

Abstract
In utero interventions aimed at restoring left ventricular hemodynamic forces in fetuses with prenatally diagnosed hypoplastic left heart syndrome failed to stimulate ventricular myocardial growth during gestation, suggesting chamber growth during development may not rely upon fluid forces. We therefore hypothesized that ventricular hypertrophy during development may depend upon fundamental Ca2+-dependent growth pathways that function independent of hemodynamic forces. To test this hypothesis, zebrafish embryos were treated with inhibitors or activators of Ca2+ signaling in the presence or absence of contraction during the period of chamber development. Abolishment of contractile function alone in the setting of preserved Ca2+ signaling did not impair ventricular hypertrophy. In contrast, inhibition of L-type voltage-gated Ca2+ influx abolished contraction and led to reduced ventricular hypertrophy, whereas increasing L-type voltage-gated Ca2+ influx led to enhanced ventricular hypertrophy in either the presence or absence of contraction. Similarly, inhibition of the downstream Ca2+-sensitive phosphatase calcineurin, a known regulator of adult cardiac hypertrophy, led to reduced ventricular hypertrophy in the presence or absence of contraction, whereas hypertrophy was rescued in the absence of L-type voltage-gated Ca2+ influx and contraction by expression of a constitutively active calcineurin. These data suggest ventricular cardiomyocyte hypertrophy during chamber formation is dependent upon Ca2+ signaling pathways that are unaffected by heart function or hemodynamic forces. Disruption of Ca2+-dependent hypertrophy during heart development may therefore represent one mechanism for impaired chamber formation that is not related to impaired blood flow.
Keywords: calcium, hypertrophy, development, congenital heart defects, hypoplastic left heart syndrome
1. INTRODUCTION
Hypoplastic left heart syndrome (HLHS) is a devastating congenital heart malformation that accounts for approximately 25% of cardiac deaths within the first year of life [1]. The cardinal feature of HLHS is a small, underdeveloped left ventricle that is unable to support the systemic circulation. In addition, restrictive flow defects of the aorta, aortic valve, and/or mitral valve are also commonly present, leading to reduced blood flow through the developing ventricle [2]. The combination of restricted ventricular blood flow and impaired ventricular growth led to the classic belief that the primary embryologic insult in HLHS was the formation of a left-sided flow limiting lesion, and ventricular underdevelopment was thought to be a secondary consequence of reduced ventricular blood flow [3, 4]. This pathogenetic model spawned the design of fetal interventions for HLHS, based on the hypothesis that correction of left ventricular flow dynamics during development would alleviate the harmful effects of altered blood flow on the maturing myocardium and rescue left ventricular growth [4, 5]. However, human fetal interventions aimed at relieving outflow obstruction in select fetuses with aortic stenosis and evolving HLHS failed to improve left ventricular growth during gestation, suggesting the myocardial growth defect in HLHS may not depend upon altered blood flow or hemodynamic forces [5]. Alternative explanations for the ventricular chamber defect in HLHS are therefore warranted.
The morphologic period of chamber formation occurs between 22 and 28 days after fertilization in the human when the primordial heart tube loops and balloons into a structure with expanded precursor chambers. This transformation is initiated by regional increases in the volume of outer curvature ventricular cardiomyocytes, followed by increases in cellular proliferation [6]. Thus, coordinated increases in cell size, termed ‘hypertrophy’ represent the first necessary prerequisite for chamber expansion. At the cellular level, ventricular cardiomyocytes from HLHS hearts appear disorganized with scant cytoplasm and are reduced in size and number, suggesting they suffer from developmental defects in both hypertrophy and proliferation [7]. Given that hypertrophy precedes proliferation during chamber expansion, the chamber defect observed in HLHS may be caused by a primary error in developmental hypertrophy which then disrupts the downstream sequence of cellular proliferation and chamber growth.
