

Abstract
Traumatic brain injury (TBI) is a major cause of death and disability. However, the molecular events contributing to the pathogenesis are not well understood. Mitochondria serve as the powerhouse of cells, respond to cellular demands and stressors, and play an essential role in cell signaling, differentiation, and survival. There is clear evidence of compromised mitochondrial function following TBI, however, the underlying mechanisms and consequences are not clear. MicroRNAs (miRNAs) are small non-coding RNA molecules that regulate gene expression post-transcriptionally, and function as important mediators of neuronal development, synaptic plasticity, and neurodegeneration. Several miRNAs show altered expression following TBI, however, the relevance of mitochondria in these pathways is unknown. Here, we present evidence supporting the association of miRNA with hippocampal mitochondria, as well as changes in mitochondria-associated miRNA expression following a controlled cortical impact (CCI) injury in rats. Specifically, we found that the miRNA processing proteins Argonaute (AGO) and Dicer are present in mitochondria fractions from uninjured rat hippocampus, and immunoprecipitation of AGO associated miRNA from mitochondria suggests the presence of functional RNA-induced silencing complexes. Interestingly, RT-qPCR miRNA array studies revealed that a subset of miRNA is enriched in mitochondria relative to cytoplasm. At 12 hour following CCI, several miRNAs are significantly altered in hippocampal mitochondria and cytoplasm. In addition, levels of miR-155 and miR-223, both of which play a role in inflammatory processes, are significantly elevated in both cytoplasm and mitochondria. We propose that mitochondria-associated miRNAs may play an important role in regulating the response to TBI.
Keywords: microRNA, mitochondria, traumatic brain injury, controlled cortical impact, hippocampus, inflammation, miR-142-3p, miR-142-5p, miR-146a, miR-155, miR-223, miR-150
Introduction
Traumatic brain injury (TBI) is a major cause of death and disability affecting an estimated 1.7 million people annually in the United States alone (Faul M, 2010). The personal and health consequences associated with TBI are substantial with an estimated annual financial burden of over $75 billion in direct medical costs and other indirect costs (Finkelstein et al., 2006). At the present time, no effective treatment exists due, in part, to the widespread impact of numerous complex secondary biochemical and pathophysiological events occurring at different time points following the initial injury (McIntosh et al., 1998; Rosenfeld et al., 2012). These secondary injury events include, but are not limited to edema, excitotoxicity, inflammation, oxidative stress/damage, activation of necrotic and apoptotic cell death-signaling events, and impaired mitochondrial function (Rink et al., 1995; Clark et al., 2000; Sullivan et al., 2002; Lifshitz et al., 2004; Raghupathi, 2004; Singh et al., 2006; Ziebell and Morganti-Kossmann, 2010). Mitochondria play an essential role in maintaining cellular homeostasis by responding to cellular energy demands and participating directly in a wide range of cellular events including cell signaling, metabolism, and survival, in addition to cell death pathways (Saraste, 1999; McBride et al., 2006). Importantly, it has been well documented that the degree of mitochondrial dysfunction and recovery following TBI is a critical determinant of subsequent cell survival or death (Sullivan et al., 2002; Lifshitz et al., 2003; Lifshitz et al., 2004; Singh et al., 2006; Pandya et al., 2009; Patel et al., 2009). These vital roles for mitochondria in cellular function and survival have resulted in increased efforts to identify the molecular events associated with mitochondrial impairment in TBI.
Mitochondria respond rapidly to a wide range of stressors and cellular requirements. Several studies have documented the localization of microRNA (miRNA) processing machinery and various miRNA in mitochondrial fractions isolated from a variety of cells and tissues (Lung et al., 2006; Kren et al., 2009; Bian et al., 2010; Bandiera et al., 2011; Barrey et al., 2011; Huang et al., 2011; Mercer et al., 2011; Das et al., 2012; Hu et al., 2012; Sripada et al., 2012). MiRNAs are short, non-coding RNA molecules that bind to recognition elements in mRNA and regulate gene expression post-transcriptionally either by mRNA degradation or translational repression (Bartel, 2004; Chen and Rajewsky, 2007; Bartel, 2009; Huntzinger and Izaurralde, 2011). Disruption of miRNA function contributes to many disease states including cancer, cardiovascular disease, and neurodegeneration (Calin and Croce, 2006; Nelson et al., 2008a; Hata, 2013). The biological actions of miRNA are thought to require the cytoplasmic processing by Dicer, interaction with Argonaute (AGO) proteins and assembly into RNA-induced silencing complexes (RISC) (Liu et al., 2008; Bartel, 2009), which promotes and stabilizes miRNA base pair annealing with target mRNA. Recently, mitochondria have been shown to interact with AGO-miRNA containing processing bodies (P-bodies), and that mild uncoupling of mitochondria decreases cellular miRNA functional efficiency (Huang et al., 2011). The ability of a single miRNA or miRNA family to regulate the post-transcriptional expression of hundreds of genes makes them ideal candidates for coordinating complex gene expression programs, including modifying a cell’s response to stressors or mounting a protective or pathological response following TBI. Recent experimental studies have reported altered expression levels of several miRNAs in both cortex and hippocampus following controlled cortical impact (CCI), fluid percussion injury, and contusive spinal cord injury (Lei et al., 2009; Liu et al., 2009; Redell et al., 2009; Redell et al., 2010; Strickland et al., 2011; Truettner et al., 2011; Hu et al., 2012; Yunta et al., 2012; Hu et al., 2013; Truettner et al., 2013; Di et al., 2014; Liu et al., 2014; Sabirzhanov et al., 2014; Sun et al., 2014). However, the signaling events regulating cellular miRNA activity and/or function in response to TBI are largely unknown.
In the present study, we demonstrate the presence of miRNA protein machinery in mitochondrial fractions of rat hippocampal tissue. In addition, miRNAs were found in AGO-containing complexes co-immunoprecipitated from purified mitochondria supporting a role for mitochondria in regulating miRNA activity. Importantly, a subset of miRNAs was preferentially enriched in hippocampal mitochondria preparations under normal conditions, and several miRNAs were found to decrease following a severe TBI. These studies point towards a novel role for mitochondrial regulation of miRNA expression in response to TBI.
Materials and Methods
Surgical procedures for traumatic brain injury
All procedures and protocols were approved by the University of Kentucky Institutional Animal Care and Use Committee. Young adult male Sprague–Dawley rats weighed 250–300 g (Harlen Laboratories, IN) were housed for 7 days prior to experimentation to allow them to acclimate to the environment. All animals were maintained in a temperature-controlled vivarium room with free access to food and water. The surgical and controlled cortical impact (CCI) injury procedures have been described in detail previously (Sullivan et al., 1999). Briefly, animals were anesthetized with isoflurane (4%) and placed in a stereotaxic frame (David Kopf, Tujunga, CA) prior to brain surgery. Isoflurane (2.5%) was delivered via a nose cone throughout the surgical procedure. Using sterile procedures, the skin was retracted and a 6-mm craniotomy was made lateral to the sagittal suture and centered between bregma and lambda. The skull-cap was carefully removed without disruption of the underlying dura. The exposed brain was then injured using a pneumatically controlled impacting device consisting of a 5 mm diameter rod tip that compressed the cortex at 3.5 m/s velocity to a depth of 2.0 mm. The use of these injury parameters results in a severe injury to the cortex overlying the hippocampal formation (Baldwin et al., 1997; Davis et al., 2008; Gilmer et al., 2009). Following CCI, the Surgicel (Johnson and Johnson) was laid over the dura and the skull-cap was replaced. A thin coat of dental acrylica was then spread over the craniotomy site and allowed it to dry before closing the overlying skin with surgical staples. The core body temperatures of the animals were maintained at 37 °C throughout the surgical procedures and recovery period.
At 12 hr post-injury, animals were euthanatized with CO2 and the hippocampus from both the injured and naïve animals (n=5 animals per group) were rapidly removed. Any potential effect of a unilateral CCI on miRNA changes in the contralateral hippocampus has not been investigated. Therefore, the hippocampi from naïve, uninjured animals were used to obtain control tissue.
