.0 ± 0.4 | 7,200 ± 1,400 |
| 2d | NA | 839 ± 22 | ±0.055 ± 0.007 | 111 ± 5 | 15,000 ± 2,000 |
| 3d | NA | 2,620 ± 150 | ±0.044 ± 0.020 | 428 ± 12 | 60,000 ± 27,000 |
| 4d | NA | 1,510 ± 100 | 0.40 ± 0.03 | 126 ± 3 | 3,800 ± 400 |
| 8ed | NA | 9,140 ± 260 | 290 ± 7 | 805 ± 36 | 32 ± 1 |
| 9d | NA | 13.3 ± 0.3 | 3.15± 0.08 | 6.5 ± 0.3 | 4.2 ± 0.1 |
| 14a | Me | 0.46 ± 0.04 | 0.098 ± 0.029 | 1.22 ± 0.04 | 4.7 ± 1.4 |
| 14b | c-Pr | 50.5 ± 1.4 | 0.20 ± 0.03 | 103 ± 5 | 250 ± 30 |
| 14c | i-Pr | 500 ± 10 | 1.85 ± 0.09 | 561 ± 21 | 270 ± 10 |
| 14d | c-Bu | 293 ± 9 | 0.62 ± 0.05 | 233 ± 13 | 480 ± 40 |
| 14e | s-Bu | 2,290 ± 80 | 10.6 ± 0.5 | 2,170 ± 40 | 220 ± 10 |
| 14f | c-C5H11 | 1,020 ± 50 | 2.58 ± 0.22 | 677 ± 26 | 400 ± 40 |
| 14g | 2-pentyl | 5,240 ± 140 | 14.7 ± 0.3 | 3,110 ± 120 | 360 ± 10 |
| 14h | 3-pentyl | 8,530 ± 420 | 22.5 ± 1.1 | 1,990 ± 70 | 380 ± 30 |
| 14j | i-Bu | 252 ± 4 | 0.55 ± 0.02 | 30.9 ± 2.6 | 450 ± 20 |
| 14k | 2-methylbutyl | 477 ± 7 | 0.99 ± 0.05 | 112 ± 5 | 480 ± 20 |
| 141 | neo-pentyl | 161 ± 12 | 0.37 ± 0.06 | 26.2 ± 1.1 | 440 ± 80 |
| 14m | 3-methylbutyl | 266 ± 9 | 0.40 ± 0.12 | 80.9 ± 2.0 | 670 ± 200 |
| 14n | CH2CH2-c-Pr | 420 ± 15 | 1.07 ± 0.06 | 144 ± 7 | 390 ± 30 |
| 14o | CH2CH2-c-Bu | 141 ± 4 | 0.31 ± 0.09 | 74.5 ± 4.4 | 460 ± 130 |
| 14p | CH2CH2-c-C5H9 | 24.3 ± 0.1 | 0.41 ± 0.27 | 92.0 ± 4.0 | 60 ± 4.1 |
| 17g | 2-pentyl | 2,240 ± 80 | 36.7 ± 2.7 | 1,860 ± 40 | 61 ± 5 |
| 18g | 2-pentyl | 156 ± 6 | 3.84 ± 0.20 | 1,080 ± 50 | 41 ± 3 |
| 19g | 2-pentyl | 7.30 ± 0.09c | 1.24 ± 0.05 | 180 ± 3c | 5.9 ± 0.3 |
| 20g | 2-pentyl | 134 ± 8 | 1.90 ± 0.11 | 379 ± 24 | 71 ± 6 |
| 21g | 2-pentyl | 39.0 ± 2.1 | 2.15 ± 0.27 | 26.0 ± 0.3 | 18 ± 3 |
a NA signifies not applicable.
Measured at 23 ± 1°C, pH 7.7, 0.1% (v/v) DMSO. Recombinant sources of AgAChE are rAgAChE-WT and rAgAChE-G119S.
Enzymatic sensitivity ratio is calculated as ki(WT)/ki(G119S). Standard error in the ratio is calculated according to a standard propagation of error formula.20
Data reported previously.12
Amongst the β-branched dimethylcarbamates examined (15j-l), only 15j with an isobutyl side chain proved moderately toxic towards G3. Interestingly, this compound proved even more toxic to Akron strain than it was to G3 (cf. LC50 = 151 and 253 μg/mL respectively). Amongst γ-branched compounds, unlike corresponding carboxamides, no clear trend was observed in G3 and Akron toxicity. The optimal LC50 was achieved for carbamates 15n (R = CH2CH2-c-Pr, G3 LC50 = 191 μg/mL). 15m, 15o, and 15p exhibited poor toxicity towards both strains of An. gambiae. Finally, as was seen for the carboxamides, variation of the N-substituent reduced toxicity. Only 23g (ethylmethylcarbamate) showed moderate or better toxicity to G3 strain, but had reduced toxicity for Akron strain. Compound 25g (pyrrolidinylcarbamate), and 26g (morpholinocarbamate) did not show any toxicity to G3 strain An. gambiae.
Looking at both series, three conclusions can be drawn. First, for a given C5-substituent, there is no general trend for dimethylcarboxamide vs dimethylcarbamate toxicity. In some cases (b, d, f, g, h, j, l, m) the dimethylcarboxamide 14 is more toxic, and in others (c, e, h, p) the dimethylcarbamate 15 is more toxic. Second, among the exocyclic N-substituents dimethyl explored, proved optimum, though ethylmethyl analogs 18g and 23g did show significant toxicity. Third, both in the α- and γ-branched series, excellent G3 toxicity and low cross-resistance can be attained. The resistance ratios (RR) for 14c, 14g and 15j are all less than 1.0, which signifies greater susceptibility of resistant strain An. gambiae towards these novel insecticides. This property would certainly be favorable for a new anticholinesterase-based mosquitocide.
4. Inhibition of WT and G119S AgAChE by isoxazol-3-yl carbamates and 3-oxoisoxazole-2(3H)-carboxamide
Carbamates are pseudo-irreversible inhibitors of AChE; they inhibit the enzyme by carbamoylating the active site serine. We have previously used the Ellman Assay to monitor the time-dependent inhibitory activity of carbamates.7,8,12,19 We employed the same method to determine the inhibition of AChE for our heterocyclic carbamates and carboxamides. Enzyme velocities (ν/ν0) at a fixed inhibitor concentration were measured as a function of time t. Plots of ln(ν/ν0) vs incubation time t were constructed and the slope provided the pseudo first-order rate constant kobs (min−1) for inactivation. For each inhibitor, kobs values were determined at three or more inhibitor concentrations [I]. Finally, the linear fit slope of plots of kobs vs [I] provided the second order rate constant ki (mM−1min−1) for inactivation. Note that non-saturating [I] were chosen to give linear kobs vs [I] plots.7,8,12 The inhibition data and the resistance ratios for commercial carbamates are given in Table 3. As expected from their high toxicological resistance ratios (RR), the commercial aryl carbamates 1-4 inhibited the susceptible enzyme (WT AChE) much more rapidly than the resistant enzyme (G119S AChE), giving enzymatic sensitivity ratios (SR) greater than 3800-fold. In contrast, the SR values for pyrazol-4-yl methylcarbamate 8e and aldicarb (9) were only 32- and 4.2-fold, which correlate well with their low toxicological resistance ratios (0.8 and 0.5, respectively).
On the basis of low toxicological resistance ratios seen for many isoxazol-3-yl dimethylcarbamates and dimethylcarboxamides (Tables 1, 2), we predicted low SR values for these compounds. However, as will be seen below, this expectation was only partially realized. The ki values for N,N-alkyl-3-oxoisoxazole-2(3H)-carboxamides are presented in Table 3. Compound 14a (R = Me) showed very slow inactivation of AgAChE-WT (0.46 mM−1 min−1), which could be a consequence of high desolvation penalty (low lipophilicity). Dimethylcarboxamides bearing larger R groups at C5 showed much greater WT AgAChE ki values, ranging from 24.3 to 8,530 mM−1 min−1 (14p and 14h, respectively). In general, the largest AgAChE inactivation rate constants were realized with α- branched alkyl substitution, in particular 14e-h. These dimethylcarboxamides also exhibited the largest G119S inactivation constants (2.58 to 22.5 mM−1 min−1); the rate constants for 14g and 14h were approximately 5- and 7-fold higher than that of aldicarb (9). Although the WT/G119S enzymatic sensitivity ratios for these compounds are still very large (360- to 380-fold), they are 10-fold lower than that of 1-4.
Comparison of cycloalkyl analogs 14b, d, f with the corresponding open chain -branched analogs 14c, e, g, h shows that in each case, the open chain compounds offered significantly more rapid inactivation of WT or G119S AgAChE than the cycloalkyl analogs (cf 14c vs 14b, 14e vs 14d, and 14g or 14h vs 14f). It is possible that the flexibility of the side chain gives 14c, 14e, 14g, and 14h a better fit in the active site of the enzyme as compared to 14b, 14d, and 14f. With regard to AgAChE/hAChE selectivity, the α-branched carboxamides did not offer more than 4-fold selectivity. The three β-branched dimethylcarboxamides investigated (14j-l) exhibited reduced ki values compared to those with α-branching. Unfortunately, selectivity for AgAChE over hAChE within this series remained much lower than desired, reaching a maximum of 8-fold for 14j. By analogy to compounds 5 to 7, it was hoped that a γ-branched alkyl group would confer high inactivation selectivity. However within the γ-branched dimethylcarboxamide series (14m-p), the maximum AgAChE inactivation selectivity achieved was 3-fold (14n). With regard to WT and G119S inactivation, an approximate two-fold increase in ki was seen for 14n (R = CH2CH2-c-Pr) compared to 14m (R = CH2CH2-i-Pr). However, increasing size of the carbocycle in the series 14n, 14o, 14p reduced both WT and G119S ki values.
Variation of the exocyclic nitrogen substituents, while retaining a 2-pentyl group at C5, had dramatic effects on both WT and G119S AgAChE ki values. The relatively conservative replacement of the dimethylamino group of 14g with an azetidine moiety (17g) reduced WT AgAChE ki value by 50%, but more than doubled the G119S AgAChE ki value. Replacement of one of the methyl groups of 14g with an ethyl group (18g) reduced the WT AgAChE ki value 30-fold, and the G119S AgAChE ki value nearly 4-fold, suggesting steric inhibition of binding or carbamoylation in the acyl pocket of AgAChE. Consistent with this hypothesis, pyrrolidino- and morpholino-carboxamides 20g and 21g offered more rapid inactivation than the diethylcarboxamide 19g, but all three were much slower than dimethyl carboxamide 14g.
Turning to the ki values of the dimethylcarbamates, compound 15a (like its carboxamide counterpart 14a) exhibited extremely slow inactivation of all three enzymes; again low lipophilicity may play a role (Table 4). Among α-, β-, and γ-branched dimethylcarbamates, the highest WT and G119S AgAChE values were seen for the α-branched compounds (e.g. 15e-h), as was the case for dimethylcarboxamides 14 (Table 3). The highest G119S AgAChE ki values in the dimethylcarbamate series are in the range of the dimethylcarboxamides; in particular 15h, 15e, and 15g have G119S ki values ranging from 3- to 10-fold of that of aldicarb (9). However, the α-branched dimethylcarbamates 15e-h exhibited at least 10-fold slower inactivation of WT AgAChE than the corresponding dimethylcarboxamides 14e-h. Consequently, the enzymatic sensitivity ratios of the dimethylcarbamates are roughly 10-fold lower than that of the dimethylcarboxamides (cf. SR values in Tables 3 and 4). As was seen for dimethylcarboxamides, WT and G119S AgAChE ki values for open chain α-branched dimethylcarbamates were higher than those of their cycloalkyl homologs (cf. 15c vs 15b, 15e vs 15d, 15h, g vs 15f).
Table 4.
Inactivation rate constants (ki) of isoxazol-3-yl dialkylcarbamates for rAgAChE (WT and G119S), and rhAChE (h)
| Compound | R | WT ki (mM−1 min−1)a | G119S ki (mM−1 min−1)a | h ki (mM−1 min−1)a | WT/G119S SRb |
|---|---|---|---|---|---|
| 15a | Me | 0.0323 ± 0.0082c | 0.0224 ± 0.0125 | 0.118 ± 0.019 | 1.4 ± 0.9 |
| 15b | c-Pr | 3.02 ± 0.13 | 0.30 ± 0.03 | 2.02 ± 0.27 | 10 ± 1 |
| 15c | i-Pr | 7.71 ± 0.20 | 0.50 ± 0.04 | 4.46 ± 0.12 | 15 ± 1 |
| 15d | c-Bu | 6.84 ± 0.20 | 0.44 ± 0.04 | 1.86 ± 0.10 | 16 ± 2 |
| 15e | s-Bu | 323 ± 6 | 20.4 ± 0.6 | 60.1 ± 1.8 | 16 ± 1 |
| 15f | c-pentyl | 64.7 ± 2.7 | 1.94 ± 0.04 | 4.62 ± 0.07 | 33 ± 2 |
| 15g | 2-pentyl | 416 ± 7 | 30.6 ± 0.5 | 50.9 ± 1.0 | 14 ± 1 |
| 15h | 3-pentyl | 221 ± 7 | 10.5 ± 0.7 | 9.04 ± 0.31 | 21 ± 2 |
| 15i | 3-heptyl | 2.85 ± 0.11 | 0.26 ± 0.01 | 2.13 ± 0.09 | 11 ± 1 |
| 15j | i-Bu | 10.6 ± 0.5 | 0.87 ± 0.06 | 1.59 ± 0.08 | 12 ± 1 |
| 15k | 2-methylbutyl | 7.64 ± 0.21 | 0.50 ± 0.05 | 1.70 ± 0.09 | 15 ± 2 |
| 15l | neo-pentyl | 0.12 ± 0.03 | 0.05 ± 0.01 | 0.44 ± 0.04 | 2.4 ± 0.7 |
| 15m | 3- methylbutyl | 3.12 ± 0.16 | 0.37 ± 0.03 | 1.91 ± 0.04 | 8.3 ± 0.8 |
| 15n | CH2CH2-c-Pr | 23.5 ± 1.1 | 2.01 ± 0.05 | 8.26 ± 0.26 | 12 ± 1 |
| 15o | CH2CH2-c-Bu | 5.08 ± 0.23 | 0.62 ± 0.03 | 7.24 ± 0.90 | 8.2 ± 0.6 |
| 15p | CH2CH2-c-C5H9 | 0.60 ± 0.05 | 0.12 ± 0.03 | 13.1 ± 0.4 | 5.2 ± 1.2 |
| 23g | 2-pentyl | 24.5 ± 0.5 | 3.37 ± 0.20 | 9.14 ± 1.97 | 7.3 ± 0.5 |
| 25g | 2-pentyl | 12.4 ± 0.1c | 2.24 ± 0.08c | 1.62 ± 0.07c | 5.5 ± 0.2 |
| 26g | 2-pentyl | 18.6 ± 0.3c | 2.30 ± 0.10c | 2.68 ± 0.06c | 8.1 ± 0.4 |
Measured at 23 ± 1°C, pH 7.7, 0.1% (v/v) DMSO. Recombinant sources of AgAChE are rAgAChE-WT and rAgAChE-G119S.
Enzymatic sensitivity ratio (SR) is calculated as ki(WT)/ki(G119S). Standard error in the ratio is calculated according to a standard propagation of error formula.20
ki values extrapolated from single point incubation at various inhibitor concentration, e.g. t = 10 min.
Among the β-branched dimethylcarbamates, the bulky neopentyl compound 15l was much less inhibitory at all three enzymes than the isobutyl (15j) and 2-methylbutyl (15k) compounds. The γ-branched dimethylcarbamates 15m-p were again examined in the hope that they (like 5-7) would exhibit good WT Ag/h selectivity. However, as can be seen by inspection of the AgAChE and hAChE ki values in Table 3, the highest selectivity (24-fold) was obtained for the α-branched compound 15h. Within the γ-branched series 15m-p, the highest values of WT and G119S AgAChE ki were again seen for R = CH2CH2-c-Pr (15n). Finally, with regard to variation of the exocyclic N-substituents, increasing steric bulk reduced ki values at all three enzymes (cf 23g, 25g, 26g vs 15g) again suggesting steric crowding in the acyl pocket of AChE.