The mechanisms of normal cardiomyocyte hypertrophy during development remain poorly understood. However, the mechanisms of pathologic pressure overload hypertrophy have been carefully examined in adults and have been found to result from alterations in Ca2+ signaling pathways that lead to activation of quiescent growth pathways [8]. The dominant growth pathway identified in the adult heart is the Ca2+/calcineurin pathway which culminates in an increase in cardiac mass through re-expression of genes associated with fetal hearts [9]. Despite the emphasis placed on the re-expression of the fetal gene program by hypertrophic adult myocardium [10], little is known about the cellular and molecular biology of normal myocardial growth during embryonic development. However, in support of conservation of growth pathways between adult and fetal hearts, prior studies have shown reduction in components of the Ca2+/calcineurin pathway in tissue from HLHS hearts [11], as well as impaired chamber formation in hearts lacking L-type voltage-gated Ca2+ entry [12]. We therefore hypothesized that the dominant Ca2+ signaling pathways active during pathological hypertrophy in adult cardiomyocytes also regulate developmental hypertrophy in embryonic cardiomyocytes. We further predicted that these growth pathways were not directly modulated by hemodynamic forces or blood flow given the apparent unresponsiveness of HLHS myocardium to these stimuli.
In cardiac muscle, variations in intracellular Ca2+ are involved in electromechanical coupling, leading to heart contraction and blood flow, and also serve as signaling mediators, leading to alterations in gene transcription. In most organisms, the functional and signaling roles of Ca2+ in the heart are unable to be dissociated given the requirement of heart function for survival. Zebrafish represent ideal organisms for studying the interaction between Ca2+-dependent growth pathways and hemodynamic forces, as embryos can survive through all stages of heart development without the requirement for blood flow [13]. We therefore established a zebrafish model to define the role of Ca2+ signaling in developmental myocardial hypertrophy in the ventricle uncoupled from heart function and fluid forces.
2. METHODS
2.1 Zebrafish Strains
Zebrafish (Danio rerio) strains utilized included Tübingen wild-type, Tg(cmlc::DsRed2-nuc) [14] (kindly provided by Dr Kenneth Poss, Duke University, with permission from Dr C. Geoffrey Burns, Harvard Medical School), Tg(cmlc2::GFP) [15] (kindly provided by Dr Deborah Yelon, UC San Diego, with permission from Dr Huai-Jen Tsai, National Taiwan University), and Tg(Flk1::EGFP)s843 [16] (kindly provided by Dr Kenneth Poss with permission from Dr Suk-Won Jin, University of North Carolina). Zebrafish were maintained following published protocols [17]. All zebrafish experiments were approved by the Institutional Animal Care and Use Committee of Duke University.
2.2 Pharmacology and Drugs
Blebbistatin (5 uM), nisoldipine (10 uM), cyclosporine A (CsA, 10 μg/ml), FK506 (1 μg/ml), and BayK8644 (20 uM) (all from Sigma-Aldrich, St. Louis, MO) were dissolved in DMSO and diluted to final working concentration in embryo media without antibiotics. Embryos (24 hpf) were chemically and manually dechorionated using pronase. Drug solutions were applied to embryos at 24 hpf and re-applied every 6 hours during the period of drug treatment. Media containing an equivalent volume of DMSO was used as control. Embryos were incubated at 28.5°C in the dark to prevent light degradation of drugs.
2.3 Antisense Morpholino Knockdown and RNA Rescue Analysis
Morpholino oligonucleotides (Gene Tools, Philomath, OR) were diluted and injected at 2–4 ng per embryo at the one cell stage. The tnnt2 morpholino (CATGTTTGCTCTGATCTGACACGCA) was used as previously described [18]. A cocktail of two morpholino oligonucleotides against cacna1c ([CCCGTTCCTAGACAGACGAAACAGA] and [GGATCTTGCACTCACCTACGAACCA]) was used as previously described [19]. Gene Tools standard control morpholino (CCTCTTACCTCAGTTACAATTTATA) was used as negative control. cRNA rescue constructs were coinjected with the cacna1c morpholinos at 800 pg per embryo as previously described.[19] Rescue constructs used were wildtype cacna1c (CaV1.2WT cRNA), Timothy Syndrome cacna1c (CaV1.2TS cRNA), and constitutively active calcineurin (caCN cRNA).