Isolation of mitochondria
For studies examining miRNA expression, the nuclear (P1), cytosolic (S2) and purified total mitochondria (MT) fractions were isolated from rat hippocampus according to a previously published ficoll density gradient procedure (Pandya et al., 2009). Briefly, tissue homogenates were prepared in ice-cold mitochondrial isolation buffer (215 mM mannitol, 75 mM sucrose, 0.1% bovine serum albumin (BSA), 20 mM HEPES, 1 mM EGTA; pH adjusted to 7.2 with KOH), followed by centrifugation two times at 1,300 g for 3 min to obtain P1. The resulting supernatant was centrifuged at 13,000 g for 10 min to obtain crude mitochondrial pellets (P2) and the supernatant was saved as S2. The P2 pellets were re-suspended in isolation buffer and placed in a nitrogen cell disruption chamber (1200 psi, 10 min, at 4 °C) to rupture and release synaptosomal mitochondria (Brown et al., 2004). This fraction was further purified using a discontinuous ficoll gradient (7.5% layered over 10% ficoll) and centrifugation at 100,000 g (in SW 55Ti rotors) at 4 °C for 30 min. The resulting MT pellet was washed and centrifuged at 10,000 g at 4 °C for 10 min in mitochondrial isolation buffer without EGTA and finally resuspended to achieve a concentration of ~10 mg/ml in mitochondrial isolation buffer without EGTA for the miRNA expression studies. The protein content of the above listed fractions was analyzed using BCA protein assay kit.
Isolation of mitochondrial sub-fractions from ficoll-purified mitochondria
A mitochondria (MT) fraction was obtained from adult naïve rat brain as described above, and further processed to generate mitochondrial sub-fractions according to a previously published method (Atorino et al., 2003) with slight modifications. Briefly, 5 mg of MT was subjected to hypotonic swelling in 1.5 ml of 5 mM HEPES/KOH, pH 7.4 for 20 min on ice to rupture the outer mitochondrial membrane. The solution was then centrifuged at 1900 g at 4 °C for 15 min to obtain a mitoplast pellet (MP) and mitochondrial supernatant (MS) containing broken outer mitochondrial membrane and inter-membrane space (IMS) compartments. The MS fraction was then sonicated (10 seconds X 3 times) and centrifuged 35,000 g (utilizing SW 55Ti rotors) further at 4 °C for 15 min to pellet down the outer membrane (OM), with the resulting supernatant containing only the inter-membrane space (IMS) of mitochondria. The dense MP pellets were re-suspended in 250 μl of ice-cold isolation buffer, sonicated (10 seconds X 3 times) and further centrifuged at 100,000 g (utilizing SW 55Ti rotors) at 4 °C for 30 min. The resultant membrane-enriched pellet contained the mitochondrial inner membrane (IM) and the resulting supernatant contained the mitochondrial matrix (MTX) fractions. The protein content of the mitochondrial sub-fractions was determined using BCA protein assay.
Western Blot Procedure
The purity of the mitochondria samples as well as identification of miRNA machinery proteins in the sub-mitochondrial fractions were analyzed using standard Western blotting techniques. Briefly, a total of 15 μg of protein was resolved according to molecular weight by SDS–PAGE using either Criterion 4–20% Tris–HCl (10–250 kD) or Criterion 3–8% Tri-acetate (25–250 kD) gels (Bio-Rad, Hercules, CA). The gels were transblotted onto polyvinylidene difluoride membranes, blocked with 5% nonfat dry milk for an hour, and then incubated at 4 °C overnight with the primary antibody of interest. The primary antibodies used included anti-Dicer mAb (1:500 dilution; cat # sc-136979; Santa Cruz Biotechnology, Inc.), anti-Histone 3 rabbit mAb (H3; 1:25,000 dilution; cat # 4499; Cell Signaling Technology Inc.), anti-pyruvate dehydrogenase rabbit mAb (PDH; 1:10,000 dilution; cat # 3205; Cell Signaling Technology Inc.), anti-voltage-dependent anion channel rabbit polyAb (VDAC; 1:10,000 dilution; cat # PA1-954A; Thermo Scientific Inc.), anti-complex IV mAb (CoxIV; 1:200,000 dilution; Cat # A21348; Life Technologies Inc.), anti-beta tubulin rabbit polyAb (1:100,000 dilution; cat # ab6046; Abcam Inc.), anti-beta actin mAb (1:75,000 dilution; cat # A5316; Sigma-Aldrich), anti-CD45 rabbit polyAb (1:2,000 dilution; cat # ab10558; Abcam Inc.), anti-iNOS (1:10,000 dilution; cat # 610332; BD Transduction Laboratories), and anti-Argonaute 2 mAb (2A8; 1:1000 dilution). The 2A8 Ago2 antibody has been described previously (Nelson et al., 2007). Peroxidase-conjugated affinipure secondary antibodies included goat anti-mouse IgG and goat anti-rabbit IgG (cat # 115-035-166 and 111-035-144, respectively; Jackson ImmunoResearch laboratories, Inc.).
Primary rat brain cell culture
Primary cortical neuronal, astrocyte and microglial cultures were generated from Sprague–Dawley rat pups at E18 as described previously (Wang et al., 2014).
RNA isolation from mitochondria and cytosol
Total RNA isolation using TRIZOL LS reagent (Life Technologies, USA) from cytosolic fractions followed the modified procedure described previously (Nelson et al., 2008b; Wang et al., 2008a). Total mitochondrial RNA isolation employed the same procedure with minor modifications (double extraction) to accommodate RNA isolation from limited sample quantity. Briefly, mitochondria were lysed in 250 μl RIPA buffer (10 mM Tris-Cl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS) in the presence of protease inhibitors (Pierce, cat. # 78439) and RNasin (0.2 U/μl, Promega, cat. # N2615). TRIZOL LS was then added to the mitochondria lysate. After chloroform extraction and centrifugation, a 450-μl clear aqua-phase supernatant was saved. The remaining 150–200 μl supernatant (which may contain some protein interface) was collected separately, and was brought up to 250 μl using RIPA buffer. This supernatant was then subjected to a second round of TRIZOL LS extraction following the same procedure. The aqua-phase supernatants from both extractions were combined, and total RNA precipitated using iso-propanol with glycogen (Roche, cat. # 10901393001) as carrier. Isolated RNA concentrations were quantified using NanoDrop1000 Spectrophotometer (NanoDrop Technologies, Inc.).
TaqMan® miRNA RT-qPCR array
Total RNA isolated from mitochondria and cytosol of each hippocampal tissue was subjected to TaqMan® miRNA RT-qPCR array using Rodent Card A (Life Technologies). Because of the relatively low levels of mitochondrial RNA and the benefit of uniformity, a preamplification step using Megaplex™ PreAmp Primers (Life Technologies) was carried out to enhance the detection ability for low-expression miRNAs. TaqMan® miRNA RT-qPCR array was performed as described previously (Wang et al., 2014). Briefly, equal amount of total RNA (50 ng) was reverse transcribed using the TaqMan® MicroRNA Reverse Transcription Kit with Megaplex™ Primer Pools, Rodent Pool A (Life Technologies). The resulting cDNA was pre-amplified using Megaplex™ PreAmp Primers, Rodent Pool A (Life Technologies) before performing RT-qPCR Array. PCR was carried out on the ViiA™ 7 Real-Time PCR System and qPCR cycle threshold (Ct) raw data processed with ViiA 7 RUO software (Life Technologies).
TaqMan® single-tube miRNA RT-qPCR
Individual miRNA was subjected to single-tube TaqMan® assay (Life Technologies) according to manufacturer’s instructions. TaqMan® assays were designed to follow MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines (Bustin et al., 2009). An equal quantity (10–50 ng) of RNA was used in the TaqMan® assays. All single-tube TaqMan® assays were performed without a pre-amplification except for the evaluation of miR-142-3p, miR-142-5p, and miR-146a, in which the pre-amplification step was performed as in the array assays. A total of 3-5 biological replicates with 3 technical triplicates were performed for individual experiments.