5. Discussion
To gain insight into the unexpectedly high inactivation rates constants (ki) of the dimethylcarboxamides relative to the dimethylcarbamates, we modeled inhibitor 14c in the active site of mouse AChE, and compared it to the experimental X-ray structure of mouse AChE (mAChE) covalently bound by 27, a potent trifluoromethylketone (TFK) inhibitor (Figure 5).21
Figure 5.

Serine hydrolase inhibitors related to dimethylcarboxamides 14.13,22,23
Flexible ligand docking of the dimethylcarboxamide tetrahedral covalent intermediate adduct derived from 14c and mAChE was performed in ICM using default settings for ‘covalent’ docking mode (ICM-Docking module, Molsoft).24,25 The choice of mAChE was motivated by three factors: 1) availability of a high-resolution crystal structure of its complex with 27 (PDB ID 2H9Y);26 2) high sequence identity (88%) of mAChE and hAChE;27 and 3) our observation of fast inactivation of hAChE by the dimethylcarboxamides (cf. Table 3). The choice to model the tetrahedral intermediate rather than the carbamoylated AChE reflects our assumption that formation of the tetrahedral intermediate is likely the rate-limiting step of enzyme inactivation, which then leads quickly to the carbamoylated enzyme. Figure 6 shows the tetrahedral adduct formed after the attack of active site serine (S203) on the carbonyl of TFK 27 (Figure 6A) and 14c (Figure 6B). In both cases, the oxyanion formed is stabilized through hydrogen bonding with oxyanion hole residues G121, G122 and A204. Note that the corresponding active site serine and oxyanion residues in AgAChE are S199, G118, G119, and A200 respectively (see Supplementary data Figure S1). The overlay of the two structures in Figure 6C highlights the similar pose of the two molecules in the active site of AChE: the NMe2 group of 14c overlays the trifluoromethyl group of 27 in the acylpocket. It is well known that the CF3 group is similar in volume to an isopropyl group;28 as Figure 6C illustrates, it is also similar in size to NMe2. Lastly, the C5-i-Pr substituent of 14c occupies the same locus as the trimethylammonium group of 27, in the choline-binding site. It is possible that the C5-alkyl substituents of 14c and analogs could have van der Waals interactions with W86, in place of the cation- interaction experienced by 27. Given these similarities in the predicted binding pose of 14c and 27 to mouse AChE, the high ki values seen at hAChE (and by extension AgAChE) are more readily understood.
Figure 6.

Experimental and modeled tetrahedral adducts of mouse AChE active site serine S203 with electrophilic carbonyl derivatives. (A) X-ray structure of the tetrahedral adduct of 27 with S203 (PDB ID 2H9Y).26 (B) Computational modeling of the tetrahedral adduct of dimethylcarboxamide 14c with S203 of mouse AChE. (C) Overlay of structures (A) and (B). In both cases the backbone NH moieties of oxyanion hole residues G121, G122 and A204 are within hydrogen bonding range of the anionic oxygen. Image created with Molsoft Browser Pro version 3.7-a.
As mentioned earlier, the X-ray crystal structure of 14d (Figure 4B) also provides some insight into the reactivity of the dimethylcarboxamides. The extent of resonance between the endocyclic N (N1) and the amide carbonyl carbon (C8) appears to be low based on the out-of-plane twisting of the dimethylcarbamoyl group. The magnitude of amide twist can be expressed as the τ value29,30 along a given N-C bond. As expected, the endocyclic amide bond N1-C7 has a low value (-12.6°), as does the dimethylamino amide bond N2-C8 (8.6°). However, the bond from N1 to the reactive carbonyl carbon C8 has a much larger twist, with a τ value of 47.5°. As mentioned earlier, this twist appears to be caused by steric interaction of C9 and O2, and the ensuing lack of resonance donation of N1 into the C8 carbonyl should serve to increase the electrophilicity of the dimethylcarboxamides. We note that N1 in 14d is also quite pyramidalized (the sum of angles at N1 is only 346°). Additional details on this structure, and for that of 16j are provided in the Supplementary Material. Close analogs of these carboxamides have been cited in the literature as effective serine hydrolase inhibitors. Isoxazolonyl carboxamide22 28 and 1,2,4-triazole carboxamide23 29 are potent hormone-sensitive lipase (HSL) inhibitors (Figure 5). Like vertebrate AChE, HSL belongs to the family of serine hydrolases with a α/β hydrolase fold.31 Since they originated from the same ancestor, the catalytic residues are preserved in these enzymes. Triazamate 30, a carbamoyl triazole AChE inhibitor aphicide, has been approved for plant protection in many European countries.32 Based on the structural similarity of these compounds to 14d, it seems likely that the serine hydrolase inhibitory power of 28-30 derives in part from a similar out-of-plane twist of the exocyclic amide moiety.
Finally, it would seem obvious to expect that G3 toxicity and WT AgAChE ki values would be highly correlated. However, as we have shown for aryl methylcarbamates, ADME can be very influential,7 and no general correlation is seen between G3 LC50 and AgAChE ki values in either the isoxazole carboxamide or carbamate series. For example, the α-branched compounds 14h and 15e have almost equal toxicities towards G3 An. gambiae (38 and 41 μg/mL respectively). Despite the similar toxicities, 15e is 26-fold less inhibitory than 14h (323 and 8,530 mM−1 min−1, respectively). Similarly, 14j and 14m have similar AgAChE (WT) ki values (252 and 266 mM−1 min−1, respectively), but the G3 LC50 value of 14j is 2.4 fold higher than of 14m (201 and 73 μg/mL, respectively). Further, despite the fact that these two compounds have similar G119S AgAChE ki values, 14m is considerably more toxic to Akron strain than 14j. In all these cases, differential pharmacokinetics and metabolism may be dominant factors in toxicity. Nevertheless, we can offer the following observations. Firstly, C5-methyl dimethylcarboxamide 14a and dimethylcarbamate 15a had very poor toxicity and very slow enzyme inactivation rate constants, likely as a consequence of low lipophilicity. The calculated CLogP values for these compounds are 0.70 and 0.20, as compared to 2.2 and 1.7 for the s-Bu substituted compounds 14e and 15e.33 It is also possible that if the C5-substituent is insufficiently large, the carbamoyl group will not be positioned properly to react with the active site serine. Secondly, as previously mentioned, open chain α-branched dimethylcarbamates were more toxic to G3 strain than the corresponding cycloalkyl analogs (cf 15c vs 15b, 15e vs 15d and 15g vs 15f), and this trend correlates well with differences in enzyme inactivation ki values. Third, increasing the size of the exocyclic N-substituent reduced toxicity in both dimethylcarboxamide (14g vs 17-21g) and dimethylcarbamate (15g vs 23, 25g, 26g), and decreased enzyme inactivation ki values accompany these decreased toxicities. Fourth, for those compounds having excellent (14d, 14g, 14h, 14m, and 15e) and good (14c, 14e, 14f, 14n, 15c, 15j, and 15n) toxicity to Akron strain An. gambiae, we examined the corresponding G119S AgAChE ki values. All these compound give measurable ki values at the G119S enzyme, ranging from 13% (14m) to 700% (14h) of the ki value of aldicarb (9). The fact that Akron/G3 toxicological resistance ratios (RR) for these compounds are much smaller than the WT/G119S enzyme sensitivity ratios (SR) may reflect enhanced bioactivation or compromised detoxification in Akron, relative to G3. Nevertheless it seems likely that the Akron-toxic properties of these compounds indeed stem from engagement of the G119S AgAChE target. This clearly advantageous property would be even more valuable, if it could be achieved in compound class that offers excellent selectivity against inhibition of hAChE. Work to achieve this goal is in progress.
6. Experimental
6.1 Chemistry
NMR spectra were obtained on JEOL Eclipse-plus 500 MHz MHz spectrometer at 500 (1H) and 126 (13C) MHz or Unity-plus 400 at 400 (1H) and 101 (13C) MHz. The chemical shifts are reported in δ (ppm), and coupling constants are given in Hz. High-resolution ESI mass spectra were obtained on an Agilent 6220 accurate mass TOF LC/MS. X-ray data collection routine, unit cell refinement, and data processing were carried out with the program CrysAlisPro. The structure was solved using SHELXS-2013 and refined using SHELXL-2013 via OLEX2. THF for moisture sensitive reactions was distilled from sodium-benzophenone. Other dry solvents were purchased from EMD Millipore and were used without any further purification. Column chromatography was performed using Silica gel (ZEOprep 60 ECO 40-63 μ) was purchased from AIC. Reagents were purchased mainly from Sigma Aldrich and were used as received.
6.2 General procedure for the synthesis of acyl Meldrum's acids
Method A
Adapted from the procedure of Sørensen et al.14 To a solution of Meldrum's acid (1 equiv) in dichloromethane at 0 °C was added pyridine (2 equiv) drop wise, and the resulting solution was stirred for 15 minutes. The corresponding acid chloride (1 equiv) was added to this reaction mixture. Thereafter, the reaction was stirred for 1.5 h at 0 °C, and for an additional 1.5 h at room temperature. The reaction was quenched with 2 M hydrochloric acid, and extracted with dichloromethane. The combined organic layers were dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography using a gradient from 5-10% ethyl acetate in hexane, 1% acetic acid to yield acyl Meldrum's acids (11a-e, j, l,p).
Method B
Adapted from the procedure of Sørensen et al.14. Meldrum's acid (1 equiv) and corresponding acid (1 equiv) were dissolved in DMF, and cooled to 0 °C. To this solution was added diethyl cyanophosphonate (1.1 equiv) and triethylamine (3.1 equiv) drop wise. The mixture was allowed to stir at 0 °C for 30 minutes followed by 16 h at room temperature. The reaction was quenched with 2 M hydrochloric acid and extracted twice with ethyl acetate. The combined organic extracts were washed with brine, and dried over sodium sulfate. The solution was filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography using a gradient from 5-10% ethyl acetate in hexane, 1% acetic acid to yield acyl Meldrum's acids (11f-i, k, m, n, o).
6.2.1 5-(1-hydroxyethylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11a)
Prepared using the general procedure above (Method A) from Meldrum's acid (500 mg, 3.47 mmol), acetyl chloride (0.25 mL, 3.47 mmol), pyridine (0.71 mL, 6.94 mmol), and CH2Cl2 (4.2 mL). Aqueous workup, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11a as a white solid (536 mg, 83%). 1H NMR (500 MHz, CDCl3) δ 15.12 (s, 1H), 2.67 (s, 3H), 1.73 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 194.8, 170.3, 160.6, 105.1, 92.0, 27.0, 23.7.
6.2.2 5-(cyclopropyl(hydroxy)methylene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11b)
Prepared using the general procedure above (Method A) from Meldrum's acid (500 mg, 3.46 mmol), cyclopropyl chloride (0.32 mL, 3.46 mmol), pyridine (0.56 mL, 6.93 mmol), and CH2Cl2 (5.0 mL). Aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11b as a yellowish oil (462 mg, 63%). 1H NMR (500 MHz, CDCl3) δ 15.41 (s, 1H), 3.54-3.44 (m, 1H), 1.74 (s, 6H), 1.49-1.41 (m, 2H), 1.32-1.20 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 198.1, 170.7, 161.4, 104.8, 91.2, 26.9, 15.7, 14.3.
6.2.3 5-(1-hydroxy-2-methylpropylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11c)
Prepared using the general procedure above (Method A) from Meldrum's acid (2.00 g, 13.9 mmol), isobutyryl chloride (1.46 mL, 13.9 mmol), pyridine (2.24 mL, 27.7 mmol), and CH2Cl2 (17.0 mL). Aqueous workup, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11c as a yellowish oil (1.77 g, 60%). 1H NMR (500 MHz, CDCl3) δ 15.53 (s, 1H), 4.08 (hept, J = 6.8 Hz, 1H), 1.73 (s, 6H), 1.23 (d, J = 6.8 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 202.6, 171.1, 160.1, 104.8, 90.3, 33.2, 26.9, 19.2.
6.2.4 5-(cyclobutyl(hydroxy)methylene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11d)
Prepared using the general procedure above (Method A) from Meldrum's acid (500 mg, 3.47 mmol), cyclobutane carbonyl chloride (0.40 mL, 3.47 mmol), pyridine (0.71 mL, 6.94 mmol), and CH2Cl2 (4.2 mL). Aqueous work up and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11d as a yellowish oil (604 mg, 77%). 1H NMR (500 MHz, CDCl3) δ 15.53 (s, 1H), 4.47 (quintet, J = 8.5 Hz, 1H), 2.42-2.27 (m, 4H), 2.12-2.01 (m, 1H), 1.96-1.86 (m, 1H), 1.72 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 198.3, 170.8, 160.0, 104.8, 89.8, 39.3, 26.8, 25.4, 17.9. HRMS (ESI) calcd for C11H14O5 [M-H]− 225.0768, found 225.0759.
6.2.5 5-(1-hydroxy-2-methylbutylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11e)
Prepared using the general procedure above (Method A) from Meldrum's acid (3.00 g, 20.8 mmol), 2-methyl butyryl chloride (2.58 mL, 20.8 mmol), pyridine (3.36 mL, 41.6 mmol), and CH2Cl2 (25.0 mL). Aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11e as a yellowish oil (2.44 g, 52%). 1H NMR (500 MHz, CDCl3) δ 15.48 (s, 1H), 3.97 (hextet, J = 6.9 Hz, 1H), 1.81-1.74 (m, 1H), 1.73 (s, 6H), 1.54 (doublet of quintet, J = 14.6, 7.3 Hz, 1H), 1.21 (d, J = 6.8 Hz, 3H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 202.2, 170.9, 160.3, 104.8, 91.3, 39.5, 27.3, 27.0, 26.8, 17.1, 11.9. HRMS (ESI) calcd for C11H16O5 [M+Na]+ 251.0895, found 251.1618.
6.2.6 5-(cyclopentyl(hydroxy)methylene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11f)
Prepared using the general procedure above (Method B) from Meldrum's acid (500 mg, 3.47 mmol), cyclopentanoic acid (0.38 mL, 3.47 mmol), diethyl cyanophosphonate (0.58 mL, 3.82 mmol), triethylamine (1.52 mL, 10.7 mmol), and DMF (7.2 mL). Aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11f as an off white solid (721 mg, 86%); mp 68.8-70.7 °C. 1H NMR (500 MHz, CDCl3) δ 15.51 (s, 1H), 4.24-4.17 (m, 1H), 2.08-1.98 (m, 2H), 1.88-1.75 (m, 5H), 1.73 (s, 7H), 1.71-1.66 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 201.6, 171.0, 160.4, 104.8, 90.8, 43.9, 31.1, 26.9, 26.6. HRMS (ESI) calcd for C12H16O5 [M-H]− 239.0925, found 239.0935.
6.2.7 5-(1-hydroxy-2-methylpentylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11g)
Prepared using the general procedure above (Method B) from Meldrum's acid (3.00 g, 20.8 mmol), 2-methyl valeric acid (2.60 mL, 20.8 mmol), diethyl cyanophosphonate (3.47 mL, 22.9 mmol), triethylamine (8.99 mL, 64.6 mmol), and DMF (25.0 mL). Aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11g as a yellowish oil (3.96 g, 79%). 1H NMR (500 MHz, CDCl3) δ 15.46 (s, 1H), 4.05 (hextet, J = 6.8 Hz, 1H), 1.71 (s, 6H), 1.50-1.24 (m, 4H), 1.20 (d, J = 6.8 Hz, 3H), 0.89 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 202.2, 170.9, 160.2, 104.7, 91.1, 37.8, 36.2, 26.9, 26.7, 20.6, 17.5, 14.1. HRMS (ESI) calcd for C12H18O5 [M-H]− 241.1081, found 241.1091.