2.4 Video Recording
Tg(cmlc2::GFP) embryos treated with drugs or morpholinos were anesthetized in 0.016% tricaine at 48 hpf and imaged using a Leica DM RAZ microscope equipped with a fluorescence imaging system. Videos were captured using a standard CCD camera at 20 frames/s.
2.5 Immunohistochemistry
For experiments to determine cardiomyocyte cell volume, Tg(cmlc::DsRed2-nuc) embryos were stained with anti-DsRed polyclonal antibody and anti-Zn5 monoclonal antibody as previously described [20]. This protocol allowed visualization of cardiomyocyte borders in green and nuclei in red. For experiments to determine endocardial development, Tg(Flk1::EGFP)s843 embryos were stained with anti-GFP polyclonal antibody and anti-MF20 antibody as previously described [20]. This protocol allowed visualization of myocardium in red and endocardium in green.
2.6 Cell Volume Measurements
Antibody stained embryos were embedded in 4% low melt agarose. A vibratome was used to create 50 μm floating sections taken perpendicular to the long-axis of the embryo. The section containing all or most of the heart was identified using fluorescence microscopy and mounted for confocal microscopy. Three dimensional Z-stacks of the hearts were generated using a Zeiss LSM510 confocal microscope with a 40X objective. ImageJ software (NIH) was then used to calculate the volume of select cardiomyocytes. Cells were chosen for measurement only when their outlines were clearly visible within the xy plane of view. For cell measurements at the 24 hpf stage, outflow tract cells that will contribute to the future ventricle were measured. Given that myocardial cells at the 24 hpf stage possess a rounded morphology [21, 22], cell volume was estimated by measuring the radial length and width of the cell in the xy plane and the radial height of the cell in the z plane. The volume of an ellipsoid was then calculated using the three elliptic radii with the equation 4/3πRx*Ry*Rz. For cell measurements at the 48 hpf stage, outer curvature ventricular cardiomyocytes were measured following the period of volume expansion. Given that outer curvature ventricular cardiomyocytes at the 48 hpf stage possess a flattened and elongated morphology [21, 22], cell volume was estimated by treating the cells as thin cuboidal sheets and multiplying the cross-sectional area of the cell by the thickness of the cell in the z dimension (area*thickness). The nuclei of cells at all stages were treated as ellipsoids and nuclear volume was calculated using the aforementioned elliptical measurements. To standardize the measurement process, 10 cardiomyocytes located within the first two cell layers of the outer curvature were measured from each representative heart by two investigators who were blinded to the treatment group of the heart. The 10 cardiomyocyte cell volumes were then averaged into a single value so that the weighted value of each heart was equivalent within a dataset. Between 6 and 20 hearts were imaged and measured per treatment group, with the sample size for statistical analysis considered as the number of hearts measured (not the total number of cells measured).
2.7 Statistics
Differences in cell volume between groups were compared using a 2-tailed Student’s t test or ANOVA, where appropriate. Data are expressed as mean ± SEM where applicable. A P value less than 0.05 was considered a statistically significant difference.