Data analysis
Raw PCR Ct values were first determined using ViiA 7 RUO software (Life Technologies) with the same parameter settings across all samples (threshold set at 0.15 with automatic baseline). Data corresponding to failed QC as determined by ViiA 7 RUO software were excluded from the analysis. In addition, miRNAs with a PCR Ct value >35 were considered as undetectable. DataAssist® software (Life Technologies) was then used to analyze miRNA array data. Different array data from cytosol and mitochondria fractions were normalized separately. The Global Mean Normalization method (Mestdagh et al., 2009) was applied to normalize the pooled dataset of both Naïve and TBI. The Ct values presented in this report, including the Supplemental data, are normalized data. The miRNA expression levels between treatments (TBI vs Naïve) or mitochondria versus cytosol fractions from naïve rat brain samples were expressed as fold changes using the following equation: fold change=2^ −(CtTBI − CtNaive), or fold change=2^ −(CtMito − CtCyto), and were evaluated using an unpaired Student t-test. Benjamini-Hochberg or permutation tests were used to control False Discovery Rate (FDR), and all significant data points (p<0.05) had a FDR< 5%. The data from the TaqMan® single-tube miRNA RT-qPCR assays were compared using unpaired student t-tests with a p<0.05 considered as statistically significant.
Results
Mitochondrial Purity
To examine the purity (and contamination) of our preparations, total mitochondria (MT) fractions were isolated from uninjured rat hippocampus and the proteins subjected to immunoblotting to test for the presence of cytosolic and nuclear protein components. As shown in Figure 1A, PDH, a mitochondrial matrix protein, was detected only in fractions containing mitochondria. Neither β-tubulin, a cytosolic protein marker, nor the nuclear protein marker, Histone H3, was detected in purified mitochondrial samples. The purity of the mitochondria preparation was further examined using RT-qPCR for the presence of cytosolic GAPDH mRNA (Figure 1B). Using this highly sensitive approach, GAPDH mRNA levels were found to be ~50-fold lower in the mitochondria preparation compared to the cytosolic fraction. Together, these experiments provide evidence that the mitochondria preparation used in our experiments was relatively free of cytoplasmic and nuclear contaminants.
Figure 1. Purity of isolated mitochondria.

Western blot analysis and RT-qPCR were used to examine the purity of isolated mitochondria. The results suggested that the mitochondria preparations were relatively free from cytosolic or nuclear contaminations. A. Rat hippocampal cytosolic, mitochondrial, and nuclear proteins were subjected to Western blot analysis using β-tubulin (cytosol marker), PDH (mitochondria marker), and Histone H3 (nuclear marker) antibodies. 1: Total brain homogenate; 2: Total hippocampal homogenate; 3: Cytosolic fraction; 4: Nuclear fraction; 5 and 7: Blank; 6: Mitochondrial fraction with 2X ficoll gradient; 8: Mitochondrial fraction with 1X Ficoll gradient. B. TaqMan® RT-qPCR for GAPDH in cytosol and mitochondria preparations. Cyto: cytosol; Mito: mitochondria.
Presence of AGO2-miRNA complexes in hippocampus mitochondria
The localization of AGO and Dicer in mitochondrial fractions of naïve rat brain was examined using immunoblotting with antibodies against AGO and Dicer. Western blot analysis demonstrated that both miRNA machinery proteins were enriched in mitochondrial matrix and outer membrane (Figure 2). The presence of miRNA machinery proteins in the mitochondrial fractions prompted us to examine whether these fractions also contain AGO-associated miRNA complexes. The anti-AGO antibody 2A8 (Nelson et al., 2007) was used to co-immunoprecipitate (co-IP) AGO protein complexes (Wang et al., 2010) from isolated rat hippocampus mitochondrial (Mito-IP2A8) and cytosolic (Cyto-IP2A8) fractions. Western blot analysis of the IP products showed that the anti-AGO antibody 2A8 (IP2A8), but not normal mouse serum (IPNMS), effectively immunoprecipitated AGO2 from both the cytosolic and mitochondrial fractions (Figure 3 inset). Total RNA was extracted from the co-IP complexes and subjected to miRNA RT-qPCR TaqMan® assays for miR-124a and miR-107, two brain-enriched miRNAs (Wang et al., 2014), and miR-23a, which has been reported to be present in mitochondria (Das et al., 2012), as well as the small nuclear RNA U6. The RT-qPCR results demonstrated that these test miRNAs, but not U6, were co-immunoprecipitated with and enriched in mitochondrial AGO complexes (Fig. 3). As expected, IP using normal mouse serum (IPNMS, data not shown) in the same preparation did not result in appreciable levels of these miRNAs.
Figure 2. MiRNA machinery proteins Dicer and AGO2 were present in rat brain mitochondria.
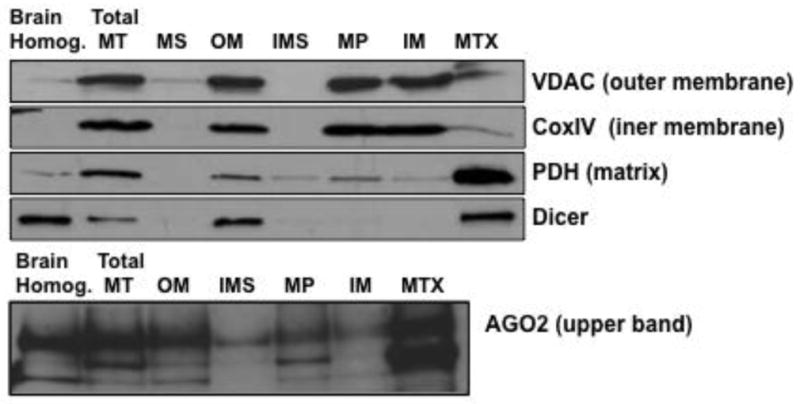
Isolated mitochondria and mitochondrial subfractions were analyzed for Dicer and AGO2 using Western blotting. Antibodies against marker proteins for the mitochondria subfractions included VDAC (outer membrane), CoxIV (inner membrane), and PDH (matrix). Brain Homog.: total brain homogenate; Total MT: total mitochondria lysate; MS: mitochondrial supernatant containing broken outer membrane and inter-membrane space compartments; OM: outer membrane; IMS: inter-membrane space; MP: mitoplast; IM: inner membrane; MTX: mitochondria matrix.
Figure 3. Enrichment of mitochondrial miRNA in AGO complexes.

Co-immunoprecipitation (co-IP) with anti-AGO 2A8 antibody was performed using isolated total mitochondrial lysate. Corresponding cytosols were also subjected to co-IP. The co-IP RNAs were extracted and analyzed using RT-qPCR TaqMan® assays for miR-23a, miR-107, and miR-124a, and a control small nuclear RNA U6. MiRNAs were specifically enriched in anti-AGO 2A8 antibody co-immunoprecipitates (IP2A8), while co-IP using normal mouse serum (IPNMS, data not shown) did not result in any specific signal. As expected, U6, which does not associate with AGO complexes was not detected in co-IP RNA samples. Inset: Western blot analysis of the IP products using anti-AGO 2A8 antibody (IP2A8) or normal mouse serum (IPNMS). TL: total lysate.
Profiling of naïve hippocampal cytosolic and mitochondrial miRNAs
The TaqMan® RT-qPCR Array Rodent MicroRNA Card A (V2.0) (Life Technologies) was employed to analyze the global expression levels of miRNA associated with the mitochondria and, separately, the cytosolic fractions. A pre-amplification step using Megaplex™ PreAmp Primers was added to enhance the detection of low-expression miRNAs for both mitochondrial and cytosolic fractions. Quantitative PCR was performed using the ViiA™ 7 Real-Time PCR System (Life Technologies), and the data collected and processed using ViiA 7 RUO software (Life Technologies). Data that failed QC were excluded. In addition, individual miRNAs with a cycle threshold (Ct) value of 35 or higher (Ct>35) were considered as undetectable and excluded from analysis. Using these criteria, 285 out of 381 miRNAs in the Rodent Array Card A were detected in both cytosolic and mitochondrial fractions (Suppl. File 1). The overall miRNA expression levels in the cytosol were generally higher relative to mitochondria with an average Ct difference of 1.3, which is equivalent to a 2.5 fold higher level (Suppl. File 1). The largest differential expression was observed with rno-miR-333, where expression levels in cytosol were over 100-fold higher than that found in mitochondria.