6.2.8 5-(2-ethyl-1-hydroxybutylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11h)
Prepared using the general procedure above (Method B) from Meldrum's acid (2.00 g, 13.9 mmol), 2-ethylbutyric acid (1.75 mL, 13.9 mmol), diethyl cyanophosphonate (2.32 mL, 15.3 mmol), triethylamine (5.99 mL, 43.0 mmol), and DMF (28.8 mL). Aqueous workup and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11h as a yellowish oil (1.65 g, 49%). 1H NMR (500 MHz, CDCl3) δ 15.43 (s, 1H), 4.02-3.93 (m, 1H), 1.73 (s, 6H), 1.77-1.68 (m, 2H), 1.68-1.58 (m, 2H), 0.92 (t, J = 7.5 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 201.6, 170.7, 160.5, 104.7, 93.0, 46.1, 26.9, 25.9, 11.8. HRMS (ESI) calcd for C12H18O5 [M-H]− 242.1116, found 242.1096.
6.2.9 5-(2-ethyl-1-hydroxyhexylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11i)
Prepared using the general procedure above (Method B) from Meldrum's acid (2.00 g, 13.9 mmol), 2-ethylhexanoic acid (2.21 mL, 13.9 mmol), diethyl cyanophosphonate (2.32 mL, 15.3 mmol), triethylamine (5.99 mL, 43.0 mmol), and DMF (28.8 mL). Aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11i as a yellowish oil (2.82 g, 75%). 1H NMR (500 MHz, CDCl3) δ 15.42 (s, 1H), 4.04 (tt, J = 8.7, 5.6 Hz, 1H), 1.73 (s, 6H), 1.72-1.52 (m, 4H), 1.34-1.18 (m, 4H), 0.91 (t, J = 7.5 Hz, 3H), 0.87 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 201.8, 170.8, 160.5, 104.7, 92.8, 44.6, 32.5, 29.6, 26.9, 26.9, 26.4, 22.9, 14.0, 11.8. HRMS (ESI) calcd for C14H22O5 [M-H]− 269.1394, found 269.1421.
6.2.10 5-(1-hydroxy-3-methylbutylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11j)
Prepared using the general procedure above (Method A) from Meldrum's acid (300 mg, 2.08 mmol), isovaleryl chloride (0.25 mL, 2.08 mmol), pyridine (0.34 mL, 4.16 mmol), and CH2Cl2 (2.5 mL). Aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11j as a yellowish oil (274 mg, 58%). 1H NMR (500 MHz, CDCl3) δ 15.30 (s, 1H), 2.98 (d, J = 7.1 Hz, 2H), 2.26-2.14 (m, 1H), 1.73 (s, 6H), 1.01 (d, J = 6.7 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 197.7, 170.7, 160.4, 104.8, 92.0, 44.0, 27.5, 27.0, 22.7. HRMS (ESI) calcd for C11H16O5 [M-H]− 227.0925, found 227.0939.
6.2.11 5-(1-hydroxy-3-methylpentylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11k)
Prepared using the general procedure above (Method B) from Meldrum's acid (300 mg, 2.08 mmol), 3-methylvaleric acid (0.26 mL, 2.08 mmol), diethyl cyanophosphonate (0.35 mL, 2.29 mmol), triethylamine (0.86 mL, 6.45 mmol), and DMF (4.3 mL). Aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11k as a yellowish oil (393 mg, 78%). 1H NMR (500 MHz, CDCl3) δ 15.31 (s, 1H), 3.08 (dd, J = 13.4, 7.2 Hz, 1H), 2.91 (dd, J = 13.4, 7.2 Hz, 1H), 2.04-1.93 (m, 1H), 1.72 (s, 6H), 1.44 (doublet of quintet, J = 14.8, 7.4, 1H), 1.30 (doublet of quintet, J = 14.8, 7.4 Hz, 1H), 0.96 (d, J = 6.7 Hz, 3H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 198.1, 170.7, 160.4, 104.8, 92.1, 42.2, 33.7, 29.8, 26.9, 19.2, 11.4. HRMS (ESI) calcd for C12H18O5 [M-H]− 241.1081, found 241.1093.
6.2.12 5-(1-hydroxy-3,3-dimethylbutylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11l)
Prepared using the general procedure above (Method A) from Meldrum's acid (500 mg, 3.47 mmol), 3,3-dimethylbutanoyl chloride (0.49 mL, 3.47 mmol), pyridine (0.71 mL, 6.94 mmol), and CH2Cl2 (4.0 mL). Aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11l as an off white solid (377 mg, 45%). 1H NMR (500 MHz, CDCl3) δ 15.38 (s, 1H), 3.12 (s, 2H), 1.73 (s, 6H), 1.07 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 196.9, 170.6, 160.5, 104.4, 93.0, 46.4, 34.0, 30.0, 26.8.
6.2.13 5-(1-hydroxy-4-methylpentylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11m)
Prepared using the general procedure above (Method B) from Meldrum's acid (500 mg, 3.47 mmol), 4-methylvaleric acid (0.44 mL, 3.47 mmol), diethyl cyanophosphonate (0.58 mL, 3.82 mmol), triethylamine (1.52 mL, 10.7 mmol), and DMF (7.2 mL). Aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11m as a yellowish oil (646 mg, 77%). 1H NMR (500 MHz, CDCl3) δ 15.29 (s, 1H), 3.11-3.03 (m, 2H), 1.74 (s, 6H), 1.72-1.62 (m, 1H), 1.61-1.55 (m, 2H), 0.95 (d, J = 6.5 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 198.8, 170.7, 160.3, 104.9, 91.3, 35.1, 34.1, 28.3, 26.9, 22.4. HRMS (ESI) calcd for C12H18O5 [M-H]− 241.1154, found 241.073.
6.2.14 5-(3-cyclopropyl-1-hydroxypropylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11n)
Prepared using the general procedure above (Method B) from Meldrum's acid (350 mg, 2.40 mmol), 3-cyclobutylpropanoic acid34 (0.27 g, 2.40 mmol), diethyl cyanophosphonate (0.45 mL, 2.64 mmol), triethylamine (1.04 mL, 7.44 mmol), and DMF (4.0 mL). Aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11n as a yellowish oil (38 mg, 65%). 1H NMR (400 MHz, CDCl3) δ 15.26 (s, 1H), 3.15 (t, J = 7.3 Hz, 2H), 1.70 (s, 6H), 1.58 (q, J = 7.3 Hz, 2H), 0.48-0.39 (m, 3H), 0.08 (q, J = 5.7, 5.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 198.1, 170.6, 160.2, 104.8, 91.4, 36.0, 31.4, 26.9, 10.8, 4.7. HRMS (ESI) calcd for C12H16O5 [M-H]− 239.0925, found 239.0917.
6.2.15 5-(3-cyclobutyl-1-hydroxypropylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11o)
Prepared using the general procedure above (Method B) from Meldrum's acid (1.05 g, 7.26 mmol), 3-cyclobutylpropanoic acid34 (939 mg, 7.26 mmol), diethyl cyanophosphonate (1.2 mL, 7.99 mmol), triethylamine (3.14 mL, 22.51 mmol), and DMF (13.0 mL). Aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11o as a yellowish oil (1.31 g, 70%). 1H NMR (400 MHz, CDCl3) δ 15.24 (s, 1H), 2.94 (m, 2H), 2.33 (hept, J = 7.9 Hz, 1H), 2.11-1.98 (m, 2H), 1.90-1.70 (m, 4H), 1.71 (s, 6H), 1.63 (pd, J = 9.1, 1.8 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 198.4, 170.6, 160.2, 104.8, 91.3, 35.8, 33.9, 33.3, 28.0, 26.9, 18.3. HRMS (ESI) calcd for C13H18O5 [M-H]− 253.1081, found 253.1077.
6.2.16 5-(3-cyclopentyl-1-hydroxypropylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (11p)
Prepared using the general procedure above (Method A) from Meldrum's acid (1.50 g, 10.4 mmol), cyclopentane propionyl chloride (1.59 mL, 10.4 mmol), pyridine (2.12 mL, 20.8 mmol), and CH2Cl2 (13.0 mL). However, the reaction was stirred at room temperature for 3.5 h instead of 1.5 h for this particular substrate. Aqueous workup and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 11p as a yellowish oil (2.42 mg, 87%). 1H NMR (500 MHz, CDCl3) δ 15.27 (s, 1H), 3.09-3.04 (m, 2H), 1.91-1.75 (m, 3H), 1.73 (s, 6H), 1.72-1.67 (m, 2H), 1.66-1.57 (m, 2H), 1.57-1.47 (m, 2H), 1.20-1.08 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 198.6, 170.7, 160.3, 104.9, 91.3, 40.1, 35.4, 32.6, 27.0, 25.3. HRMS (ESI) calcd for C14H20O5 [M-H]− 267.1238, found 267.1228.
6.3 General procedure for the synthesis of N,O-diBoc-protected β-keto hydroxamic acids
Method C
Adapted from the procedure of Sørensen et al.14 To a solution of acyl Meldrum's acid (196 mg, 0.915 mmol) in toluene (8.5 mL) was added N,O-bis(tert-butoxycarbonyl)hydroxylamine (213 mg, 0.915 mmol). The resulting reaction mixture was stirred at 65 °C for 16 h, and then cooled to room temperature. The solvent was concentrated in vacuo, and the residue was purified on silica gel chromatography using 5% ethyl acetate in hexane to yield the intermediate β-keto hydroxamic acid (12b, c, e, j, k, m-o) as colorless oil. In the case of 12e, and 12m-o, this procedure gave the desired hydroxamic acids as roughly 9:1 mixtures with residual BocNHOBoc. Rather than achieve analytical purity for these compounds, the crude products were used directly in the next step. Thus synthetic details and characterization data for 12e, and 12m-o are not reported below.
Method D
To a solution of acyl Meldrum's acid (314 mg, 1.69 mmol, 1.8 equiv) in dry toluene (4.5 mL) at room temperature was added N,O-bis(tert-butoxycarbonyl)hydroxylamine (219 mg, 0.938 mmol, 1.0 equiv), and the reaction mixture was immersed in an oil bath preheated to 90 °C. The reaction was monitored for the complete consumption of N,O-bis(tertbutoxycarbonyl)hydroxylamine. Leaving the reaction for longer time can lead to loss in yield and competing side reactions. After concentration in vacuo, the residue was purified on silica gel chromatography using 5% ethyl acetate in hexane to yield β-keto hydroxamic acid (12a, d, f-i, l, p) as colorless oil.
6.3.1 tert-butyl tert-butoxycarbonyloxy(3-oxobutanoyl)carbamate (12a)
Prepared using the general procedure above (Method D) from acyl Meldrum's acid (11a) (314 mg, 1.69 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (219 mg, 0.94 mmol), and toluene (4.5 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12a as a clear oil (212 mg, 72%). 1H NMR (500 MHz, CDCl3) of keto tautomer δ 4.11-3.85 (m, 2H), 2.26 (s, 3H), 1.54 (s, 9H), 1.50 (s, 9H). 13C NMR (126 MHz, CDCl3) of keto tautomer δ199.9, 163.3, 151.0, 149.7, 86.5, 86.1, 52.6, 30.1, 27.9, 27.6. The keto/enol ratio for this compound was approximately 6:1.
6.3.2 tert-butyl tert-butoxycarbonyloxy(3-cyclopropyl-3-oxopropanoyl)carbamate (12b)
Prepared using the general procedure above (Method C) from acyl Meldrum's acid (11b) (600 mg, 2.82 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (659 mg, 2.82 mmol), and toluene (26.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12b as a clear oil (503 mg, 52%). 1H NMR of keto tautomer (500 MHz, CDCl3) δ 4.27-3.98 (m, 2H), 2.02-1.96 (m, 1H), 1.53 (s, 9H), 1.50 (s, 9H), 1.15-1.06 (m, 2H), 0.98-0.90 (m, 2H). 13C NMR of keto tautomer (126 MHz, CDCl3) δ 202.2, 163.6, 151.0, 149.5, 86.3, 85.9, 52.5, 27.9, 27.6, 20.8, 11.6. The keto/enol ratio for this compound was approximately 33:1.
6.3.3 tert-butyl tert-butoxycarbonyloxy(4-methyl-3-oxopentanoyl)carbamate (12c)
Prepared using the general procedure above (Method C) from acyl Meldrum's acid (11c) (196 mg, 0.92 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (213 mg, 0.92 mmol), and toluene (8.5 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12c as a clear oil (100 mg, 32%). 1H NMR (500 MHz, CDCl3) of keto tautomer δ 4.23-3.89 (m, 2H), 2.73 (hept, J = 6.9 Hz, 1H), 1.54 (s, 9H), 1.49 (s, 9H), 1.13 (d, J = 6.9 Hz, 6H). 13C NMR (126 MHz, CDCl3) for keto/enol tautomeric mixture δ 206.0, 187.8, 168.7, 163.8, 151.5, 151.0, 149.7, 87.6, 86.3, 86.2, 85.8, 85.3, 50.1, 41.1, 34.9, 28.0, 27.9, 27.7, 27.6, 19.9, 18.1. The keto/enol ratio for this compound was approximately 4:1.
6.3.4 tert-butyl tert-butoxycarbonyloxy(3-cyclobutyl-3-oxopropanoyl)carbamate (12d)
Prepared using the general procedure above (Method D) from acyl Meldrum's acid (11d) (522 mg, 2.30 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (298 mg, 1.28 mmol), and toluene (6.4 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12d as a clear oil (crude) contaminated with residual N,O-bis(tert-butoxycarbonyl)hydroxylamine as the impurity (389 mg, 85% (crude)). 1H NMR (500 MHz, CDCl3) of keto tautomer δ 4.13-3.77 (m, 2H), 3.37 (p, J = 9.0 Hz, 1H), 2.37-2.22 (m, 2H), 2.21-2.10 (m, 2H), 2.01-1.90 (m, 1H), 1.88-1.77 (m, 1H), 1.54 (s, 9H), 1.49 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 203.2, 184.7, 168.6, 163.6, 151.5, 151.0, 149.6, 149.4, 88.2, 86.4, 86.2, 85.9, 85.3, 53.6, 49.6, 45.6, 40.1, 27.9, 27.7, 26.1, 24.5, 18.3, 17.9. HRMS (ESI) calcd for C17H27NO7 [M+Na]+ 380.1680, found 380.1706. The keto/enol ratio for this compound was approximately 5:1.
6.3.5 tert-butyl tert-butoxycarbonylox(3-cyclopentyl-3-oxopropanoyl)carbamate (12f)
Prepared using the general procedure above (Method D) from acyl Meldrum's acid (11f) (93.3 mg, 0.39 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (50.0 mg, 0.22 mmol), and toluene (1.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12f as a clear oil (59.0 mg, 73%). 1H NMR (500 MHz, CDCl3) of keto tautomer δ 4.22-3.89 (m, 2H), 2.98 (p, J = 8.0 Hz, 1H), 1.91-1.76 (m, 4H), 1.70-1.63 (m, 2H), 1.62-1.56 (m, 2H), 1.54 (s, 9H), 1.49 (s, 9H). 13C NMR (126 MHz, CDCl3) of keto/enol tautomeric mixture δ 204.7, 186.6, 168.5, 163.7, 151.5, 151.0, 149.6, 149.4, 88.4, 86.3, 86.2, 85.8, 85.3, 51.7, 51.2, 45.9, 30.9, 28.9, 28.0, 27.9, 27.7, 27.6, 26.0, 26.0. HRMS (ESI) calcd for C18H29NO7 [M+Na]+ 394.1836, found 394.1868. The keto/enol ratio for this compound was approximately 6:1.
6.3.6 tert-butyl tert-butoxycarbonyloxy(4-methyl-3-oxoheptanoyl)carbamate (12g)
Prepared using the general procedure above (Method D) from acyl Meldrum's acid (11g) (278 mg, 1.15 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (149 mg, 0.64 mmol), and toluene (3.1 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12g as a clear oil (163 mg, 68%). 1H NMR (500 MHz, CDCl3) of keto tautomer δ 4.22-3.88 (m, 2H), 2.65 (h, J = 6.7 Hz, 1H), 1.73-1.62 (m, 2H), 1.54 (s, 9H), 1.49 (s, 9H), 1.40-1.26 (m, 2H), 1.11 (d, J = 7.0 Hz, 3H), 0.89 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) of keto/enol tautomer δ 205.9, 187.3, 168.6, 163.7, 151.5, 151.0, 149.7, 149.4, 88.7, 86.3, 86.2, 85.8, 85.3, 50.6, 46.2, 40.4, 36.6, 34.8, 28.0, 27.9, 27.7, 27.6, 20.6, 20.3, 18.2, 15.9, 14.2, 14.1. HRMS (ESI) calcd for C18H31NO7 [M+H2O] 391.21, found 391.25. The keto/enol ratio for this compound was approximately 6:1.