3. RESULTS
3.1 Analysis of Cardiomyocyte Hypertrophy During Zebrafish Chamber Expansion
Given that an arrest in ventricular cardiomyocyte hypertrophy may underlie the chamber defect in HLHS [7], we sought to develop a model for studying cardiomyocyte hypertrophy during chamber expansion in zebrafish. The morphologic period of chamber formation occurs between 24 and 48 hpf in the two-chambered zebrafish heart when the primordial heart tube loops and balloons into a structure with an expanded atrium and expanded ventricle. Although prior zebrafish studies have measured changes in myofibril content and the two-dimensional cross-sectional area of ventricular cardiomyocytes as surrogates for hypertrophy, none have strictly measured changes in cell volume during chamber emergence [21, 23]. We therefore first quantified zebrafish outer curvature ventricular cardiomyocyte cell volume during chamber formation using confocal microscopy with three-dimensional volumetric image analysis (see Supplemental Videos 1 and 2 for representative three-dimensional heart models at 24 hpf and 48 hpf). Experiments demonstrated that outer curvature ventricular cardiomyocytes increased in volume by 3- to 4-fold between 24 and 48 hpf in zebrafish (Figure 1), consistent with volume changes reported previously during the period of chamber formation in the chick heart [6]. Further, this change in cellular dimension was confirmed to be caused primarily by cytoplasmic expansion, similar to the cytoarchitectural changes observed during developmental hypertrophy in the mouse heart [22]. These experiments validated zebrafish as powerful organisms for studying cardiomyocyte hypertrophy during ventricular chamber formation.
Figure 1.
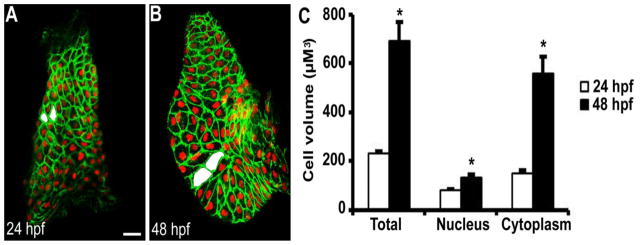
Three-dimensional volumetric analysis of ventricular cardiomyocyte hypertrophy during zebrafish chamber formation. (A) Linear heart tube at 24 hpf prior to chamber expansion (n=10). (B) Expanded ventricle at 48 hpf (n=8). (C) Quantitation of cell volumes demonstrated outer curvature ventricular cardiomyocytes increased 3- to 4-fold in volume during chamber formation primarily due to cytoplasmic expansion. Examples of representative measured cells are shaded in white. * P < 0.001 vs 24 hpf. Scale bar = 20 μM.
3.2 Contraction and Blood Flow are Not Necessary for Developmental Hypertrophy
Relief of left ventricular outflow obstruction, intended to increase blood flow through the left ventricle, did not stimulate the growth of HLHS myocardium during gestation in humans [5]. We therefore sought to prove experimentally whether blood flow was required to stimulate cardiomyocyte hypertrophy during early chamber formation in the zebrafish ventricle. To accomplish this goal, zebrafish hearts were rendered non-contractile during chamber formation using pharmacologic and genetic excitation-contraction uncouplers that do not interfere with voltage-gated Ca2+ entry or electrical conduction. First, zebrafish embryos were treated with the non-muscle myosin IIA inhibitor, blebbistatin, between 24 hpf and 48 hpf [24]. This treatment led to loss of contractile function and blood flow during the period of chamber formation (see Supplemental Videos 3 and 4 for representative control and blebbistatin-treated hearts at 48 hpf). Analysis of developmental hypertrophy showed there was no change in ventricular cardiomyocyte cell volume at 48 hpf between control hearts and hearts that developed in the absence of blood flow due to blebbistatin treatment (Figure 2, A, B, and E). Next, we used morpholino antisense technology to knock-down troponin t (tnnt2) translation by injection of tnnt2-specific morpholino into single-cell stage embryos. As shown in prior studies, this results in a non-contractile heart in > 95% of injected embryos but does not interfere with normal electrical conduction or Ca2+ transients (see Supplemental Video 5 for representative tnnt2 morphant heart at 48 hpf) [18, 25]. Similar to the blebbistatin-treated embryos, there was no change in cardiomyocyte cell volume at 48 hpf in the tnnt2 morphant embryos when compared to embryos injected with control morpholino (Figure 2, C, D, and E). These findings are consistent with prior reports demonstrating grossly normal ventricular morphology and cardiomyocyte cross-sectional area in zebrafish mutants lacking blood flow [18, 21, 25, 26]. Taken together, these results suggest that contraction and blood flow are not necessary stimuli for cardiomyocyte hypertrophy during chamber formation.