Subset of miRNAs is enriched in naïve hippocampal mitochondria
Further analysis of the mitochondrial and cytosolic miRNA array data revealed contrasting miRNA expression pattern in samples from naïve rat hippocampi. While the levels of most miRNAs were found to be much higher in the cytosol, a subset of miRNAs including mmu-miR-741, mmu-miR-142-5p, mmu-miR-302a, mmu-miR-142-3p, rno-miR-339-3p, mmu-miR-10a, mmu-miR-146a, mmu-miR-202-3p, mmu-miR-150, mmu-miR-339-5p, and rno-miR-344-5p were found to be at least 1.5 fold higher in mitochondria (Figure 4, Suppl. File 2). Among these mitochondria-enriched miRNAs, miR-142-5p, miR-142-3p, and miR-146a were significantly higher (p<0.05, FDR < 5%) compared to levels in the cytosol. Although not statistically significant, miR-150 also displayed enrichment in mitochondria relative to the cytosol.
Figure 4. Differential miRNA enrichment in cytosol and mitochondria.

Volcano plot of RT-qPCR array data displayed differential miRNA enrichment in cytosol and mitochondria. Each data point was plotted as the level of fold change (Log2(Fold Change—Mito vs Cyto)) on the X-axis and corresponding statistical level of significance (−log10(p value)) on the y-axis. Fold change=2^ −(CtMito − CtCyto). Mitochondria-enriched miRNAs with a more than 2 fold change were colored in red; likewise, cytosol-enriched miRNAs that having a change more than 2 fold were colored in green. Data points above the red-dashed-line are statistically significant (p < 0.05). The numeric data set of this plot is provided in Suppl. File 2.
As a follow-up to the array data described above, TaqMan® single-tube RT-qPCR assays for miR-142-3p, miR-142-5p, miR-146a, miR-150 were carried out on RNA samples isolated from mitochondria and cytosol of naïve rat hippocampus (Figure 5A). We also examined the levels of miR-19b, a miRNA that did not display significantly different levels between cytosolic and mitochondria fractions, and U6, a small nuclear RNA that does not associate with mitochondria. The results of the single-tube TaqMan® assays confirmed that miR-142-3p, miR-142-5p, and miR-146a levels were significantly higher (p<0.05) in mitochondrial RNA samples relative to the cytosol. As observed in the array data, miR-150 also showed higher trending levels in mitochondria relative to cytosol although not statistically significant (p=0.11). In comparison, miR-19b levels were not enriched in the mitochondria, and the small nuclear RNA U6 was approximately 13-fold less abundant in mitochondria relative to cytosol RNA. We also examined these same miRNAs in mitochondria and cytosolic fractions isolated from cultured primary rat brain astrocytes. The TaqMan® assays confirmed that miR-142-3p, miR-142-5p, and miR-146a levels were 2.30, 2.42 and 2.26 fold higher, respectively, in astrocyte mitochondria than that found in cytosol (Figure 5B).
Figure 5. Subset of miRNAs was enriched in mitochondria.

Single-tube TaqMan® assays further validated that a subset of miRNAs was enriched in mitochondria. TaqMan® assays for the indicated miRNAs and U6 were performed using rat hippocampal mitochondrial (Mito) and cytosolic (Cyto) preparations (A), or rat cortical astrocyte mitochondrial and cytosolic preparations (B). The results demonstrated that miR-142-3p, miR-142-5p, and miR-146a are enriched in mitochondrial fractions, which is consistent with the array findings.
Altered expression of hippocampal mitochondria-associated miRNA following severe TBI
TBI resulted in altered expression levels of miRNA in both the hippocampal mitochondria and cytosol fractions at 12 hr following injury (Suppl. File 1). The TaqMan® RT-qPCR Array revealed that the differential overall expression levels of miRNAs in cytosol relative to mitochondria increased from 2.5 fold in naïve samples to 4.2 fold in injured hippocampus (Suppl. File 1). The levels of most mitochondria-associated miRNAs were decreased at this early time point following TBI (Table 1, Suppl. Files 1 and 2). In particular, mitochondria-enriched miRNAs such as miR-142-3p, miR-142-5p, and miR-146a were decreased more than two fold following TBI, while these miRNAs increased in cytosol. Follow-up single-tube TaqMan® assays were used to confirm the initial array data (single-tube assays are presented in parentheses next to the array data in Table 1). The single-tube assays revealed that the mitochondria-enriched miRNAs, miR-142-3p and miR-142-5p were indeed significantly reduced in mitochondrial fractions while levels in cytosol were significantly increased (see Table 1 for p values). The levels of another mitochondria-enriched miRNA, miR-146a, were not significantly decreased in mitochondria (p=0.058), but were significantly higher (p=0.004) in cytosol.
Table 1. Changes in mitochondria-associated and cytosolic miRNAs at 12 hr following TBI.
List of miRNAs with significant changes based on the RT-qPCR array analysis. Data and p values presented in parentheses were follow-up studies using single-tube TaqMan assay analysis. Red and green colors designate miRNA that significantly increased or decreased, respectively, in each fraction. Fold difference of each miRNA was calculated following the formula: Fold Change=2^ −(CtTBI − CtNaive). Permutation test was used to control False Discovery Rate (FDR). All significant data point (p<0.05) has a FDR< 5%.
| Mitochondria | Cytosol | |||
|---|---|---|---|---|
|
|
||||
| miRNA | Fold Change (TBI vs Naïve) | P Value | Fold Change (TBI vs Naïve) | P Value |
| mmu-miR-155 | 7.30 | 0.023 | 5.2 | 0.05 |
| mmu-miR-223 | 4.63 | 0.021 | 14.9 | 3.2E-06 |
| mmu-miR-15a | 0.66 | 0.043 | 1.06 | 0.885 |
| rno-miR-421 | 0.65 | 0.048 | 1.13 | 0.705 |
| mmu-miR-532-3p | 0.56 | 0.014 | 1.05 | 0.881 |
| rno-miR-532-5p | 0.56 | 0.019 | 1.11 | 0.646 |
| mmu-miR-344 | 0.54 | 0.045 | 0.97 | 0.885 |
| mmu-miR-187 | 0.52 | 0.0008 | 0.76 | 0.140 |
| mmu-miR-434-5p | 0.52 | 0.038 | 0.90 | 0.727 |
| mmu-miR-142-5p | 0.28 (0.5) | 0.031 (0.004) | 2.02 (1.95) | 0.168 (0.022) |
| mmu-miR-1 | 0.23 | 0.017 | 0.63 | 0.458 |
| mmu-miR-10a | 0.17 | 0.033 | 1.39 | 0.767 |
| mmu-miR-743a | 0.06 | 0.009 | 2.30 | 0.552 |
| mmu-miR-142-3p | 0.47 (0.49) | 0.176 (0.0004) | 2.15 (1.73) | 0.112 (0.008) |
| mmu-miR-292-3p | 5.18 | 0.140 | 8.61 | 0.074 |
| rno-miR-224 | 1.87 | 0.571 | 5.11 | 0.058 |
| mmu-miR-224 | 1.67 | 0.621 | 3.77 | 0.015 |
| mmu-miR-200c | 0.82 | 0.694 | 1.67 | 0.068 |
| mmu-miR-682 | 2.33 | 0.245 | 1.55 | 0.024 |
| mmu-miR-146a | 0.44 (0.61) | 0.134 (0.058) | 1.47 (1.40) | 0.251 (0.004) |
The levels of several mitochondria-associated miRNAs were found to increase following TBI. Among them, miR-155 and miR-223 levels were significantly elevated to 7.3 and 4.63 fold higher, respectively, in mitochondria fractions of injured relative to naïve (uninjured) rats (Table 1). To further validate these changes, single tube TaqMan® RT-qPCR was carried out for miR-155, miR-223, and a control miRNA, miR-19b, which did not change after injury. The levels of miR-155 and miR-223 were then normalized against the levels of miR-19b. RT-qPCR of hippocampal cytosolic and mitochondrial RNA preparations (n = 5 animals in each group) showed that miR-155 and miR-223 were indeed significantly elevated following TBI (Figure 6), confirming the results of the array experiments. The fold changes observed in the single-tube RT-qPCR assays were much greater compared to the array data, which may be due to methodological differences, as well as differences in data normalization. Notably, in the single-tube RT-qPCR assay, the pre-amplification step was omitted, and the data normalized using miR-19b.