6.3.7 tert-butyl tert-butoxycarbonyloxy(4-ethyl-3-oxohexanoyl)carbamate (12h)
Prepared using the general procedure above (Method D) from acyl Meldrum's acid (11h) (942 mg, 3.9 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (504 mg, 2.16 mmol), and toluene (11.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12h as a clear oil (536 mg, 66%). 1H NMR of keto/enol tautomeric mixture (500 MHz, CDCl3) δ 13.13 (s, 1H), 6.14 (s, 1H), 4.29-3.80 (m, 2H), 2.49-2.42 (m, 1H), 2.00-1.92 (m, 1H), 1.75-1.55 (m, 4H), 1.55 (s, 9H), 1.54 (s, 9H), 1.53 (s, 9H), 1.49 (s, 9H), 0.91-0.84 (m, 6H). 13C NMR of keto/enol tautomer (126 MHz, CDCl3) δ 205.4, 185.9, 168.4, 163.7, 151.5, 151.0, 149.6, 149.3, 90.4, 86.3, 86.2, 85.7, 85.3, 55.0, 51.3, 50.2, 28.0, 27.9, 27.7, 27.6, 25.7, 23.3, 12.1, 11.5. HRMS (ESI) calcd for C18H31NO7 [M+Na]+ 396.1993, found 396.197. The keto/enol ratio for this compound was approximately 2:1.
6.3.8 tert-butyl tert-butoxycarbonyloxy(4-ethyl-3-oxooctanoyl)carbamate (12i)
Prepared using the general procedure above (Method D) from acyl Meldrum's acid (11i) (54.6 mg, 0.20 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (26.1 mg, 0.11 mmol), and toluene (0.9 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12i as a clear oil (30.2 mg, 66%). 1H NMR of keto/enol tautomeric mixture (500 MHz, CDCl3) δ 13.14 (s, 0.5H), 6.13 (s, 0.5H), 4.41-3.63 (m, 2H), 2.50 (p, J = 6.5, 5.9 Hz, 1H), 2.07-2.00 (m, 0.5H), 1.80-1.57 (m, 4H), 1.55 (s, 4.5H), 1.54 (s, 9H), 1.53 (s, 4.5H), 1.49 (s, 9H), 1.48-1.35 (m, 2H), 1.34-1.15 (m, 7H), 0.91 - 0.84 (m, 9H). 13C NMR of keto/enol tautomer (126 MHz, CDCl3) δ 205.5, 186.1, 168.5, 163.7, 151.5, 151.0, 149.6, 149.3, 90.3, 86.3, 86.2, 85.7, 85.3, 53.6, 51.3, 48.6, 32.5, 30.2, 29.7, 29.3, 28.1, 27.9, 27.7, 27.7, 26.1, 23.9, 23.0, 22.9, 14.1, 14.1, 12.1, 11.6. HRMS (ESI) calcd for C20H35NO7 [M+Na]+ 424.2306, found 424.2343. The keto/enol ratio for this compound was approximately 2:1.
6.3.9 tert-butyl tert-butoxycarbonyloxy(5-methyl-3-oxohexanoyl)carbamate (12j)
Prepared using the general procedure above (Method C) from acyl Meldrum's acid (11j) (239 mg, 1.05 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (244 mg, 1.05 mmol), and toluene (9.7 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12j as a clear oil (125 mg, 33%). 1H NMR (500 MHz, CDCl3) of keto tautomer δ 4.16-3.74 (m, 2H), 2.41 (d, J = 6.8 Hz, 2H), 2.22-2.11 (m, 1H), 1.54 (s, 9H), 1.49 (s, 9H), 0.93 (d, J = 6.7 Hz, 6H). 13C NMR (126 MHz, CDCl3) for keto/enol tautomeric mixture δ 201.6, 182.3, 168.3, 163.3, 151.3, 150.8, 149.5, 149.2, 90.5, 86.2, 86.0, 85.7, 85.2, 52.2, 51.5, 45.2, 27.9, 27.8, 27.5, 27.5, 26.6, 24.1, 22.4. HRMS (ESI) calcd for C17H29NO7 [M+Na]+ 382.1836, found 382.869. The keto/enol ratio for this compound was approximately 4:1.
6.3.10 tert-butyl tert-butoxycarbonyloxy(5-methyl-3-oxoheptanoyl)carbamate (12k)
Prepared using the general procedure above (Method C) from acyl Meldrum's acid (11k) (380 mg, 1.57 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (366 mg, 1.57 mmol), and toluene (14.5 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12k as a clear oil (191 mg, 33%). 1H NMR (500 MHz, CDCl3) of keto tautomer δ 4.25-3.66 (m, 2H), 2.59-2.44 (m, 1H), 2.39-2.18 (m, 1H), 1.95 (m, J = 13.4, 6.7 Hz, 1H), 1.54 (s, 9H), 1.49 (s, 9H), 1.42-1.27 (m, 1H), 1.26 - 1.12 (m, 1H), 0.90 (d, J = 6.7 Hz, 3H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) for keto/enol tautomeric mixture δ 201.8, 182.6, 168.2, 163.4, 151.3, 150.8, 149.5, 149.2, 90.6, 86.2, 86.1, 85.7, 85.2, 52.2, 49.7, 43.3, 32.8, 30.2, 29.4, 29.3, 27.9, 27.8, 27.5, 27.5, 19.3, 19.0, 11.3, 11.2. HRMS (ESI) calcd for C18H31NO7 [M+Na]+ 396.1993, found 396.2007. The keto/enol ratio for this compound was approximately 4:1.
6.3.11 tert-butyl tert-butoxycarbonyloxy(5,5-dimethyl-3-oxohexanoyl)carbamate (12l)
Prepared using the general procedure above (Method D) from acyl Meldrum's acid (11l) (362 mg, 1.49 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (194 mg, 0.83 mmol), and toluene (4.2 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12l as a clear oil (242 mg, 78%). 1H NMR (500 MHz, CDCl3) of keto tautomer δ 4.14-3.67 (m, 2H), 2.44 (s, 2H), 1.54 (s, 9H), 1.49 (s, 9H), 1.03 (s, 9H). 13C NMR (126 MHz, CDCl3) of keto/enol tautomeric mixture δ 201.4, 182.0, 168.4, 163.5, 151.5, 151.0, 149.7, 149.3, 91.8, 86.3, 86.2, 85.8, 85.3, 54.9, 53.6, 49.9, 31.9, 30.9, 30.0, 29.6, 28.0, 27.9, 27.7, 27.7. The keto/enol ratio for this compound was approximately 2:1.
6.3.12 tert-butyl tert-butoxycarbonyloxy(5-cyclopentyl-3-oxopentanoyl)carbamate (12p)
Prepared using the general procedure above (Method D) from acyl Meldrum's acid (11p) (2.32 g, 8.65 mmol), N,O-bis(tert-butoxycarbonyl)-hydroxylamine (2.02 g, 8.65 mmol), and toluene (80.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (5% ethyl acetate in hexane) yielded 12p as a clear oil (1.50 g, 43%). 1H NMR (500 MHz, CDCl3) of keto tautomer δ 4.26-3.54 (m, 2H), 2.57-2.50 (m, 2H), 1.81-1.70 (m, 3H), 1.65-1.57 (m, 6H), 1.54 (s, 9H), 1.49 (s, 9H), 1.15-1.00 (m, 2H). 13C NMR (126 MHz, CDCl3) δ of keto tautomer 202.2, 163.4, 150.8, 149.5, 86.2, 85.8, 51.8, 41.9, 39.4, 32.4, 29.4, 27.8, 27.5, 25.1. HRMS (ESI) calcd for C20H33NO7 [M+Na]+ 422.2149, found 422.2172. The keto/enol ratio for this compound was approximately 6:1.
6.4 General procedure for the synthesis of 5-substituted isoxazol-3-ols 13a-p
Method E
Adapted from the procedure of Sørensen et al.14 β-keto hydroxamic acid (496 mg, 1.33 mmol) was dissolved in methanol (3.0 mL), and added drop wise to concentrated HCl (5.0 mL) preheated to 50 °C. The reaction mixture was stirred for 2.5 h at 50 °C prior to being brought to room temperature. The reaction mixture was concentrated in vacuo. The residual reaction mixture was partitioned between water and dichloromethane. The aqueous layer was extracted twice with dichloromethane and the combined organic layers were dried with sodium sulfate. The solvent was evaporated, and the subsequent residue was purified on silica gel chromatography using a gradient from 5-10% ethyl acetate in hexane, 1% acetic acid to yield isoxazol-3-ols.
6.4.1 5-methylisoxazol-3-ol (13a)
Prepared using the general procedure above (Method E) from β-keto hydroxamic acid (12a) (66.5 mg, 0.21 mmol), concentrated HCl (1.0 mL), and methanol (0.5 mL). Concentration of the reaction mixture in vacuofollowed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13a as a white solid (15.5 mg, 75%). 1H NMR (500 MHz, CDCl3) 11.57 (s, 1H), 5.67 (s, 1H), 2.33 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 171.4, 170.4, 94.0, 13.0.
6.4.2 5-cyclopropylisoxazol-3-ol (13b)
Prepared using the general procedure above (Method E) from β-keto hydroxamic acid (12b) (2.69 g, 7.83 mmol), concentrated HCl (25.0 mL), and methanol (16.0 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13b as an off white solid (464 mg, 47%). 1H NMR (500 MHz, CDCl3) δ 10.94 (s, 1H), 5.57 (s, 1H), 1.95-1.86 (m, 1H), 1.08-1.00 (m, 2H), 0.97-0.91 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 175.6, 171.5, 91.1, 8.6, 8.3.
6.4.3 5-isopropylisoxazol-3-ol (13c)
Prepared using the general procedure above (Method E) from β-keto hydroxamic acid (12b) (973 mg, 2.82 mmol), concentrated HCl (12.0 mL), and methanol (6.0 mL). Concentration of the reaction mixture in vacuofollowed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13c as a white solid (276 mg, 77%). 1H NMR (500 MHz, CDCl3) δ 10.17 (s, 1H), 5.64 (s, 1H), 2.94 (hept, J = 6.9 Hz, 1H), 1.27 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 179.7, 171.2, 91.5, 27.7, 20.6.
6.4.4 5-cyclobutylisoxazol-3-ol (13d)
Prepared using the general procedure above (Method E) from β-keto hydroxamic acid (12d) (318 mg, 0.89 mmol), concentrated HCl (3.1 mL), and methanol (2.0 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13d as a semi-solid (75.0 mg, 61%). 1H NMR (500 MHz, CDCl3) δ 5.68 (s, 1H), 3.50 (p, J = 8.5 Hz, 1H), 2.40-2.31 (m, 2H), 2.30-2.20 (m, 2H), 2.10-2.01 (m, 1H), 2.01-1.91 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 177.4, 171.2, 91.9, 32.6, 27.8, 18.9. HRMS (ESI) calcd for C7H9NO2 [M+H]+ 140.0706, found 140.0693.
6.4.5 5-sec-butylisoxazol-3-ol (13e)
Prepared using the general procedure above (Method E) from crude β-keto hydroxamic acid (12e) (1.96 g, 5.53 mmol), concentrated HCl (20.0 mL), and methanol (15.0 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13e as an off white solid (620 mg, 77%); mp 37.0-38.0 °C. 1H NMR (500 MHz, CDCl3) δ 11.41 (s, 1H), 5.64 (s, 1H), 2.74 (h, J = 7.0 Hz, 1H), 1.74-1.64 (m, 1H), 1.58 (doublet of quintet, J = 14.1, 7.2 Hz, 1H), 1.24 (d, J = 7.0 Hz, 3H), 0.90 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 178.8, 171.2, 92.1, 34.5, 28.2, 18.1, 11.4. HRMS (ESI) calcd for C7H11NO2 [M+H]+ 142.0863, found 142.086.
6.4.6 5-cyclopentylisoxazol-3-ol (13f)
Prepared using the general procedure above (Method E) from β-keto hydroxamic acid (12f) (1.77 g, 4.76 mmol), concentrated HCl (25.0 mL), and methanol (10.0 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13f as a white solid (508 mg, 70%); mp 71.3-73.4 °C. 1H NMR (500 MHz, CDCl3) δ 10.42 (s, 1H), 5.63 (s, 1H), 3.06 (quintet, J = 7.9 Hz, 1H), 2.09-1.96 (m, 2H), 1.80-1.61 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 178.4, 171.2, 91.8, 38.0, 31.6, 25.4. HRMS (ESI) calcd for C8H11NO2 [M+H]+ 154.0863, found 154.0855.
6.4.7 5-(pentan-2-yl)isoxazol-3-ol (13g)
Prepared using the general procedure above (Method E) from β-keto hydroxamic acid (12g) (2.85 g, 7.63 mmol), concentrated HCl (15.0 mL), and methanol (16.0 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13g as a colorless oil (889 mg, 75%). 1H NMR (500 MHz, CDCl3) δ 11.20 (s, 1H), 5.62 (s, 1H), 2.82 (h, J = 7.0 Hz, 1H), 1.64 (ddt, J = 13.5, 9.4, 6.6 Hz, 1H), 1.50 (ddt, J = 13.5, 9.4, 6.6 Hz, 1H), 1.38-1.26 (m, 2H), 1.23 (d, J = 7.1 Hz, 3H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 179.0, 171.2, 91.8, 37.4, 32.7, 20.2, 18.6, 13.9. HRMS (ESI) calcd for C8H13NO2 [M+H]+ 156.0946, found 156.1485.
6.4.8 5-(pentan-3-yl)isoxazol-3-ol (13h)
Prepared using the general procedure above (Method E) from β-keto hydroxamic acid (12h) (496 mg, 7.63 mmol), concentrated HCl (5.0 mL), and methanol (3.0 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13h an off white solid (115 mg, 81%); mp 49.0-50.7 °C. 1H NMR (500 MHz, CDCl3) δ 10.00 (s, 1H), 5.65 (s, 1H), 2.55 (tt, J = 8.3, 5.8 Hz, 1H), 1.72-1.56 (m, 4H), 0.87 (t, J = 7.4 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 177.7, 171.1, 93.0, 42.2, 26.3, 11.7. HRMS (ESI) calcd for C8H13NO2 [M+H]+ 156.1019, found 156.1013.
6.4.9 5-(heptan-3-yl)isoxazol-3-ol (13i)
Prepared using the general procedure above (Method E) from β-keto hydroxamic acid (12i) (2.29 g, 5.70 mmol), concentrated HCl (22.0 mL), and methanol (12.0 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13i as a colorless oil (396 mg, 40%). 1H NMR (500 MHz, CDCl3) δ 5.66 (s, 1H), 2.62 (quintet, J = 7.4 Hz, 1H), 1.74-1.53 (m, 4H), 1.39-1.12 (m, 6H), 0.95-0.81 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 178.1, 171.2, 93.0, 40.6, 33.2, 29.4, 26.8, 22.7, 14.1, 11.7. HRMS (ESI) calcd for C10H17NO2 [M+H]+ 184.1332, found 184.1339.
6.4.10 5-isobutylisoxazol-3-ol (13j)
Prepared using the general procedure above (Method E) from β-keto hydroxamic acid (12j) (2.98 g, 8.67 mmol), concentrated HCl (25.0 mL), and methanol (18.0 mL). Concentration of the reaction mixture in vacuofollowed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13j as an off white solid (804 mg, 66%). 1H NMR (500 MHz, CDCl3) δ 11.80 (s, 1H), 5.66 (s, 1H), 2.50 (d, J = 7.1 Hz, 2H), 2.06-1.94 (m, 1H), 0.95 (d, J = 6.7 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 173.8, 171.3, 93.8, 36.3, 27.6, 22.3.