Figure 2.

Contraction and blood flow are not required for ventricular hypertrophy during chamber formation. Zebrafish hearts were rendered non-contractile during chamber formation by treatment with blebbistatin or tnnt2 morpholino (MO). Of note, Ca2+ transients and electrical conduction are preserved with both treatments. No difference in outer curvature ventricular cardiomyocyte cell volume was found between hearts treated with (A) DMSO (n=8) and (B) blebbistatin (n=11) between 24 hpf and 48 hpf. Similarly, no difference in outer curvature cardiomyocyte cell volume was found between zebrafish embryos injected with (C) control MO (n=7) and (D) tnnt2 MO (n=6) when measured at 48 hpf. Examples of representative measured cells are shaded in white. Scale bar = 20 μM.
3.3 Ca2+ Signaling via the CaV1.2 L-type Ca2+ Channel is Necessary for Developmental Hypertrophy
Since mechanical forces did not appear to control early cardioimyocyte hypertrophy during development we next tested whether Ca2+ signaling via the CaV1.2 L-type Ca2+channel was necessary. Ca2+ signaling serves a dominant role in adult cardiomyocyte hypertrophy [9, 27] and zebrafish mutants lacking the CaV1.2 L-type voltage-gated Ca2+channel demonstrated impaired ventricular chamber formation in prior study [12]. We therefore abolished L-type voltage-gated Ca2+ signaling in zebrafish by exposure to the CaV1.2 channel blocker, nisoldipine, between 24 hpf and 48 hpf (Figure 3, B), or by injection with morpholinos that target the CaV1.2 α-subunit (cacna1c MO, Figure 3, D). Both CaV1.2 inhibitors again rendered zebrafish hearts non-contractile during the period of chamber formation, although now in the absence of L-type voltage-gated Ca2+ transients (see Supplemental Videos 6 and 7 for representative nisoldipine and cacna1c morphant hearts at 48 hpf). While the previous experiments demonstrated that lack of blood flow alone had no apparent effect on developmental hypertrophy, zebrafish ventricles that developed in the absence of CaV1.2 -mediated Ca2+ influx appeared grossly dysmorphic, and outer curvature ventricular cardiomyocyte cell volume was significantly reduced (Figure 3, E). These experiments suggest Ca2+ entry through the CaV1.2 L-type Ca2+ channel is required for normal cardiomyocyte hypertrophy during chamber formation, independent of the mechanical effects of Ca2+ influx on the heart.
Figure 3.

Ca2+ signaling via the CaV1.2 L-type Ca2+ channel is required for ventricular hypertrophy during chamber formation. L-type voltage-gated Ca2+ signaling was inhibited during chamber formation by treatment with nisoldipine or cacna1c morpholino (MO). Both treatments rendered hearts non-contractile during development in the absence of L-type voltage-gated Ca2+ transients. Ventricular chamber formation was grossly abnormal and outer curvature ventricular cardiomyocyte cell volume was significantly reduced in hearts treated with (B) nisoldipine (n=9) vs (A) DMSO (n=15) between 24 hpf and 48 hpf. More pronounced findings were observed in hearts injected with (D) cacna1c MO (n=19) vs (C) control MO (n=7) when measured at 48 hpf. Examples of representative measured cells are shaded in white. * P < 0.05 vs DMSO or control MO. Scale bar = 20 μM.