Figure 6. MiR-155 and miR-223 were highly elevated following TBI.
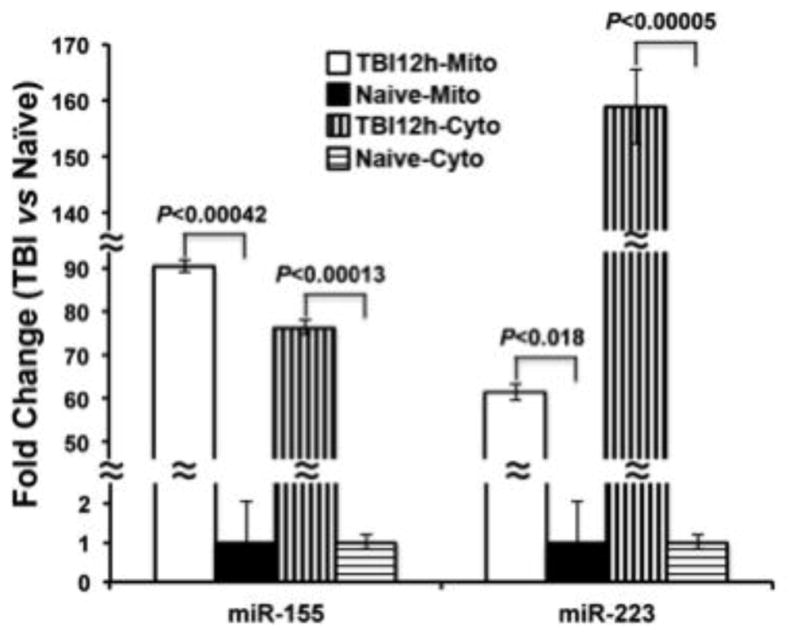
Single-tube TaqMan® assays for miR-155 and miR-223 using mitochondrial (Mito) and cytosolic (Cyto) preparations from TBI and naïve rat hippocampi demonstrated a strong induction of these two miRNAs following TBI in both mitochondrial and cytosolic fractions. Fold change was calculated relative to the levels observed in naïve samples.
MiR-155 and miR-223 are recognized as two hematopoietic cell type specific miRNAs that are associated with hematopoiesis and leukocyte activation (Fazi et al., 2005; Landgraf et al., 2007; Rodriguez et al., 2007; Vigorito et al., 2007; Johnnidis et al., 2008). To examine the possibility that hematopoietic cell infiltration in injured rat hippocampus at 12 hr following TBI were what caused the miRNA expression we observed in our samples, we examined CD45 (pan leukocyte cell marker) expression in naïve and TBI hippocampal cytosolic protein preparations. CD45 levels did not increase in the samples at this early time point following TBI, indicating that the changes of miR-155 and miR-223 were not attributable to hematopoietic cell infiltration in response to injury (Suppl. Figure 1).
We further examined the expression of miRNAs in rat brain cell preparations that were enriched for particular cell types. TaqMan® miRNA RT-qPCR was used to detect miR-155, miR-223, miR-146a, miR-142-3p, miR-142-5p, miR-150, and miR-124a expression in enriched cortical neuron, astrocyte, and microglia cultures. The outcomes of this experiment demonstrated that miR-155, miR-223, miR-146a, miR-142-3p, miR-142-5p, and miR-150 (data not shown) were detected in all three cell types but showed the highest expression in microglia and astrocytes (Fig 7), cell types that can mount immune responses. In contrast, levels of miR-124a were highest in the cortical neuronal cells as expected (Wang et al., 2008b; Wang et al., 2014).
Figure 7. MiR-155, miR-223, miR-146a, miR-142-3p, miR-142-5p, and miR-150 are detected in rat brain cells.

TaqMan® assays for miR-155, miR-223, miR-146a, miR-142-3p, miR-142-5p, and miR-150 (not shown) were performed using total RNAs isolated from untreated primary cortical neuron, astrocyte, and microglia enriched cultures. These miRNAs were readily detected in all three primary cell types with high expression levels in the astrocyte and microglial cells. As a control, a neuronal enriched miRNA, miR-124a, was tested in parallel. Expression levels of miRNAs are presented as fold change relative to levels in primary cortical neurons. The one exception is miR-124a, the levels of which are expressed relative to levels in primary astrocytes. Average Ct values of neuronal RNA samples were 28.2, 26.6, 28.0, 23.9, 28.9, 23.1, and 19.1 for miR-155, miR-223, miR-150, miR-146a, miR-142-3p, miR-142-5p, and miR-124a, respectively.
MiR-155 is known to participate in the regulation of inflammatory responses (Kurowska-Stolarska et al., 2011; Moore et al., 2013), which also occur at early time points following TBI. Incubation of rat primary microglia and astrocyte cell cultures with lipopolysaccharide (LPS) resulted in strong elevation of miR-155 levels in both cell types (p<0.001 for both microglia and astrocytes) (Fig 8). These experiments are consistent with prior findings that miR-155 is responsive to inflammatory mediators, such as LPS, in macrophages, monocytes, as well as microglia. Unlike miR-155, miR-223 was not elevated in either microglia and astrocyte cultures treated with LPS, suggesting that up-regulation of these two mRNAs in hippocampus following TBI may be driven by different signaling events.
Figure 8. MiR-155 responds to LPS treatments in rat astrocyte and microglia.

TaqMan® assays for miR-155 (A) and miR-223 (B). There was a significant elevation of miR-155, but not miR-223, in response to LPS treatment in both astrocyte and microglia cell cultures; These results suggest that the up-regulation of miR-155 and miR-223 in hippocampus following TBI may be driven by different signaling pathways. C. Western blot analysis showed that iNOS was induced in response to LPS treatment in primary microglial cell cultures. Fold change was calculated relative to the levels detected in untreated control cells.
Discussion
Mitochondria play a critical role in providing cellular energy and respond to a number of extracellular and intracellular environmental events. However, the signaling events that regulate miRNA function, especially their potential interactions with cellular organelles are largely unknown. Emerging research points to a possible interaction between miRNA and mitochondria (Maniataki and Mourelatos, 2005; Kren et al., 2009; Bian et al., 2010; Bandiera et al., 2011; Barrey et al., 2011; Huang et al., 2011; Das et al., 2012; Ernoult-Lange et al., 2012; Latronico and Condorelli, 2012; Sripada et al., 2012), suggesting a potential novel pathway regulating miRNA function.
Prior studies have reported detection of miRNAs, ranging from 3 (Mercer et al., 2011) to 428 (Sripada et al., 2012), within or associated with mitochondria from various tissues and cell types (Lung et al., 2006; Kren et al., 2009; Bian et al., 2010; Bandiera et al., 2011; Barrey et al., 2011; Huang et al., 2011; Mercer et al., 2011; Das et al., 2012; Sripada et al., 2012). In the present study, the miRNA TaqMan® qPCR array detected 285 miRNAs in highly purified naïve hippocampal mitochondria preparations. In addition, although the average expression level of most of these miRNAs was 2.5-fold lower than the same miRNA in the cytosol, several miRNAs were preferentially enriched in the mitochondria preparations. Among them, mitochondria-enriched miR-142-3p, miR-142-5p, miR-146a, and miR-150 were further validated using RT-qPCR conducted on mitochondrial RNA isolated from cultured primary rat astrocytes. Previous studies have shown that miR-146a is enriched in mitochondria from human 143B cells (Mercer et al., 2011) as well as mouse liver (Bian et al., 2010). Interestingly, all of these mitochondria-associated miRNAs are known to be involved in immune function and inflammatory responses (Landgraf et al., 2007; Xiao et al., 2007; Sonkoly et al., 2008; Sun et al., 2010; Vasilatou et al., 2010). Taken together, these studies suggest the possible existence of a regulatory process by which certain miRNA or miRNA families are partitioned to mitochondria based on common functional domains.