6.4.11 5-(2-methylbutyl)isoxazol-3-ol (13k)
Prepared using the general procedure above (Method E) from -keto hydroxamic acid (12k) (3.09 g, 8.27 mmol), concentrated HCl (30.0 mL), and methanol (17.0 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13k as an oil (871 mg, 68%). 1H NMR (500 MHz, CDCl3) δ 10.87 (s, 1H), 5.66 (s, 1H), 2.62 (dd, J = 14.9, 6.1 Hz, 1H), 2.45 (dd, J = 14.9, 7.9 Hz, 1H), 1.84-1.71 (m, J = 6.6 Hz, 1H), 1.46-1.33 (m, 1H), 1.23 (dq, J = 21.1, 7.5 Hz, 2H), 0.92 (d, J = 6.7 Hz, 3H), 0.91 (t, J = 7.5 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.9, 171.3, 94.0, 34.4, 33.9, 29.2, 19.2, 11.4. HRMS (ESI) calcd for C8H13NO2 [M+H]+ 156.1019, found 156.1013.
6.4.12 5-neopentylisoxazol-3-ol (13l)
Prepared using the general procedure above (Method E) from β-keto hydroxamic acid (12l) (2.29 g, 6.40 mmol), concentrated HCl (20.0 mL), and methanol (14.0 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13l as an off white solid (664 mg, 74%). 1H NMR (500 MHz, CDCl3) δ 5.67 (s, 1H), 2.52 (s, 2H), 0.98 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 173.1, 171.1, 94.9, 41.6, 31.7, 29.6. HRMS (ESI) calcd for C8H13NO2 [M+H]+ 156.1019, found 156.1021.
6.4.13 5-isopentylisoxazol-3-ol (13m)
Prepared using the general procedure above (Method E) from crude β-keto hydroxamic acid (12m) (1.64 g, 4.39 mmol), concentrated HCl (11.0 mL), and methanol (9.0 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13m as a white solid (629 mg, 92%); mp 53.0-54.8. 1H NMR (500 MHz, CDCl3) δ 9.64 (s, 1H), 5.65 (s, 1H), 2.65-2.61 (m, 2H), 1.66-1.49 (m, 3H), 0.93 (d, J = 6.5 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 175.0, 171.3, 93.0, 36.1, 27.7, 25.4, 22.3. HRMS (ESI) calcd for C8H13NO2 [M+H]+ 156.1019, found 156.1011.
6.4.14 5-(2-cyclopropylethyl)isoxazol-3-ol (13n)
Prepared using the general procedure above (Method E) from crude β-keto hydroxamic acid (1.45 g), concentrated HCl (21.0 mL), and methanol (9.5 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13n as a slight yellow crystalline solid (290 mg, 34% over 2 steps). 1H NMR (400 MHz, CDCl3) δ 11.73 (s, 1H), 5.67 (s, 1H), 2.73 (t, J = 7.6 Hz, 2H), 1.56 (q, J = 7.3 Hz, 2H), 0.79-0.63 (m, 1H), 0.54 -0.35 (m, 2H), 0.06 (dt, J = 5.9, 4.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 174.5, 171.3, 93.2, 32.5, 27.6, 10.6, 4.6. HRMS (ESI) calcd for C8H11NO2 [M+H]+ 154.0863, found 154.0856.
6.4.15 5-(2-cyclobutylethyl)isoxazol-3-ol (13o)
Prepared using the general procedure above (Method E) from crude β-keto hydroxamic acid (1.18 g), concentrated HCl (7.5 mL), and methanol (16.5 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13o as a white crystalline solid (340 mg, 42% over 2 steps). 1H NMR (500 MHz, CDCl3) δ 11.81 (s, 1H), 5.63 (s, 1H), 2.52 (t, J = 7.7 Hz, 2H), 2.28 (hept, J = 8.1 Hz, 1H), 2.10-1.98 (m, 2H), 1.92-1.76 (m, 2H), 1.73 (q, J = 7.6 Hz, 2H), 1.60 (pd, J = 9.0, 2.4 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 174.6, 171.3, 93.0, 35.4, 34.3, 28.0, 25.1, 18.4. HRMS (ESI) calcd for C9H13NO2 [M+H]+M 168.1019, found 168.1022.
6.4.16 5-(2-cyclopentylethyl)isoxazol-3-ol (13p)
Prepared using the general procedure above (Method E) from β-keto hydroxamic acid (12p) (1.50 g, 3.75 mmol), concentrated HCl (13.0 mL), and methanol (8.0 mL). Concentration of the reaction mixture in vacuo followed by an aqueous work up, and silica gel chromatography (5-10% ethyl acetate in hexane, 1% acetic acid) afforded 13p as a white solid (521 mg, 77%); mp 67.0-68.0 °C. 1H NMR (500 MHz, CDCl3) δ 5.65 (s, 1H), 2.67-2.59 (m, 2H), 1.84-1.75 (m, 3H), 1.70-1.64 (m, 2H), 1.64-1.49 (m, 4H), 1.17-1.01 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 174.9, 171.3, 93.0, 39.6, 33.6, 32.6, 26.7, 25.3. HRMS (ESI) calcd for C10H15NO2 [M+H]+ 182.1176, found 182.1182.
6.5 General procedure for the synthesis of N,N-dimethyl-3-oxoisoxazole-2(3H)-carboxamide 14a-h,j-p
Method F
To solution of isoxazolol (62.0 mg, 0.40 mmol) in toluene (2.0 mL) was added N,N-dimethylcarbamoyl chloride (0.19 mL, 2.00 mmol) under nitrogen atmosphere. The resulting reaction mixture was refluxed overnight. The reaction mixture was cooled to room temperature and solvent was evaporated in vacuo. The resulting residue was purified by flash column chromatography on silica gel using a gradient of 20-30% ethyl acetate in hexane to yield carboxamide as the major product and carbamate as the minor product.
6.5.1 N,N,5-trimethyl-3-oxoisoxazole-2(3H)-carboxamide (14a)
Prepared using the general procedure above (Method F) from 5-methylisoxazol-3-ol (13a) (45.7 mg, 0.46 mmol), N,N-dimethylcarbamoyl chloride (0.21 mL, 2.31 mmol), and toluene (2.5 mL). Concentration of the reaction mixture in vacuofollowed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14a as an off white solid (66.3 mg, 84%). 1H NMR (500 MHz, CDCl3) δ 5.41 (s, 1H), 3.10 (bs, 6H), 2.31 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 174.5, 167.9, 150.2, 96.9, 38.8, 37.0, 13.9. HRMS (ESI) calcd for C7H10N2O3 [M+H]+ 171.0691, found 171.0525.
6.5.2 5-cyclopropyl-N,N-dimethyl-3-oxoisoxazole-2(3H)-carboxamide (14b)
Prepared using the general procedure above (Method F) from 5-cyclopropylisoxazol-3-ol (13b) (20.5 mg, 0.16 mmol), N,N-dimethylcarbamoyl chloride (0.08 mL, 0.82 mmol), and toluene (1.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14b as a clear oil (4.60 mg, 14%). 1H NMR (500 MHz, CDCl3) δ 5.27 (s, 1H), 3.07 (bs, 6H), 1.91-1.81 (m, 1H), 1.19-1.00 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 180.0, 168.3, 150.4, 92.9, 38.8, 36.9, 9.3, 9.3. HRMS (ESI) calcd for C9H12N2O3 [M+H]+ 197.0921, found 197.0933.
6.5.3 5-isopropyl-N,N-dimethyl-3-oxoisoxazole-2(3H)-carboxamide (14c)
Prepared using the general procedure above (Method F) from 5-isopropylisoxazol-3-ol (13c) (136 mg, 1.07 mmol), N,N-dimethylcarbamoyl chloride (0.49 mL, 5.34 mmol), and toluene (6.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14c as a white solid (129 mg, 61%); mp 94.0-95.5 °C. 1H NMR (500 MHz, CDCl3) δ 5.37 (s, 1H), 3.10 (bs, 6H), 2.88 (hept, J = 6.7 Hz, 1H), 1.29 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 183.2, 168.0, 150.3, 94.1, 38.8, 37.1, 28.2, 19.9. HRMS (ESI) calcd for C9H14N2O3 [M+H]+ 198.1004, found 199.1083.
6.5.4 5-cyclobutyl-N,N-dimethyl-3-oxoisoxazole-2(3H)-carboxamide (14d)
Prepared using the general procedure above (Method F) from 5-cyclobutylisoxazol-3-ol (13d) (63.0 mg, 0.45 mmol), N,N-dimethylcarbamoyl chloride (0.21 mL, 2.26 mmol), and toluene (2.2 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14d as a white solid (75.0 mg, 79%); mp 62.5-64.3 °C. 1H NMR (500 MHz, CDCl3) δ 5.42 (s, 1H), 3.44 (quintet, J = 8.6 Hz, 1H), 3.10 (bs, 6H), 2.42-2.33 (m, 2H), 2.32-2.22 (m, 2H), 2.13-2.02 (m, 1H), 2.02-1.90 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 180.9, 167.9, 150.3, 94.5, 38.8, 37.0, 32.8, 27.1, 18.9. HRMS (ESI) calcd for C10H14N2O3 [M+H]+ 211.1077, found 211.1068.
6.5.5 5-sec-butyl-N,N-dimethyl-3-oxoisoxazole-2(3H)-carboxamide (14e)
Prepared using the general procedure above (Method F) from 5-sec-butylisoxazol-3-ol (13e) (96.0 mg, 0.68 mmol), N,N-dimethylcarbamoyl chloride (0.31 mL, 3.38 mmol), and toluene (3.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14e as clear oil (104 mg, 73%). 1H NMR (500 MHz, CDCl3) δ 5.35 (s, 1H), 3.09 (bs, 6H), 2.67 (hextet, J = 6.9 Hz, 1H), 1.71 (doublet of quintets, J = 14.3, 7.3 Hz, 1H), 1.59 (doublet of quintets, J = 14.3, 7.3 Hz, 1H), 1.25 (d, J = 7.0 Hz, 8H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 182.7, 168.1, 150.3, 94.7, 38.8, 35.0, 27.5, 17.3, 11.3. HRMS (ESI) calcd for C10H16N2O3 [M+H]+ 213.1234, found 213.1238.
6.5.6 5-cyclopentyl-N,N-dimethyl-3-oxoisoxazole-2(3H)-carboxamide (14f)
Prepared using the general procedure above (Method F) from 5-cyclopentylisoxazol-3-ol (13f) (21.0 mg, 0.14 mmol), N,N-dimethylcarbamoyl chloride (0.06 mL, 0.69 mmol), and toluene (1.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14f as a white solid (21.0 mg, 67%); mp 69.0-71.8 °C. 1H NMR (500 MHz, CDCl3) δ 5.36 (s, 1H), 3.10 (bs, 6H), 3.04-2.96 (m, 1H), 2.14-1.99 (m, 2H), 1.86-1.60 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 182.3, 168.0, 150.3, 94.4, 38.4, 31.2, 25.5. HRMS (ESI) calcd for C11H16N2O3 [M+Na]+ 225.1234, found 225.1212. Due to signal broadening (slow chemical exchange), the carboxamide methyls could not be detected by 13C NMR, though they were clearly visible in the 1H NMR spectrum (see Figure 2 for explanation).
6.5.7 N,N,-dimethyl-3-oxo-5-(pentan-2-yl)isoxazole-2(3H)-carboxamide (14g)
Prepared using the general procedure above (Method F) from 5-(pentan-2-yl)isoxazol-3-ol (13g) (98.0 mg, 0.63 mmol), N,N-dimethylcarbamoyl chloride (0.17 mL, 1.59 mmol), and toluene (3.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14g as a clear oil (110 mg, 78%). 1H NMR (500 MHz, CDCl3) δ 5.35 (s, 1H), 3.10 (bs, 6H), 2.75 (hextet, J = 6.9 Hz, 1H), 1.71-1.64 (m, 1H), 1.56-1.47 (m, 1H), 1.44-1.31 (m, 2H), 1.26 (d, J = 7.0 Hz, 3H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 182.9, 168.1, 150.3, 94.6, 36.7, 33.3, 20.2, 17.8, 14.0. HRMS (ESI) calcd for C11H18N2O3 [M+H]+ 227.139, found 227.1385. Due to signal broadening (slow chemical exchange), the carboxamide methyls could not be detected by 13C NMR, though they were clearly visible in the 1H NMR spectrum (see Figure 2 for explanation).
6.5.8 N,N,-dimethyl-3-oxo-5-(pentan-3-yl)isoxazole-2(3H)-carboxamide (14h)
Prepared using the general procedure above (Method F) from 5-(pentan-3-yl)isoxazol-3-ol (13h) (62.1 mg, 0.40 mmol), N,N-dimethylcarbamoyl chloride (0.18 mL, 2.00 mmol), and toluene (2.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20 -30% ethyl acetate in hexane) yielded 14h as a clear oil (73.4 mg, 81%). 1H NMR (500 MHz, CDCl3) δ 5.36 (s, 1H), 3.10 (bs, 6H), 2.52-2.44 (m, 1H), 1.74-1.57 (m, 4H), 0.94 (t, J = 7.4 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 181.8, 168.2, 150.3, 95.7, 42.6, 37.0, 25.6, 11.6. HRMS (ESI) calcd for C11H18N2O3 [M+Na]+ 249.121, found 249.1213.
6.5.9 5-isobutyl-N,N-dimethyl-3-oxoisoxazole-2(3H)-carboxamide (14j)
Prepared using the general procedure above (Method F) from 5-isobutylisoxazol-3-ol (13j) (49.0 mg, 0.35 mmol), N,N-dimethylcarbamoyl chloride (0.16 mL, 1.74 mmol), and toluene (1.7 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14j as a clear oil (40.0 mg, 54%). 1H NMR (500 MHz, CDCl3) δ 5.39 (s, 1H), 3.10 (bs, 6H), 2.47 (d, J = 7.1 Hz, 2H), 2.12-1.97 (m, 1H), 1.00 (d, J = 6.7 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 177.7, 168.0, 150.3, 96.7, 36.9, 27.1, 22.4. Due to signal broadening (slow chemical exchange), the carboxamide methyls could not be detected by 13C NMR, though they were clearly visible in the 1H NMR spectrum (see Figure 2 for explanation). HRMS (ESI) calcd for C10H16N2O3 [M+H]+ 213.1509, found 213.1261.
6.5.10 N,N,-dimethyl-5-(2-methylbutyl)-3-oxoisoxazole-2(3H)-carboxamide (14k)
Prepared using the general procedure above (Method F) from 5-(2-methylbutyl)isoxazol-3-ol (13k) (29.0 mg, 0.19 mmol), N,N-dimethylcarbamoyl chloride (0.09 mL, 0.94 mmol), and toluene (1.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14k as a clear oil (19.3 mg, 46%). 1H NMR (500 MHz, CDCl3) δ 5.38 (s, 1H), 3.10 (bs, 6H), 2.58 (dd, J = 15.0, 7.0 Hz, 1H), 2.39 (dd, J = 15.0, 7.0 Hz, 1H), 1.80 (doublet of quintets, J = 13.5, 6.7 Hz, 1H), 1.48-1.35 (m, 1H), 1.32-1.18 (m, 1H), 0.97 (d, J = 6.7 Hz, 3H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 177.9, 168.0, 150.3, 96.7, 39.0, 37.1, 35.0, 33.3, 29.3, 19.2, 11.3. HRMS (ESI) calcd for C11H18N2O3 [M+H]+ 227.139, found 227.1403.
6.5.11 N,N,-dimethyl-5-neopentyl-3-oxoisoxazole-2(3H)-carboxamide (14l)
Prepared using the general procedure above (Method F) from 5-neopentylisoxazol-3-ol (13l) (40.0 mg, 0.28 mmol), N,N-dimethylcarbamoyl chloride (0.13 mL, 1.41 mmol), and toluene (1.4 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14l as a white solid (52.0 mg, 81%); mp 70.7-72.0. 1H NMR (500 MHz, CDCl3) δ 5.39 (s, 1H), 3.11 (bs, 6H), 2.48 (s, 2H), 1.03 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 177.0, 168.2, 150.3, 97.7, 42.0, 38.8, 36.8, 31.8, 29.6. HRMS (ESI) calcd for C11H18N2O3 [M+H]+ 227.139, found 227.1383.