3.4 Increasing CaV1.2 -Mediated Ca2+ Signaling Leads to Enhanced Developmental Hypertrophy Independent of Contraction or Blood Flow
We next performed the reciprocal experiment to determine whether increasing L-type voltage-gated Ca2+ influx could lead to enhanced developmental hypertrophy. If so, modulation of Ca2+ signaling via CaV1.2 could serve as a therapeutic avenue for stimulating hypertrophy in the developing heart. First, a genetic approach was used to increase L-type voltage-gated Ca2+ entry during chamber formation by expression of the Timothy Syndrome mutant CaV1.2 channel, which fails to inactivate and thereby supports supernormal Ca2+ entry during an action potential [19, 28]. Embryos were injected with cacna1c morpholinos with or without wildtype (CaV1.2WT) or Timothy Syndrome (CaV1.2TS) morpholino-insensitive cRNA rescue constructs. Co-injection of cacna1c morpholinos with either CaV1.2WT cRNA (Figure 4, C) or CaV1.2TS cRNA (Figure 4, D) led to rescue of contractile function in the majority of treated embryos (66.7% of cRNA rescue animals had a beating heart at 48 hpf compared with 5% of embryos treated with cacna1c morpholinos alone, P < 0.0001) suggesting proper expression, localization, and function of the rescue channels. Quantitation of cardiomyocyte cell volume was then assessed only in contractile hearts where the likelihood of expressing a rescue channel was increased > 10-fold over the likelihood of a failed injection. Results demonstrated that expression of CaV1.2TS led to a large increase in outer curvature ventricular cardiomyocyte cell volume at 48 hpf when compared with controls (Figure 4, E). Further, expression of CaV1.2WT did not alter ventricular cardiomyocyte volume, suggesting the increase in developmental hypertrophy produced with CaV1.2TS was not the result of an over-expression phenotype. These data are consistent with prior reports of cardiomegaly in patients with Timothy Syndrome [28] and confirm the ability of CaV1.2-mediated Ca2+ signaling to modulate developmental hypertrophy in both the positive and negative directions.
Figure 4.
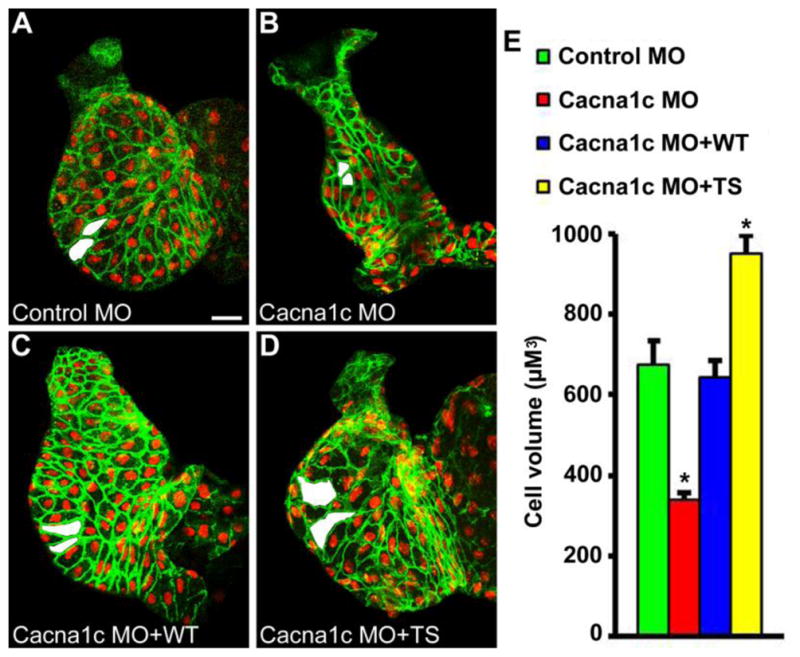
Increasing CaV1.2-mediated Ca2+ signaling during chamber formation leads to enhanced developmental hypertrophy. Voltage-gated Ca2+ entry via the CaV1.2 L-type Ca2+ channel was increased during development by expression of the Timothy Syndrome mutant CaV1.2 channel. Co-injection of cacna1c morpholinos (MO) with (D) CaV1.2TS cRNA (TS, n=7) led to increased outer curvature ventricular cardiomyocyte cell volume at 48 hpf when compared to embryos injected with (A) control MO (n=7), (B) cacna1c MO alone (n=19), or (C) cacna1c MO with CaV1.2WT cRNA (WT, n=8). Examples of representative measured cells are shaded in white. * P < 0.01 vs control MO. Scale bar = 20 μM.