The 12hr post-injury time point we examined corresponds to a dynamic period when mitochondrial dysfunction, oxidative damage, and cell death are ongoing in the hippocampus (Baldwin et al., 1997; Lifshitz et al., 2003; Ansari et al., 2008; Davis et al., 2008; Gilmer et al., 2009). Therefore, it is of interest that mitochondria-associated miRNAs in the hippocampus displayed significant changes at 12 hr following TBI. Importantly, mitochondria-enriched miRNAs, such as miR-142-3p, miR-142-5p, and miR-146a were all decreased at this early time point, while levels of the same miRNA increased in the cytosol. Any mechanistic explanation for this shift in expression levels at this early time point is unclear. The ficoll isolation procedure we used captures only intact mitochondria that are at different stages of compromised function. Therefore, we believe that the mitochondria to cytosol shift reflects a true mechanistic event in response to TBI-related cellular stressors. It is interesting to note that mitochondria in the hippocampus respond to TBI differently than cortical mitochondria, especially at early time points (Lifshitz et al., 2003; Lifshitz et al., 2004). In particular, cortical mitochondria are structurally and functionally more compromised compared to mitochondria from the hippocampus, which is consistent with differences in cell death responses between these two brain regions (Dietrich et al., 1994; Hicks et al., 1996; Lifshitz et al., 2003; Lifshitz et al., 2004). It is possible that the structural and functional changes that occur in the mitochondria are responsible for the observed shift in miRNA expression patterns. If this indeed is occurring, it will be important to determine the functional consequences as well as the cell type(s) impacted by this shift in miRNA expression patterns. This differential temporal and regional response to TBI also guided our decision to examine changes in mitochondria associated miRNA from the hippocampal formation at 12 hr following injury. Our observations suggest that mitochondria-associated miRNAs in the hippocampus are highly sensitive to cellular changes and respond to cellular stressors by translocating from the mitochondrial to the cytosolic compartment. Alternatively, these specific miRNAs may be up-regulated in the cytosol after TBI independent of what is occurring in the mitochondrial compartment. Regardless, the observation that levels of mitochondria-enriched miRNA decreased after TBI suggests a potential mechanism by which miRNA function may be regulated in cells under conditions of duress.
The levels of miR-155 and miR-223, both of which play a role in several biological/pathological processes including immunity, cancer, and inflammatory responses (Sonkoly et al., 2008; Quinn and O’Neill, 2011; Higgs and Slack, 2013; Vigorito et al., 2013; Seddiki et al., 2014; Taibi et al., 2014), were significantly elevated in both the mitochondria as well as cytosolic fractions. MiR-223 levels have previously been shown to be elevated in rat total hippocampal tissue, or cerebral cortex homogenates at different times following TBI (Lei et al., 2009; Redell et al., 2009). Interestingly, miR-155 levels were found to increase in mitochondria isolated from mouse liver after streptozotocin treatment, which is known to produce Type 1 diabetes in animal model and impair mitochondria function (Bian et al., 2010). We further tested the potential signaling pathways that trigger elevated miR-155 and miR-223 by treating primary astrocyte and microglia cells with LPS, which activates the Toll-like receptor signaling pathway leading to an inflammatory response (Hoshino et al., 1999). The results of this experiment demonstrated that miR-155, but not miR-223, was significantly up-regulated in both astrocyte and microglia following LPS treatment. One explanation for this differential effect of LPS is that the up-regulation of miR-155 and miR-223 in the hippocampus following TBI may be driven by different injury-related signaling events.
Recent studies have reported changes in several miRNAs in the hippocampus following TBI (Lei et al., 2009; Redell et al., 2009; Redell et al., 2010; Truettner et al., 2011; Hu et al., 2012; Truettner et al., 2013; Liu et al., 2014; Sabirzhanov et al., 2014; Sun et al., 2014). The results of our experiments revealed an increase in miR-223 in both mitochondria and cytosol, which is consistent with the results of previous studies using the same injury model and whole tissue homogenates (Lei et al., 2009; Redell et al., 2009) However, we did not detect significant changes in levels of several other miRNAs, (such as miR-107 and miR-21) that have been reported to respond to TBI. It is difficult to compare the results of the present study examining changes in mitochondrial versus cytoplasmic miRNA levels with existing studies that examined miRNA changes in whole tissue homogenates. In addition, we chose the 12hr post-injury recovery period as it captures an early time point when there is a maximal loss of bioenergetics and structural integrity in hippocampal mitochondria following the injury (Lifshitz et al., 2003; Singh et al., 2006; Gilmer et al., 2009). Finally, differences in the injury model and the miRNA assay systems used may contribute to some of the variations in the results observed across studies. Therefore, it will be important to examine changes in mitochondrial and cytosolic miRNA expression patterns at other time points using the same injury model and miRNA detection system.
TBI induces secondary signaling events that are biochemically relevant to functional recovery (McIntosh et al., 1998; Rosenfeld et al., 2012). Overall, the results of our study suggest that mitochondria-associated miRNAs actively participate in the response to TBI. However, the events contributing to mitochondria and miRNA interactions, and the mechanism(s) affecting altered mitochondria-associated miRNA levels following TBI are still largely unknown. A previous study has demonstrated a dynamic interaction between mitochondria and miRNA associated processing bodies (P-bodies) (Huang et al., 2011). Interestingly, this same study reported that treatment of mitochondria with an uncoupling agent resulted in delocalization of AGO from P-bodies, which led to a significant decrease in miRNA mediated RNAi efficiency. Although additional studies are required to reveal the crosstalk and trafficking of miRNAs between mitochondria, cytosol, and other cellular organelles/compartments, this study raises the possibility that mitochondria play a role in regulating miRNA function. Future studies exploring this dynamic interaction, and its implication in various pathophysiological processes may lead to a better understanding of secondary damage following TBI.
Supplementary Material
Highlights.