6.5.12 5-isopentyl-N,N-dimethyl-3-oxoisoxazole-2(3H)-carboxamide (14m)
Prepared using the general procedure above (Method F) from 5-isopentylisoxazol-3-ol (13m) (100 mg, 0.64 mmol), N,N-dimethylcarbamoyl chloride (0.30 mL, 3.22 mmol), and toluene (3.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14m as a clear oil (122 mg, 71%). 1H NMR (500 MHz, CDCl3) δ 5.38 (s, 1H), 3.08 (bs, 6H), 2.57 (t, J = 7.9 Hz, 2H), 1.70-1.50 (m, 3H), 0.92 (d, J = 6.5 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 178.9, 168.0, 150.3, 95.7, 38.8, 37.0, 35.1, 27.7, 26.0, 22.2. HRMS (ESI) calcd for C11H18N2O3 [M+H]+ 227.1390, found 227.1397.
6.5.13 5-(2-cyclopropylethyl)-N,N-dimethyl-3-oxoisoxazole-2(3H)-carboxamide (14n)
Prepared using the general procedure above (Method F) from 5-(2-cyclopropylethyl)isoxazol-3-ol (13n) (74 mg, 0.48 mmol), N,N-dimethylcarbamoyl chloride (0.23 mL, 2.41 mmol), and toluene (2.5 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14n as a clear liquid (78 mg, 72%). 1H NMR (400 MHz, CDCl3) δ 5.39 (s, 1H), 3.08 (bs, 6H), 2.67 (t, J = 7.9 Hz, 2H), 1.56 (q, J = 7.3 Hz, 2H), 0.80-0.65 (m, 1H), 0.53-0.40 (dq, J = 8.3, 2.67 Hz, 3H), 0.07 (q, J = 4.6 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 178.4, 167.9, 150.3, 96.0, 38.7, 37.1, 31.5, 28.2, 10.5, 4.7. HRMS (ESI) calcd for C11H16N2O3 [M+H]+ 225.1234, found 225.1231.
6.5.14 5-(2-cyclobutylethyl)-N,N-dimethyl-3-]oxoisoxazole-2(3H)-carboxamide (14o)
Prepared using the general procedure above (Method F) from 5-(2-cyclobutylethyl)isoxazol-3-ol (13o) (90 mg, 0.54 mmol), N,N-dimethylcarbamoyl chloride (0.25 mL, 2.72 mmol), and toluene (3.5 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14o as a clear liquid (87 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 5.33 (s, 1H), 3.05 (bs, 6H), 2.44 (td, J = 7.8, 0.7 Hz, 3H), 2.27 (hept, J = 7.8 Hz, 1H), 2.11-1.95 (m, 2H), 1.89-1.74 (m, 2H), 1.71 (q, J = 7.7 Hz, 2H), 1.64-1.51 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 178.5, 167.8, 150.2, 95.7, 38.7, 36.9, 35.2, 33.2, 27.9, 27.9, 25.7, 18.2. HRMS (ESI) calcd for C12H18N2O3 [M+H]+ 239.1390, found 239.1387.
6.5.15 5-(2-cyclopentylethyl)-N,N-dimethyl-3-oxoisoxazole-2(3H)-carboxamide (14p)
Prepared using the general procedure above (Method F) from 5-(2-cyclopentylethyl)isoxazol-3-ol(13p) (77.8 mg, 0.43 mmol), N,N-dimethylcarbamoyl chloride (0.20 mL, 2.15 mmol), and toluene (3.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 14p as a white solid (85.0 mg, 79%); mp 47.8-47.9 °C. 1H NMR (500 MHz, CDCl3) δ 5.39 (s, 1H), 3.11 (bs, 6H), 2.59 (t, J = 7.9 Hz, 2H), 1.87-1.74 (m, 3H), 1.71-1.65 (m, 2H), 1.65-1.61 (m, 2H), 1.58-1.49 (m, 2H), 1.18-1.01 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 178.8, 167.9, 150.3, 95.8, 39.6, 38.8, 36.9, 32.6, 32.5, 27.3, 25.2. HRMS (ESI) calcd for C13H20N2O3 [M+H]+ 253.1547, found 253.1547.
6.6 General procedure for the synthesis of synthesis of isoxazol-3-yl dimethylcarbamate 15a-p
Method G
To a dry round bottom flask was added isoxazol-3-ol (73.5 mg, 0.47 mmol) and dry tetrahydrofuran (3.0 mL) prior to cooling it to 0 °C. To this solution was added potassium tertbutoxide (1M in THF, 0.71 mL, 0.71 mmol) at 0 °C and subsequently the reaction mixture was stirred for 20 min at RT. To this solution was added N,N-dimethyl carbamoyl chloride (0.11 mL, 1.18 mmol), and the resulting solution was stirred at room temperature for additional 16 h. The solvent was concentrated and the residue was taken up in dichloromethane and washed with 0.25 M HCl hydrochloric acid, and then brine, and dried over sodium sulfate. The solvent was evaporated and the residue was purified by silica gel column chromatography using 20% ethyl acetate in hexane to afford the dimethyl carbamate as the major product.
6.6.1 5-methylisoxazol-3-yl dimethylcarbamate (15a)
Prepared using the general procedure above (Method G) from 5-methylisoxazol-3-ol (13a) (200 mg, 2.01 mmol), potassium tertbutoxide (2.62 mL, 2.62 mmol), N,N-dimethylcarbamoyl chloride (0.46 mL, 5.04 mmol), and THF (7.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15a as a clear oil (256 mg, 75%). 1H NMR (500 MHz, CDCl3) δ 6.09 (s, 1H), 3.04 (s, 3H), 2.95 (s, 3H), 2.34 (s, 3H). 13C NMR (126 MHz, CDCl3) 170.9, 166.7, 151.8, 96.2, 36.8, 36.6, 13.1. HRMS (ESI) calcd for C7H10N2O3 [M+H]+ 171.0691, found 171.0771.
6.6.2 5-cyclopropylisoxazol-3-yl dimethylcarbamate (15b)
Prepared using the general procedure above (Method F) from 5-cyclopropylisoxazol-3-ol (13b) (20.5 mg, 0.16 mmol), N,N-dimethylcarbamoyl chloride (0.08 mL, 0.82 mmol), and toluene (1.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 15b as a clear oil (25.0 mg, 78%). 1H NMR (500 MHz, CDCl3) δ 6.07 (s, 1H), 3.08 (s, 3H), 3.00 (s, 3H), 1.98 (tt, J = 8.5, 5.1 Hz, 1H), 1.07-1.01 (m, 2H), 1.01-0.95 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 176.2, 166.8, 151.9, 93.3, 37.0, 36.7, 8.8, 8.3. HRMS (ESI) calcd for C9H12N2O3 [M+H]+ 197.0921, found 197.0921.
6.6.3 5-isopropylisoxazol-3-yl dimethylcarbamate (15c)
Prepared using the general procedure above (Method G) from 5-isopropylisoxazol-3-ol (13c) (122 mg, 0.96 mmol), potassium tert-butoxide (1.24 mL, 1.25 mmol), N,N-dimethylcarbamoyl chloride (0.22 mL, 2.39 mmol), and THF (7.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15c as a clear oil (170 mg, 80%). 1H NMR (500 MHz, CDCl3) δ 6.12 (s, 1H), 3.09 (s, 3H), 3.06-2.96 (m, 1H), 3.00 (s, 3H), 1.29 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 180.2, 166.7, 152.0, 93.7, 37.0, 36.7, 27.9, 20.6. HRMS (ESI) calcd for C9H14N2O3 [M+H]+ 198.1004, found 199.1079.
6.6.4 5-cyclobutylisoxazol-3-yl dimethylcarbamate (15d)
Prepared using the general procedure above (Method G) from 5-cyclobutylisoxazol-3-ol (13d) (39.0 mg, 0.28 mmol), potassium tert-butoxide (0.42 mL, 0.42 mmol), N,N-dimethylcarbamoyl chloride (0.06 mL, 0.70 mmol), and THF (2.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15d as a clear oil (52.0 mg, 89%). 1H NMR (500 MHz, CDCl3) δ 6.17 (s, 1H), 3.57 (quintet, J = 8.5 Hz, 1H), 3.09 (s, 3H), 3.01 (s, 3H), 2.43-2.21 (m, 4H), 2.11-1.87 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 178.0, 166.8, 152.0, 94.2, 37.0, 36.8, 32.8, 27.8, 18.8. HRMS (ESI) calcd for C10H14N2O3 [M+H]+ 211.1077, found 211.1072.
6.6.5 5-sec-butylisoxazol-3-yl dimethylcarbamate (15e)
Prepared using the general procedure above (Method G) from 5-sec-butylisoxazol-3-ol (13e) (45.1 mg, 0.32 mmol), potassium tert-butoxide (0.48 mL, 0.48 mmol), N,N-dimethylcarbamoyl chloride (0.07 mL, 0.80 mmol), and THF (2.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15e as a clear oil (55.5 mg, 82%). 1H NMR (500 MHz, CDCl3) δ 6.14 (s, 1H), 3.09 (s, 3H), 3.01 (s, 3H), 2.82 (sextet, J = 7.0 Hz, 1H), 1.72 (doublet of quintet, J = 14.3, 7.2 Hz, 1H), 1.61 (doublet of quintet, J = 14.3, 7.2 Hz, 1H), 1.27 (d, J = 7.0 Hz, 3H), 0.91 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 179.4, 166.7, 152.0, 94.3, 37.0, 36.8, 34.7, 28.2, 18.1, 11.5. HRMS (ESI) calcd for C10H16N2O3 [M+H]+ 213.1234, found 213.1243.
6.6.6 5-cyclopentylisoxazol-3-yl dimethylcarbamate (15f)
Prepared using the general procedure above (Method G) from 5-cyclopentylisoxazol-3-ol (13f) (250 mg, 1.63 mmol), potassium tert-butoxide (2.12 mL, 2.12 mmol), N,N- dimethylcarbamoyl chloride (0.37 mL, 4.08 mmol), and THF (10.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15f as a clear oil (300 mg, 82%). 1H NMR (500 MHz, CDCl3) δ 6.13 (d, J = 0.8 Hz, 1H), 3.14 (quintet, J = 7.5 Hz, 1H), 3.09 (s, 3H), 3.01 (s, 3H), 2.12-1.97 (m, 2H), 1.80-1.70 (m, 4H), 1.70-1.60 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 178.9, 166.6, 151.9, 93.9, 38.1, 36.9, 36.6, 31.5, 25.2. HRMS (ESI) calcd for C11H16N2O3 [M+Na]+ 247.1053, found 247.1036.
6.6.7 5-(pentan-2-yl)isoxazol-3-yl dimethylcarbamate (15g)
Prepared using the general procedure above (Method G) from 5-(pentan-2-yl)isoxazol-3-ol (13g) (170 mg, 1.09 mmol), potassium tert-butoxide (1.42 mL, 1.42 mmol), N,N-dimethylcarbamoyl chloride (0.25 mL, 2.74 mmol), and THF (7.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15g as a clear oil (207 mg, 84%). 1H NMR (500 MHz, CDCl3) δ 6.11 (s, 1H), 3.06 (s, 3H), 2.98 (s, 3H), 2.88 (sextet, J = 7.0 Hz, 1H), 1.66 (ddt, J = 13.4, 9.5, 6.5 Hz, 1H), 1.50 (ddt, J = 13.4, 9.5, 6.5 Hz, 1H), 1.38-1.26 (m, 2H), 1.24 (d, J = 7.0 Hz, 3H), 0.87 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 179.5, 166.6, 151.9, 94.1, 37.4, 36.9, 36.7, 32.9, 20.1, 18.5, 13.9. HRMS (ESI) calcd for C11H18N2O3 [M+H]+ 227.1317, found 227.1677.
6.6.8 5-(pentan-3-yl)isoxazol-3-yl dimethylcarbamate (15h)
Prepared using the general procedure above (Method G) from 5-(pentan-3-yl)isoxazol-3-ol (13h) (73.5 mg, 0.47 mmol), potassium tert-butoxide (0.71 mL, 0.71 mmol), N,N-dimethylcarbamoyl chloride (0.11 mL, 1.18 mmol), and THF (3.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15h as a clear oil (89.3 mg, 83%). 1H NMR (500 MHz, CDCl3) δ 6.17 (s, 1H), 3.10 (s, 3H), 3.01 (s, 3H), 2.67-2.59 (m, 1H), 1.78-1.50 (m, 4H), 0.87 (t, J = 7.4 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 178.3, 166.7, 152.0, 95.1, 42.3, 37.0, 36.8, 26.3, 11.7. HRMS (ESI) calcd for C11H18N2O3 [M+H]+ 227.139, found 227.1392.
6.6.9 5-(heptan-3-yl)isoxazol-3-yl dimethylcarbamate (15i)
Prepared using the general procedure above (Method G) from 5-(heptan-3-yl)isoxazol-3-ol (13i) (230 mg, 1.25 mmol), potassium tert-butoxide (1.63 mL, 1.63 mmol), N,N-dimethylcarbamoyl chloride (0.29 mL, 3.13 mmol), and THF (8.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15i as a clear oil (194 mg, 61%). 1H NMR (500 MHz, CDCl3) δ 6.15 (s, 1H), 3.08 (s, 3H), 2.99 (s, 3H), 2.68 (quintet, J = 7.2 Hz, 1H), 1.70-1.53 (m, 4H), 1.34-1.12 (m, 4H), 0.88-0.80 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 178.5, 166.7, 151.9, 95.0, 40.7, 36.9, 36.7, 33.0, 29.3, 26.7, 22.7, 14.0, 11.6. HRMS (ESI) calcd for C13H22N2O3 [M+H]+ 255.1427, found 255.1630.
6.6.10 5-isobutylisoxazol-3-yl dimethylcarbamate (15j)
Prepared using the general procedure above (Method G) from 5-isobutylisoxazol-3-ol (13j) (252 mg, 1.78 mmol), potassium tertbutoxide (2.32 mL, 2.32 mmol), N,N-dimethylcarbamoyl chloride (0.41 mL, 4.45 mmol), and THF (10.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15j as a clear oil (248 mg, 66%). 1H NMR (500 MHz, CDCl3) δ 6.16 (s, 1H), 3.09 (s, 3H), 3.01 (s, 3H), 2.58 (d, J = 7.1 Hz, 2H), 2.09-1.97 (m, 1H), 0.96 (d, J = 6.7 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 174.4, 166.8, 152.0, 96.1, 37.0, 36.8, 36.6, 27.6, 22.4. HRMS (ESI) calcd for C10H16N2O3 [M+H]+ 213.1509, found 213.1261.
6.6.11 5-(2-methylbutyl)isoxazol-3-yl dimethylcarbamate (15k)
Prepared using the general procedure above (Method G) from 5-(2-methylbutyl)isoxazol-3-ol (13k) (300 mg, 1.93 mmol), potassium tert-butoxide (2.5 mL, 2.5 mmol), N,N-dimethylcarbamoyl chloride (0.44 mL, 4.83 mmol), and THF (10 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15k as a clear oil (359 mg, 82%). 1H NMR (500 MHz, CDCl3) δ 6.15 (s, 1H), 3.09 (s, 3H), 3.00 (s, 3H), 2.69 (dd, J = 15.0, 7.0 Hz, 1H), 2.52 (dd, J = 15.0, 7.0 Hz, 1H), 1.79 (doublet of quintet, J = 13.5, 7.2 Hz, 1H), 1.47-1.32 (m, 1H), 1.27-1.16 (m, 1H), 0.92 (d, J = 6.7 Hz, 3H), 0.90 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 174.5, 166.8, 152.0, 96.2, 37.0, 36.7, 34.6, 33.9, 29.2, 19.2, 11.4. HRMS (ESI) calcd for C11H18N2O3 [M+Na]+ 249.121, found 249.1201.