MiRNA machinery proteins Argonaute and Dicer are present in hippocampal mitochondria
Several miRNAs are enriched in hippocampal mitochondria under normal conditions
Numerous mitochondria-associated miRNAs are significantly altered following TBI
MiRNAs enriched in mitochondria decreased and cytosolic levels increased after TBI
Acknowledgments
The described work was supported by the Morton Cure Paralysis Fund and an endowment from Cardinal Hill Rehabilitation Hospital (JES), an ADC-Pilot grant by the National Center for Advancing Translational Sciences, National Institutes of Health, through grant number UL1TR000117 (WXW), PHS grants NS085830 and AG042419 (PTN) and a KSCHIRT endowed chair funds (PGS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free radical biology & medicine. 2008;45:443–452. doi: 10.1016/j.freeradbiomed.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atorino L, Silvestri L, Koppen M, Cassina L, Ballabio A, Marconi R, Langer T, Casari G. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. The Journal of cell biology. 2003;163:777–787. doi: 10.1083/jcb.200304112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin SA, Gibson T, Callihan CT, Sullivan PG, Palmer E, Scheff SW. Neuronal cell loss in the CA3 subfield of the hippocampus following cortical contusion utilizing the optical disector method for cell counting. Journal of neurotrauma. 1997;14:385–398. doi: 10.1089/neu.1997.14.385. [DOI] [PubMed] [Google Scholar]
- Bandiera S, Ruberg S, Girard M, Cagnard N, Hanein S, Chretien D, Munnich A, Lyonnet S, Henrion-Caude A. Nuclear outsourcing of RNA interference components to human mitochondria. PloS one. 2011;6:e20746. doi: 10.1371/journal.pone.0020746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrey E, Saint-Auret G, Bonnamy B, Damas D, Boyer O, Gidrol X. Pre-microRNA and mature microRNA in human mitochondria. PloS one. 2011;6:e20220. doi: 10.1371/journal.pone.0020220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Z, Li LM, Tang R, Hou DX, Chen X, Zhang CY, Zen K. Identification of mouse liver mitochondria-associated miRNAs and their potential biological functions. Cell research. 2010;20:1076–1078. doi: 10.1038/cr.2010.119. [DOI] [PubMed] [Google Scholar]
- Brown MR, Sullivan PG, Dorenbos KA, Modafferi EA, Geddes JW, Steward O. Nitrogen disruption of synaptoneurosomes: an alternative method to isolate brain mitochondria. Journal of neuroscience methods. 2004;137:299–303. doi: 10.1016/j.jneumeth.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical chemistry. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nature reviews Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nature reviews Genetics. 2007;8:93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, Melick J, Loeffert JE, Nathaniel PD, Jin KL, Graham SH. Caspase-3 mediated neuronal death after traumatic brain injury in rats. Journal of neurochemistry. 2000;74:740–753. doi: 10.1046/j.1471-4159.2000.740740.x. [DOI] [PubMed] [Google Scholar]
- Das S, Ferlito M, Kent OA, Fox-Talbot K, Wang R, Liu D, Raghavachari N, Yang Y, Wheelan SJ, Murphy E, Steenbergen C. Nuclear miRNA regulates the mitochondrial genome in the heart. Circulation research. 2012;110:1596–1603. doi: 10.1161/CIRCRESAHA.112.267732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LM, Pauly JR, Readnower RD, Rho JM, Sullivan PG. Fasting is neuroprotective following traumatic brain injury. Journal of neuroscience research. 2008;86:1812–1822. doi: 10.1002/jnr.21628. [DOI] [PubMed] [Google Scholar]
- Di Y, Lei Y, Yu F, Changfeng F, Song W, Xuming M. MicroRNAs Expression and Function in Cerebral Ischemia Reperfusion Injury. Journal of molecular neuroscience: MN. 2014 doi: 10.1007/s12031-014-0293-8. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Alonso O, Halley M. Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. Journal of neurotrauma. 1994;11:289–301. doi: 10.1089/neu.1994.11.289. [DOI] [PubMed] [Google Scholar]
- Ernoult-Lange M, Benard M, Kress M, Weil D. P-bodies and mitochondria: which place in RNA interference? Biochimie. 2012;94:1572–1577. doi: 10.1016/j.biochi.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Faul MXL, Wald MM, Coronado VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, Bozzoni I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. 2005;123:819–831. doi: 10.1016/j.cell.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Finkelstein E, Corso PS, Miller TR. The incidence and economic burden of injuries in the United States. Oxford; New York: Oxford University Press; 2006. [Google Scholar]
- Gilmer LK, Roberts KN, Joy K, Sullivan PG, Scheff SW. Early mitochondrial dysfunction after cortical contusion injury. Journal of neurotrauma. 2009;26:1271–1280. doi: 10.1089/neu.2008.0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A. Functions of microRNAs in cardiovascular biology and disease. Annual review of physiology. 2013;75:69–93. doi: 10.1146/annurev-physiol-030212-183737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks R, Soares H, Smith D, McIntosh T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta neuropathologica. 1996;91:236–246. doi: 10.1007/s004010050421. [DOI] [PubMed] [Google Scholar]
- Higgs G, Slack F. The multiple roles of microRNA-155 in oncogenesis. Journal of clinical bioinformatics. 2013;3:17. doi: 10.1186/2043-9113-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. Journal of immunology. 1999;162:3749–3752. [PubMed] [Google Scholar]
- Hu JZ, Huang JH, Zeng L, Wang G, Cao M, Lu HB. Anti-apoptotic effect of microRNA-21 after contusion spinal cord injury in rats. Journal of neurotrauma. 2013;30:1349–1360. doi: 10.1089/neu.2012.2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Yu D, Almeida-Suhett C, Tu K, Marini AM, Eiden L, Braga MF, Zhu J, Li Z. Expression of miRNAs and their cooperative regulation of the pathophysiology in traumatic brain injury. PloS one. 2012;7:e39357. doi: 10.1371/journal.pone.0039357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Mollet S, Souquere S, Le Roy F, Ernoult-Lange M, Pierron G, Dautry F, Weil D. Mitochondria associate with P-bodies and modulate microRNA-mediated RNA interference. The Journal of biological chemistry. 2011;286:24219–24230. doi: 10.1074/jbc.M111.240259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nature reviews Genetics. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, Brummelkamp TR, Fleming MD, Camargo FD. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- Kren BT, Wong PY, Sarver A, Zhang X, Zeng Y, Steer CJ. MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA biology. 2009;6:65–72. doi: 10.4161/rna.6.1.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurowska-Stolarska M, Alivernini S, Ballantine LE, Asquith DL, Millar NL, Gilchrist DS, Reilly J, Ierna M, Fraser AR, Stolarski B, McSharry C, Hueber AJ, Baxter D, Hunter J, Gay S, Liew FY, McInnes IB. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:11193–11198. doi: 10.1073/pnas.1019536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf P, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latronico MV, Condorelli G. The might of microRNA in mitochondria. Circulation research. 2012;110:1540–1542. doi: 10.1161/CIRCRESAHA.112.271312. [DOI] [PubMed] [Google Scholar]
- Lei P, Li Y, Chen X, Yang S, Zhang J. Microarray based analysis of microRNA expression in rat cerebral cortex after traumatic brain injury. Brain research. 2009;1284:191–201. doi: 10.1016/j.brainres.2009.05.074. [DOI] [PubMed] [Google Scholar]
- Lifshitz J, Sullivan PG, Hovda DA, Wieloch T, McIntosh TK. Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion. 2004;4:705–713. doi: 10.1016/j.mito.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Lifshitz J, Friberg H, Neumar RW, Raghupathi R, Welsh FA, Janmey P, Saatman KE, Wieloch T, Grady MS, McIntosh TK. Structural and functional damage sustained by mitochondria after traumatic brain injury in the rat: evidence for differentially sensitive populations in the cortex and hippocampus. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2003;23:219–231. doi: 10.1097/01.WCB.0000040581.43808.03. [DOI] [PubMed] [Google Scholar]
- Liu L, Sun T, Liu Z, Chen X, Zhao L, Qu G, Li Q. Traumatic brain injury dysregulates microRNAs to modulate cell signaling in rat hippocampus. PloS one. 2014;9:e103948. doi: 10.1371/journal.pone.0103948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu NK, Wang XF, Lu QB, Xu XM. Altered microRNA expression following traumatic spinal cord injury. Experimental neurology. 2009;219:424–429. doi: 10.1016/j.expneurol.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Fortin K, Mourelatos Z. MicroRNAs: biogenesis and molecular functions. Brain pathology. 2008;18:113–121. doi: 10.1111/j.1750-3639.2007.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lung B, Zemann A, Madej MJ, Schuelke M, Techritz S, Ruf S, Bock R, Huttenhofer A. Identification of small non-coding RNAs from mitochondria and chloroplasts. Nucleic acids research. 2006;34:3842–3852. doi: 10.1093/nar/gkl448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniataki E, Mourelatos Z. Human mitochondrial tRNAMet is exported to the cytoplasm and associates with the Argonaute 2 protein. RNA (New York, NY. 2005;11:849–852. doi: 10.1261/rna.2210805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Current biology: CB. 2006;16:R551–560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Saatman KE, Raghupathi R, Graham DI, Smith DH, Lee VM, Trojanowski JQ. The Dorothy Russell Memorial Lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. Neuropathology and applied neurobiology. 1998;24:251–267. doi: 10.1046/j.1365-2990.1998.00121.x. [DOI] [PubMed] [Google Scholar]
- Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, Filipovska A, Mattick JS. The human mitochondrial transcriptome. Cell. 2011;146:645–658. doi: 10.1016/j.cell.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestdagh P, Van Vlierberghe P, De Weer A, Muth D, Westermann F, Speleman F, Vandesompele J. A novel and universal method for microRNA RT-qPCR data normalization. Genome biology. 2009;10:R64. doi: 10.1186/gb-2009-10-6-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS, Rao VT, Durafourt BA, Bedell BJ, Ludwin SK, Bar-Or A, Antel JP. miR-155 as a multiple sclerosis-relevant regulator of myeloid cell polarization. Annals of neurology. 2013;74:709–720. doi: 10.1002/ana.23967. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Wang WX, Rajeev BW. MicroRNAs (miRNAs) in neurodegenerative diseases. Brain pathology. 2008a;18:130–138. doi: 10.1111/j.1750-3639.2007.00120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Wang WX, Wilfred BR, Tang G. Technical variables in high-throughput miRNA expression profiling: much work remains to be done. Biochimica et biophysica acta. 2008b;1779:758–765. doi: 10.1016/j.bbagrm.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, De Planell-Saguer M, Lamprinaki S, Kiriakidou M, Zhang P, O’Doherty U, Mourelatos Z. A novel monoclonal antibody against human Argonaute proteins reveals unexpected characteristics of miRNAs in human blood cells. RNA (New York, NY. 2007;13:1787–1792. doi: 10.1261/rna.646007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Sullivan PG. The optimal dosage and window of opportunity to maintain mitochondrial homeostasis following traumatic brain injury using the uncoupler FCCP. Experimental neurology. 2009;218:381–389. doi: 10.1016/j.expneurol.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Pandya JD, Rabchevsky AG. Differential effects of the mitochondrial uncoupling agent, 2,4-dinitrophenol, or the nitroxide antioxidant, Tempol, on synaptic or nonsynaptic mitochondria after spinal cord injury. Journal of neuroscience research. 2009;87:130–140. doi: 10.1002/jnr.21814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn SR, O’Neill LA. A trio of microRNAs that control Toll-like receptor signalling. International immunology. 2011;23:421–425. doi: 10.1093/intimm/dxr034. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain pathology. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redell JB, Liu Y, Dash PK. Traumatic brain injury alters expression of hippocampal microRNAs: potential regulators of multiple pathophysiological processes. Journal of neuroscience research. 2009;87:1435–1448. doi: 10.1002/jnr.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redell JB, Moore AN, Ward NH, 3rd, Hergenroeder GW, Dash PK. Human traumatic brain injury alters plasma microRNA levels. Journal of neurotrauma. 2010;27:2147–2156. doi: 10.1089/neu.2010.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. The American journal of pathology. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JV, Maas AI, Bragge P, Morganti-Kossmann MC, Manley GT, Gruen RL. Early management of severe traumatic brain injury. Lancet. 2012;380:1088–1098. doi: 10.1016/S0140-6736(12)60864-2. [DOI] [PubMed] [Google Scholar]
- Sabirzhanov B, Zhao Z, Stoica BA, Loane DJ, Wu J, Borroto C, Dorsey SG, Faden AI. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:10055–10071. doi: 10.1523/JNEUROSCI.1260-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- Seddiki N, Brezar V, Ruffin N, Levy Y, Swaminathan S. Role of miR-155 in the regulation of lymphocyte immune function and disease. Immunology. 2014;142:32–38. doi: 10.1111/imm.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2006;26:1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- Sonkoly E, Stahle M, Pivarcsi A. MicroRNAs and immunity: novel players in the regulation of normal immune function and inflammation. Seminars in cancer biology. 2008;18:131–140. doi: 10.1016/j.semcancer.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Sripada L, Tomar D, Prajapati P, Singh R, Singh AK, Singh R. Systematic analysis of small RNAs associated with human mitochondria by deep sequencing: detailed analysis of mitochondrial associated miRNA. PloS one. 2012;7:e44873. doi: 10.1371/journal.pone.0044873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland ER, Hook MA, Balaraman S, Huie JR, Grau JW, Miranda RC. MicroRNA dysregulation following spinal cord contusion: implications for neural plasticity and repair. Neuroscience. 2011;186:146–160. doi: 10.1016/j.neuroscience.2011.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Experimental neurology. 1999;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Keller JN, Bussen WL, Scheff SW. Cytochrome c release and caspase activation after traumatic brain injury. Brain research. 2002;949:88–96. doi: 10.1016/s0006-8993(02)02968-2. [DOI] [PubMed] [Google Scholar]
- Sun TY, Chen XR, Liu ZL, Zhao LL, Jiang YX, Qu GQ, Wang RS, Huang SZ, Liu L. Expression profiling of microRNAs in hippocampus of rats following traumatic brain injury. Journal of Huazhong University of Science and Technology Medical sciences = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban. 2014;34:548–553. doi: 10.1007/s11596-014-1313-1. [DOI] [PubMed] [Google Scholar]
- Sun W, Shen W, Yang S, Hu F, Li H, Zhu TH. miR-223 and miR-142 attenuate hematopoietic cell proliferation, and miR-223 positively regulates miR-142 through LMO2 isoforms and CEBP-beta. Cell research. 2010;20:1158–1169. doi: 10.1038/cr.2010.134. [DOI] [PubMed] [Google Scholar]
- Taibi F, Metzinger-Le Meuth V, Massy ZA, Metzinger L. miR-223: An inflammatory oncomiR enters the cardiovascular field. Biochimica et biophysica acta. 2014;1842:1001–1009. doi: 10.1016/j.bbadis.2014.03.005. [DOI] [PubMed] [Google Scholar]
- Truettner JS, Motti D, Dietrich WD. MicroRNA overexpression increases cortical neuronal vulnerability to injury. Brain research. 2013;1533:122–130. doi: 10.1016/j.brainres.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truettner JS, Alonso OF, Bramlett HM, Dietrich WD. Therapeutic hypothermia alters microRNA responses to traumatic brain injury in rats. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31:1897–1907. doi: 10.1038/jcbfm.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilatou D, Papageorgiou S, Pappa V, Papageorgiou E, Dervenoulas J. The role of microRNAs in normal and malignant hematopoiesis. European journal of haematology. 2010;84:1–16. doi: 10.1111/j.1600-0609.2009.01348.x. [DOI] [PubMed] [Google Scholar]
- Vigorito E, Kohlhaas S, Lu D, Leyland R. miR-155: an ancient regulator of the immune system. Immunological reviews. 2013;253:146–157. doi: 10.1111/imr.12057. [DOI] [PubMed] [Google Scholar]
- Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, Das PP, Miska EA, Rodriguez A, Bradley A, Smith KG, Rada C, Enright AJ, Toellner KM, Maclennan IC, Turner M. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27:847–859. doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Wilfred BR, Hu Y, Stromberg AJ, Nelson PT. Anti-Argonaute RIP-Chip shows that miRNA transfections alter global patterns of mRNA recruitment to microribonucleoprotein complexes. RNA (New York, NY. 2010;16:394–404. doi: 10.1261/rna.1905910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Wilfred BR, Baldwin DA, Isett RB, Ren N, Stromberg A, Nelson PT. Focus on RNA isolation: obtaining RNA for microRNA (miRNA) expression profiling analyses of neural tissue. Biochimica et biophysica acta. 2008a;1779:749–757. doi: 10.1016/j.bbagrm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008b;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Danaher RJ, Miller CS, Berger JR, Nubia VG, Wilfred BS, Neltner JH, Norris CM, Nelson PT. Expression of miR-15/107 family microRNAs in human tissues and cultured rat brain cells. Genomics, proteomics & bioinformatics. 2014;12:19–30. doi: 10.1016/j.gpb.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Yunta M, Nieto-Diaz M, Esteban FJ, Caballero-Lopez M, Navarro-Ruiz R, Reigada D, Pita-Thomas DW, del Aguila A, Munoz-Galdeano T, Maza RM. MicroRNA dysregulation in the spinal cord following traumatic injury. PloS one. 2012;7:e34534. doi: 10.1371/journal.pone.0034534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.