6.6.12 5-neopentylisoxazol-3-yl dimethylcarbamate (15l)
Prepared using the general procedure above (Method G) from 5-neopentylisoxazol-3-ol (13l) (230 mg, 1.63 mmol), potassium tert-butoxide (2.12 mL, 2.12 mmol), N,N-dimethylcarbamoyl chloride (0.37 mL, 4.07 mmol), and THF (7.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15l as a clear oil (262 mg, 71%). 1H NMR (500 MHz, CDCl3) δ 6.13 (s, 1H), 3.04 (s, 3H), 2.95 (s, 3H), 2.54 (s, 2H), 0.93 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 173.3, 166.5, 151.8, 97.0, 41.5, 36.8, 36.6, 31.6, 29.3. HRMS (ESI) calcd for C11H18N2O3 [M+NH4]+ 244.1656, found 244.1668
6.6.13 5-isopentylisoxazol-3-yl dimethylcarbamate (15m)
Prepared using the general procedure above (Method G) from 5-isopentylisoxazol-3-ol (13m) (300 mg, 1.93 mmol), potassium tert-butoxide (2.5 mL, 2.5 mmol), N,N-dimethylcarbamoyl chloride (0.44 mL, 4.83 mmol), and THF (10.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15m as a clear oil (342 mg, 80%). 1H NMR (500 MHz, CDCl3) δ 6.15 (s, 1H), 3.10 (s, 3H), 3.01 (s, 3H), 2.71 (t, J = 8.0, 7.6 Hz, 2H), 1.68-1.54 (m, 3H), 0.93 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 175.4, 166.7, 151.9, 95.1, 36.9, 36.6, 35.9, 27.5, 25.4, 22.2. HRMS (ESI) calcd for C11H18N2O3 [M+H]+ 227.139, found 227.1398.
6.6.14 5-(2-cyclopropylethyl)isoxazol-3-yl dimethylcarbamate (15n)
Prepared using the general procedure above (Method G) from 5-(2-cyclopropylethyl)isoxazol-3-ol (13n) (80 mg, 0.54 mmol), potassium tert-butoxide (1M in THF, 0.81 mL, 0.81 mmol), N,N-dimethylcarbamoyl chloride (0.125 mL, 1.35 mmol), and THF (5.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15n as a clear liquid (90 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 11.73 (s, 1H), 5.67 (s, 1H), 2.73 (t, J = 7.6 Hz, 2H), 1.56 (q, J = 7.3 Hz, 2H), 0.79-0.63 (m, 1H), 0.54-0.35 (dq, J = 8.5, 4.2 Hz 2H), 0.06 (dt, J = 5.9, 4.4 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 175.1, 166.8, 152.0, 95.5, 37.0, 36.8, 32.5, 27.8, 10.6, 4.6. HRMS (ESI) calcd for C11H16N2O3 [M+H]+ 225.1234, found 225.1229.
6.6.15 5-(2-cyclobutylethyl)isoxazol-3-yl dimethylcarbamate (15o)
Prepared using the general procedure above (Method G) from 5-(2-cyclobutylethyl)isoxazol-3-ol (13o) (50 mg, 0.33 mmol), potassium tert-butoxide (1M in THF, 0.5 mL, 0.5 mmol), N,N-dimethylcarbamoyl chloride (0.08 mL, 0.83 mmol), and THF (3.5 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15o as a clear liquid (50 mg, 64%). 1H NMR (500 MHz, CDCl3) δ 6.12 (s, 1H), 3.08 (s, 3H), 2.99 (s, 3H), 2.59 (t, J = 7.7 Hz, 2H), 2.28 (h, J = 7.7 Hz, 1H), 2.08 – 1.98 (m, 2H), 1.90 – 1.78 (m, 2H), 1.75 (q, J = 7.6 Hz, 2H), 1.59 (pd, J = 9.0, 2.5 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 175.1, 166.6, 151.8, 95.2, 36.8, 36.6, 35.2, 34.1, 27.9, 25.2, 18.3. HRMS (ESI) calcd for C12H18N2O3 [M+H]+ 239.1390, found 239.1389.
6.6.16 5-(2-cyclopentylethyl)isoxazol-3-yl dimethylcarbamate (15p)
Prepared using the general procedure above (Method G) from 5-(2-cyclopentylethyl)isoxazol-3-ol (13p) (58.4 mg, 0.32 mmol), potassium tert-butoxide (0.48 mL, 0.48 mmol), N,N-dimethylcarbamoyl chloride (0.074 mL, 0.81 mmol), and THF (1.0 mL). Aqueous work up followed by silica gel flash chromatography (20% ethyl acetate in hexane) of the residue yielded 15p as a clear oil (73.0 mg, 90%). 1H NMR (500 MHz, CDCl3) δ 6.15 (s, 1H), 3.09 (s, 3H), 3.01 (s, 3H), 2.71 (t, J = 7.9 Hz, 2H), 1.86-1.73 (m, 3H), 1.69 (q, J = 7.2 Hz, 2H), 1.65-1.57 (m, 2H), 1.56-1.48 (m, 2H), 1.20-0.99 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 175.5, 166.8, 152.0, 95.3, 39.6, 37.0, 36.8, 33.5, 32.5, 26.9, 25.3. HRMS (ESI) calcd for C13H20N2O3 [M+H]+ 253.1547, found 253.1525.
6.7 Synthesis of N-substituted derivatives of 13g(17g-26g)
6.7.1 2-(azetidine-1-carbonyl)-5-(pentan-2-yl)isoxazol-3(2H)-one (17g)
To a solution of triphosgene (63.5 mg, 0.21 mmol, 0.35 equiv.) in DCM (1.0 mL) was added a solution of 5-(pentan-2- yl)isoxazol-3-ol (13g) (94.0 mg, 0.61 mmol, 1 equiv.) in DCM (2.0 mL), and the resulting mixture was allowed to stir at room temperature for 5.3 h. To the reaction mixture, a solution of azetidine hydrochloride (85.0 mg, 0.91 mmol, 1.5 equiv) and diisopropylethylamine (0.35 mL, 1.99 mmol, 3.3 equiv.) in dichloromethane (3 mL) was added drop wise, and the reaction was stirred for an additional 30 min. The subsequent reaction mixture was washed with aqueous solution of ammonium chloride, and the organic phase was dried over sodium sulfate and concentrated. The residue was purified by silica gel chromatography eluting with 20% ethyl acetate in hexanes to yield the desired compound 17g as clear oil (59.3 mg, 41% yield). Starting material recovered (18.0 mg). 1H NMR (500 MHz, CDCl3) δ 5.39 (s, 1H), 4.37 (s, 2H), 4.19 (s, 2H), 2.73 (sextet, J = 7.0 Hz, 1H), 2.36 (quintet, J = 7.8 Hz, 2H), 1.71-1.59 (m, 1H), 1.56-1.45 (m, 1H), 1.43-1.29 (m, 2H), 1.25 (d, J = 7.0 Hz, 3H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 181.06, 165.57, 148.98, 95.50, 53.01, 49.54, 36.63, 33.18, 20.14, 17.84, 16.57, 13.94. HRMS (ESI) calcd for C12H18N2O3 [M+H]+ 239.139, found 239.1371.
6.7.2 N-ethyl-N-methyl-3-oxo-5-(pentan-2-yl)isoxazole-2(3H)-carboxamide (18g)
To a solution of triphosgene (66.2 mg, 0.22 mmol, 0.35 equiv.) in DCM (1.0 mL) was added a solution of 5-(pentan-2-yl)isoxazol-3-ol (13g) (98.0 mg, 0.63 mmol, 1 equiv.) in DCM (2.0 mL), and the resulting mixture was allowed to stir at room temperature for 2 h. To the reaction mixture, a solution of N-ethylmethyl amine (0.8 mL, 0.95 mmol, 1.5 equiv) and diisopropylethylamine (0.2 mL, 1.14 mmol, 1.8 equiv.) in dichloromethane (1.0 mL) was added drop wise, and the reaction was stirred for an additional 30 min. The subsequent reaction mixture was washed with aqueous solution of ammonium chloride, and the organic phase was dried over sodium sulfate and concentrated. The residue was purified by silica gel chromatography eluting with 20% ethyl acetate in hexanes to yield the desired compound 18g as clear oil (66.1 mg, 44% yield). 1H NMR (500 MHz, CDCl3) δ 5.35 (s, 1H), 3.47 (bs, 2H), 3.09 (bs, 3H), 2.75 (hextet, J = 7.0 Hz, 1H), 1.72 -1.61 (m, 1H), 1.56-1.47 (m, 1H), 1.45-1.31 (m, 2H), 1.26 (d, J = 7.0 Hz, 3H), 1.23 (t, J = 7.2 Hz, 3H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 182.91, 168.23, 150.03, 94.58, 44.80, 36.68, 35.97, 33.31, 20.14, 17.84, 13.96, 12.24. HRMS (ESI) calcd for C12H20N2O3 [M+H]+ 241.1547, found 241.1527.
6.7.3 N,N,-diethyl-3-oxo-5-(pentan-2-yl)isoxazole-2(3H)-carboxamide (19g)
Prepared using the general procedure above (Method F) from 5-(pentan-2-yl)isoxazol-3-ol (13g) (85.0 mg, 0.55 mmol), N,N-diethylcarbamoyl chloride (0.35 mL, 2.74 mmol), and toluene (3.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 19g as a clear oil (96.0 mg, 69%). 1H NMR (500 MHz, CDCl3) δ 5.34 (s, 1H), 3.49 (q, J = 6.5 Hz, 4H), 2.74 (hextet, J = 7.0 Hz, 1H), 1.74-1.61 (m, 1H), 1.56-1.45 (m, 1H), 1.45-1.29 (m, 2H), 1.27-1.21 (m, 9H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 182.81, 168.36, 149.62, 94.31, 42.47, 36.45, 33.07, 19.89, 17.59, 13.71, 13.30. Due to signal broadening (slow chemical exchange) in carboxamide the two CH2 and two CH3 of the N,N-diethyl overlap in 13C NMR. This is consistent with similar observations in seen 1H NMR. (see Figure 2 for explanation). HRMS (ESI) calcd for C13H22N2O3 [M+H]+ 255.1703, found 255.1687.
6.7.4 5-(pentan-2-yl)-2-(pyrrolidine-1-carbonyl)isoxazol-3(2H)-one (20g)
Prepared using the general procedure above (Method F) from 5-(pentan-2-yl)isoxazol-3-ol (13g) (65.1 mg, 0.42 mmol), 1-pyrrolidinecarbonyl chloride (0.23 mL, 2.10 mmol), and toluene (2.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 20g as a clear oil (38.7 mg, 37%). 1H NMR (500 MHz, CDCl3) δ 5.36 (s, 1H), 3.73 (t, J = 6.3 Hz, 2H), 3.57 (t, J = 6.4 Hz, 2H), 2.75 (sextet, J = 7.0 Hz, 1H), 1.94 (s, 4H), 1.72-1.61 (m, 1H), 1.56-1.47 (m, 1H), 1.45-1.31 (m, 2H), 1.26 (d, J = 7.0 Hz, 3H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 182.14, 167.78, 148.43, 94.91, 48.37, 47.31, 36.68, 33.24, 25.89, 24.48, 20.14, 17.83, 13.96. HRMS (ESI) calcd for C13H20N2O3 [M+H]+ 253.1547, found 253.1547.
6.7.5 2-(morpholine-4-carbonyl)-5-(pentan-2-yl)isoxazol-3(2H)-one (21g)
Prepared using the general procedure above (Method F) from 5-(pentan-2-yl)isoxazol-3-ol (13g) (99.4 mg, 0.64 mmol), 4-morpholinecarbamoyl chloride (0.37 mL, 3.20 mmol), and toluene (3.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 21g as a clear oil (133 mg, 78%). 1H NMR (500 MHz, CDCl3) δ 5.36 (s, 1H), 3.80-3.75 (m, 4H), 3.64-3.56 (m, 4H), 2.77 (sextet, J = 7.0 Hz, 1H), 1.73-1.61 (m, 1H), 1.57-1.48 (m, 1H), 1.45-1.30 (m, 2H), 1.27 (d, J = 7.0 Hz, 3H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 183.23, 167.55, 148.92, 94.29, 66.66, 47.73, 45.19, 36.53, 33.22, 20.00, 17.68, 13.80. HRMS (ESI) calcd for C13H20N2O4 [M+H]+ 269.1496, found 269.1476.
6.7.6 5-(pentan-2-yl)isoxazol-3-yl ethyl(methyl)carbamate (23g)
To a solution of triphosgene (66.2 mg, 0.22 mmol, 0.35 equiv.) in DCM (1.0 mL) was added a solution of 5-(pentan-2-yl)isoxazol- 3-ol (13g) (98.0 mg, 0.63 mmol, 1 equiv.) in DCM (2.0 mL), and the resulting mixture was allowed to stir at room temperature for 2 h. To the reaction mixture, a solution of N-ethylmethyl amine (0.8 mL, 0.95 mmol, 1.5 equiv) and diisopropylethylamine (0.2 mL, 1.14 mmol, 1.8 equiv.) in dichloromethane (1.0 mL) was added drop wise, and the reaction was stirred for an additional 30 min. The subsequent reaction mixture was washed with aqueous solution of ammonium chloride, and the organic phase was dried over sodium sulfate and concentrated. The residue was purified by silica gel chromatography eluting with 20% ethyl acetate in hexanes to yield the desired compound 23g as clear oil (30.0 mg, 20% yield). (Observed (Z)- and (E)- amide rotamers in 1:1 ratio, the following integral ratios correspond to the 1:1 mixture) 1H NMR (500 MHz, CDCl3) δ 6.14 (s, 2H, both (Z)- and (E)-), 3.46 (q, J = 7.2 Hz, 2H), 3.39 (q, J = 7.2 Hz, 2H), 3.05 (s, 3H), 2.98 (s, 3H), 2.90 (sextet, J = 7.0 Hz, 2H), 1.74-1.62 (m, 2H), 1.58-1.46 (m, 2H), 1.39-1.29 (m, 4H), 1.27 (d, J = 7.0 Hz, 6H), 1.21 (t, J = 7.2 Hz, 3H), 1.18 (t, J = 7.2 Hz, 3H), 0.89 (t, J = 7.3 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 179.57, 166.75, 166.72, 151.71, 151.57, 94.21, 94.16, 44.59, 44.45, 37.47, 34.51, 34.18, 32.98, 20.21, 18.62, 14.02, 13.28, 12.37. HRMS (ESI) calcd for C12H20N2O3 [M+H]+ 241.1547, found 241.1532.
6.7.7 5-(pentan-2-yl)isoxazol-3-yl diethylcarbamate (24g)
Prepared using the general procedure above (Method F) from 5-(pentan-2-yl)isoxazol-3-ol (13g) (85.0 mg, 0.55 mmol), N,N-diethylcarbamoyl chloride (0.35 mL, 2.74 mmol), and toluene (3.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 24g as a clear oil (23.0 mg, 16%). 1H NMR (500 MHz, CDCl3) δ 6.17 (s, 1H), 3.43 (q, J = 7.1 Hz, 2H), 3.37 (q, J = 7.2 Hz, 2H), 2.91 (sextet, J = 7.0 Hz, 1H), 1.69 (ddt, J = 13.6, 9.2, 6.5 Hz, 1H), 1.59-1.47 (m, 1H), 1.41-1.28 (m, 2H), 1.27 (d, J = 7.0 Hz, 3H), 1.23 (t, J = 7.2 Hz, 3H), 1.20 (t, J = 7.2 Hz, 3H), 0.90 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 179.52, 166.75, 151.39, 94.21, 42.70, 42.34, 37.48, 32.98, 20.22, 18.64, 14.24, 14.02, 13.24. HRMS (ESI) calcd for C13H22N2O3 [M+H]+ 255.1703, found 255.1687.
6.7.8 5-(pentan-2-yl)isoxazol-3-yl pyrrolidine-1-carboxylate (25g)
Prepared using the general procedure above (Method F) from 5-(pentan-2-yl)isoxazol-3-ol (13g) (65.1 mg, 0.42 mmol), 1-pyrrolidinecarbonyl chloride (0.23 mL, 2.10 mmol), and toluene (2.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 25g as a clear oil (60.4 mg, 57%). 1H NMR (500 MHz, CDCl3) δ 6.17 (s, 1H), 3.56 (t, J = 6.6 Hz, 2H), 3.46 (t, J = 6.6 Hz, 2H), 2.90 (sextet, J = 7.0 Hz, 1H), 2.01-1.84 (m, 4H), 1.74-1.63 (m, 1H), 1.58-1.47 (m, 1H), 1.41-1.28 (m, 2H), 1.26 (d, J = 7.0 Hz, 3H), 0.89 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 179.51, 166.67, 150.20, 94.11, 46.80, 46.71, 37.46, 32.97, 25.84, 25.01, 20.21, 18.62, 14.02. HRMS (ESI) calcd for C13H20N2O3 [M+H]+ 253.1547, found 253.1528.
6.7.9 5-(pentan-2-yl)isoxazol-3-yl morpholine-4-carboxylate (26g)
Prepared using the general procedure above (Method F) from 5-(pentan-2-yl)isoxazol-3-ol (13g) (99.4 mg, 0.64 mmol), 4-morpholinecarbamoyl chloride (0.37 mL, 3.20 mmol), and toluene (3.0 mL). Concentration of the reaction mixture in vacuo followed by silica gel flash chromatography (20-30% ethyl acetate in hexane) yielded 26g as a clear oil (20.0 mg, 12%). 1H NMR (500 MHz, CDCl3) δ 6.14 (s, 1H), 3.77-3.69 (m, 4H), 3.68-3.64 (m, 2H), 3.59-3.51 (m, 2H), 2.91 (h, J = 7.0 Hz, 1H), 1.74 1.64 (m, 1H), 1.58-1.48 (m, 1H), 1.39-1.29 (m, 2H), 1.28 (d, J = 7.0 Hz, 3H), 0.90 (t, J =7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 179.86, 166.47, 150.75, 94.11, 66.59, 66.48, 45.20, 44.42, 37.46, 33.01, 20.22, 18.61, 14.03. HRMS (ESI) calcd for C13H20N2O4 [M+H]+ 269.1496, found 269.1478.
6.8 General procedure for the synthesis of N-methyl-3-oxoisoxazole-2(3H-carboxamide
6.8.1 5-isobutyl-N-methyl-3-oxoisoxazole-2(3H)-carbox-amide (16j)
To a dry round bottom flask was added 5-isobutylisoxazol-3-ol (13j) (165 mg, 1.17 mmol) and dry tetrahydrofuran (7.0 mL) prior to cooling it to 0 °C. To this solution was added potassium tert-butoxide (1M in THF, 1.52 mL, 1.52 mmol) at 0 °C and subsequently the reaction mixture was stirred for 20 min at RT. To this solution was added N-methylcarbamoyl chloride (273 mg, 2.92 mmol) and the resulting solution was stirred at room temperature for additional 16 h. The solvent was concentrated and the residue was taken up in dichloromethane and washed with 0.25 M HCl hydrochloric acid, and then brine, and dried over sodium sulfate. The solvent was evaporated and the residue was purified using silica gel column chromatography (20% ethyl acetate in hexane) to yield 16j as a white semi-solid (207 mg, 90 %). 1H NMR (500 MHz, Chloroform-d) 7.83 (s, 1H), 5.56 (s, 1H), 2.96 (d, J = 4.8 Hz, 3H), 2.49 (d, J = 7.2 Hz, 2H), 2.14-1.98 (m, 1H), 0.99 (d, J = 6.7 Hz, 6H). 13C NMR (126 MHz, Chloroform-d) 174.44, 165.47, 148.45, 98.12, 36.55, 27.16, 26.48, 22.35. HRMS (ESI) calcd for C9H14N2O3 [M+H]+ 199.1077, found 199.1086.
6.9 Bioassays
6.9.1 Enzyme inhibition assays
Recombinant WT and G119S AgAChE were prepared as previously described.12 Recombinant hAChE (C1682) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Inhibition potency of carbamate and carboxamide insecticides was assessed by measuring apparent second-order rate constants ki (mM−1 min−1) for inactivation of the enzymes. We followed our previous progressive inactivation approach,8,12 in which enzymes were incubated with different concentrations of carbamates or carboxamides for differing times before measuring enzyme residual activity (v/v0). Enzymes were incubated with typically five concentrations of inhibitors (and an inhibitor-free control) for up to 6 minutes at approximately 1 min intervals. Each concentration was present in the microplate in duplicate, and each experiment was repeated. Note that for the G119S enzyme inhibition was typically very low after 10 min at 10 μM, and so incubations with these enzymes were often extended to 60 min (10 min intervals). Residual activities v/v0 are the ratio of the rate in the presence of inhibitor to a time-matched inhibitor-free control and thus correct for slow thermal inactivation of the enzyme. At each inhibitor concentration [I], plots of ln(v/v0) vs. time (t) were constructed; the slopes of these unconstrained linear fits represent the pseudo first-order rate constants (kobs) associated with those inhibitor concentrations (see Supplementary Material, Figure S2). These kobs values (obtained in at least two separate experiments) were then plotted against [I]; in each case the slope of the unconstrained linear fit is the apparent second-order rate constant ki (mM−1 min−1) for inactivation (see Supplementary Material, Figure S3). Note that the error in kobs is typically smaller than the symbol in these plots (see Figure S3). The error in ki is estimated as the standard error in the slope of this plot. Note that inhibitor concentrations [I] were chosen to be low enough to remain in the domain where a plot of kobs vs [I] was linear. For fast-inactivating carbamates, such as terbam (4) on rAgAChE-WT, we saw signs of saturation behavior above [I] = 0.2 μM. Saturation behavior is expected for a two-step mechanism of inhibition.35 Enzymatic sensitivity ratios (SR) and Ag/h selectivity for inhibition (Tables 3-4) were calculated from the measured ki values; the error in these ratios was determined using a standard propagation of error method.20
6.9.2 Mosquito contact toxicity determination
Anopheles gambiae eggs (G3 (MRA-112) and Akron (MRA-913)) were obtained from MR4, and reared in tap water with fish food for larval sustenance (Tetra Fish, Blacksburg, VA, USA). Adult female non-blood fed An. gambiae (both G3 and Akron strains) 3–5 days old, were used for filter paper assays of tarsal contact toxicity, which were performed in exposure tubes according to the 2006 World Health Organization recommendations18 with slight modification. In brief, filter papers (15 × 12 cm) were treated with 2.0 mL of various concentrations of the carbamate in ethanol, and allowed to dry overnight. For the G3 strain, batches of 20-25 mosquitoes (in triplicate) were transferred to a holding tube and allowed to adapt for one hour. Due to lower colony numbers, toxicity assays with the Akron strain used batches of 10-15 mosquitoes in duplicate. Mosquitoes were then transferred to the exposure tube (held horizontally) that contained a treated filter paper. Knockdown was noted after 1 h, and all mosquitoes were transferred back to the holding tube (held upright), and given free access to 10% (w/v) sugar water. Mortality was recorded at 24 h. Both during exposure and the 23 h period following, mosquito tubes were kept in an environmental chamber at 24 ± 1 °C and 75% RH. To determine LC50 values, typically 5-8 concentrations were examined, and mortality data were used for probit analysis using PoloPlus36 or SAS Probit.
6.10 Computational modeling
Flexible ligand docking of the dimethylcarboxamide tetrahedral covalent intermediate adduct derived from 14c and mAChE was performed in ICM using default settings for ‘covalent’ docking mode (ICM-Docking module, Molsoft).24,25 The receptor structure was prepared for docking starting from PDB entry 2H9Y following the standard conversion procedure in ICM. Two possible stereoisomers of the covalent adduct were sampled and lowest-score solution selected. To get more accurate covalent geometry of the bound ligand, the lowest-scoring ligand pose from docking was subjected to unrestrained minimization of the ligand in fixed explicit receptor (3000 steps) with MMFF94 force-field.37
Supplementary Material
Acknowledgments
We thank the MR4 as part of the BEI Resources Repository, NIAID, NIH, for providing eggs for the G3 (MRA-112) and Akron (MRA-913) strains of Anopheles gambiae; the latter was deposited by M. Akogbeto. We thank the NIH for financial support (AI082581).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data (enzyme alignments, details of X-ray crystallography, and calculation of d835 values) associated with this article can be found, in the online version, at http://dx.doi.org/.
References and notes
- 1. [24 July 2014];World Malaria Report 2013, The World Health Organization. Available: http://www.who.int/malaria/publications/world_malaria_report_2013/en/
- 2.Heppner DG. Travel Med. Infect. Dis. 2013;11:2–7. doi: 10.1016/j.tmaid.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Bayoh MN, Mathias DK, Odiere MR, Mutuku FM, Kamau L, Gimnig JE, Vulule JM, Hawley WA, Hamel MJ, Walker ED. Malaria J. 2010;9:62. doi: 10.1186/1475-2875-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Killeen GF, Smith TA, Ferguson HM, Mshinda H, Abdulla S, Lengeler C, Kachur SP. PLoS Medicine. 2007;4:e229. doi: 10.1371/journal.pmed.0040229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mutuku FM, King CH, Mungai P, Mbogo C, Mwangangi J, Muchiri EM, Walker ED, Kitron U. Malaria J. 2011;10:356. doi: 10.1186/1475-2875-10-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.N'Guessan R, Boko P, Odjo A, Knols B, Akogbeto M, Rowland M. Trop. Med. Int. Health. 2009;14:389–395. doi: 10.1111/j.1365-3156.2009.02245.x. [DOI] [PubMed] [Google Scholar]
- 7.Wong DM, Li J, Lam PCH, Hartsel JA, Mutunga JM, Totrov M, Bloomquist JR, Carlier PR. Chem. Biol. Interact. 2013;203:314–318. doi: 10.1016/j.cbi.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartsel JA, Wong DM, Mutunga JM, Ma M, Anderson TD, Wysinski A, Islam R, Wong EA, Paulson SL, Li J, Lam PC-H, Totrov MM, Bloomquist JR, Carlier PR. Bioorg. Med. Chem. Lett. 2012;22:4593–4598. doi: 10.1016/j.bmcl.2012.05.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlier PR, Anderson TD, Wong DM, Hsu DC, Hartsel J, Ma M, Wong EA, Choudhury R, Lam PC-H, Totrov MM, Bloomquist JR. Chem. Biol. Interact. 2008;175:368–375. doi: 10.1016/j.cbi.2008.04.037. [DOI] [PubMed] [Google Scholar]
- 10.Weill M, Lutfalla G, Mogensen K, Chandre F, Berthomieu A, Berticat C, Pasteur N, Philips A, Fort P, Raymond M. Nature. 2003;423:136–137. doi: 10.1038/423136b. [DOI] [PubMed] [Google Scholar]
- 11.Weill M, Malcolm C, Chandre F, Mogenson K, Berthomieu A, Marquine M, Raymond M. Insect Mol. Biol. 2004;13:1–7. doi: 10.1111/j.1365-2583.2004.00452.x. [DOI] [PubMed] [Google Scholar]
- 12.Wong DM, Li J, Chen Q-H, Han Q, Mutunga JM, Wysinski A, Anderson TD, Ding H, Carpenetti TL, Verma A, Islam R, Paulson SL, Lam PC-H, Totrov M, Bloomquist JR, Carlier PR. PLOS One. 2012;7:e46712. doi: 10.1371/journal.pone.0046712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomita K, Murakami T, Tsuji H, Matsumoto K, Fujita K. Isoxazoline derivatives, process for their preparation and insecticidal and acaracidal composition containing them. EP 94082 A1, Nov. 16, 1983. [Google Scholar]
- 14.Sørensen US, Falch E, Krogsgaard-Larsen P. J. Med. Chem. 2000;65:1003–1007. doi: 10.1021/jo991409d. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Y, Raymond MK, Tsai H, Roberts JD. The Journal of Physical Chemistry. 1993;97:2910–2913. [Google Scholar]
- 16.Deetz MJ, Forbes CC, Jonas M, Malerich JP, Smith BD, Wiest O. J. Org. Chem. 2002;67:3949–3952. doi: 10.1021/jo025554u. [DOI] [PubMed] [Google Scholar]
- 17.Dolomanov OV, Bouris LJ, Gildea RJ, Howard JAK, Puschmann H. J. Appl. Cryst. 2009;42:339–341. doi: 10.1107/S0021889811041161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guidelines for Testing Mosquito Adulticides for Indoor Residual Spraying and Treatment of Mosquito Nets. World Health Organization; Geneva: 2006. WHO/CDS/NTD/WHOPES/GCDPP/2006.3. [Google Scholar]
- 19.Ellman GL, Courtney KD, Andres VJ, Featherstone RM. Biochem. Pharm. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 20.Andraos J. J. Chem. Educ. 1996;73:150–154. [Google Scholar]
- 21.Nair HK, Lee K, Quinn DM. J. Am. Chem. Soc. 1993;115:9939–9941. [Google Scholar]
- 22.Lowe DB, Magnuson S, Qi N, Campbell A-M, Cook J, Hong Z, Wang M, Rodriguez M, Achebe F, Kluender H, Wong WC, Bullock WH, Salhanick AI, Witman-Jones T, Bowling ME, Keiper C, Clairmont KB. Bioorg. Med. Chem. Lett. 2004;14:3155–3159. doi: 10.1016/j.bmcl.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 23.Ebdrup S, Sørensen LG, Olsen OH, Jacobsen P. J. Med. Chem. 2003;47:400–410. doi: 10.1021/jm031004s. [DOI] [PubMed] [Google Scholar]
- 24.Totrov MM, Abagyan RA. Proteins, Suppl. 1997;1:215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 25.Neves MC, Totrov M, Abagyan R. Journal of Computer-Aided Molecular Design. 2012;26:675–686. doi: 10.1007/s10822-012-9547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bourne Y, Radic Z, Sulzenbacher G, Kim E, Taylor P, Marchot P. J. Biol. Chem. 2006;281:29256–29267. doi: 10.1074/jbc.M603018200. [DOI] [PubMed] [Google Scholar]
- 27.Cheung J, Rudolph MJ, Burshteyn F, Cassidy MS, Gary EN, Love J, Franklin MC, Height JJ. J. Med. Chem. 2012;55:10282–10286. doi: 10.1021/jm300871x. [DOI] [PubMed] [Google Scholar]
- 28.Meanwell NA. J. Med. Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 29.Otani Y, Nagae O, Naruse Y, Inagaki S, Ohno M, Yamaguchi K, Yamamoto G, Uchiyama M, Ohwada T. J. Am. Chem. Soc. 2003;125:15191–15199. doi: 10.1021/ja036644z. [DOI] [PubMed] [Google Scholar]
- 30.Antolak SA, Yao Z-K, Richoux GM, Slebodnick C, Carlier PR. Org. Lett. 2014;16:5204–5207. doi: 10.1021/ol502586n. [DOI] [PubMed] [Google Scholar]
- 31.Contreras JA, Karlsson M, Østerlund T, Laurell H, Svensson A, Holm C. J. Biol. Chem. 1996;271:31426–31430. doi: 10.1074/jbc.271.49.31426. [DOI] [PubMed] [Google Scholar]
- 32.Hetmanski MT, Fussell RJ, Sykes MD, Vega AB, Sharma A. Food Addit. Contam. Part A-Chem. 2004;21:447–456. doi: 10.1080/02652030410001677736. [DOI] [PubMed] [Google Scholar]
- 33.CLogP values calculated using ChemBioDraw 13.0.2. Perkin Elmer, Inc.; [Google Scholar]
- 34.Liu Y, Prashad M, Ciszewski L, Vargas K, Repi O, Blacklock T. J. Org. Proc. Res. Dev. 2008;12:183–191. [Google Scholar]
- 35.Copeland RA. In Evaluation of Enzyme Inhibitors in Drug Discovery. Wiley-Interscience; New York: 2005. pp. 214–219. [Google Scholar]
- 36.Robertson JL, Preisler HK, Russell RM. PoloPlus Probit and Logit Analysis. LeOra Software; 2002. [Google Scholar]
- 37.Halgren TA. Journal of Computer-Aided Molecular Design. 1996;17:490–512. doi: 10.1007/BF00124478. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.