Next, a pharmacologic approach was used to increase CaV1.2-mediated Ca2+ signaling during chamber formation in the presence or absence of heart function. Zebrafish embryos were exposed to the CaV1.2 agonist BayK8644 between 24 hpf and 48 hpf (Figure 5, A and B). These experiments were then repeated in hearts rendered non-contractile by co-treatment with blebbistatin (Figure 5, C and D) or by injection of tnnt2 morpholinos (Figure 5, E and F). Analysis of developmental hypertrophy demonstrated that outer curvature ventricular cardiomyocyte cell volume was increased at 48 hpf after treatment with the CaV1.2 agonist Bayk8644 in contractile and non-contractile hearts (Figure 5, G). The ability of CaV1.2 activation to modulate hypertrophy in the absence of contraction demonstrates that developmental hypertrophy is regulated by the signaling role of Ca2+ and not the mechanical or hemodynamic sequela of voltage-gated Ca2+ entry. These data further suggest Ca2+ signaling activators may be able to increase developmental hypertrophy in hemodynamic settings similar to HLHS, where developmental blood flow is often diminished.
Figure 5.

Increasing CaV1.2-mediated Ca2+ signaling during chamber formation leads to enhanced developmental hypertrophy independent of contraction and blood flow. L-type voltage-gated Ca2+ entry was increased during development by treatment with the CaV1.2 agonist BayK8644 (BayK). Experiments were repeated in the absence of contraction by co-treatment with blebbistatin or tnnt2 morpholino (MO). Treatment of contractile hearts with (B) BayK (n=13) between 24 hpf and 48 hpf led to increased developmental hypertrophy when compared with (A) DMSO (n=8). The ability of BayK treatment to increase developmental hypertrophy was reproduced in hearts rendered non-contractile through treatment with (C, D) blebbistatin (n=11 for blebbistatin, n=8 for blebbistatin+BayK) or (E, F) tnnt2 MO (n=6 for tnnt2 MO, n=7 for tnnt2 MO+BayK). Examples of representative measured cells are shaded in white. * P < 0.05 vs paired control. Scale bar = 20 μM.
3.5 Calcineurin Regulates Developmental Hypertrophy Independent of Contraction and Blood Flow
We next sought to confirm whether the major downstream Ca2+-sensitive phosphatase, calcineurin, implicated in pathologic adult hypertrophy was similarly required for developmental hypertrophy during chamber formation. Embryos were first treated with CsA, a well characterized pharmacologic calcineurin inhibitor. The previously established effects of CsA on endocardial cushion development were re-confirmed by observation of lack of atrioventricular valve formation at 72 hpf (Supplemental figure 1) [29]. Developmental hypertrophy was then assessed after CsA treatment between 24 hpf and 48 hpf (Figure 6, A and B). These experiments were then repeated in heart rendered non-contractile hearts by co-treatment with blebbistatin (Figure 6, C and D) or tnnt2 morpholino injection (Figure 6, E and F). Analysis of developmental hypertrophy demonstrated small ventricles with reduced outer curvature ventricular cardiomyocyte cell volume at 48 hpf after treatment with CsA in both contractile and non-contractile hearts (Figure 6, G). These experiments were repeated using the related calcineurin inhibitor, FK506, yielding similar results (data not shown).
Figure 6.

Calcineurin is required for ventricular hypertrophy independent of contraction and blood flow. Calcineurin was inhibited during development by treatment with cyclosporine A (CsA). Experiments were repeated in the absence of contraction by co-treatment with blebbistatin or tnnt2 morpholino (MO). Treatment of contractile hearts with (B) CsA (n=11) between 24 hpf and 48 hpf led to small ventricles with reduced cardiomyocyte cell volume when compared with (A) DMSO (n=8). Similar results were achieved in hearts rendered non-contractile during development through treatment with (C, D) blebbistatin (n= 11 for blebbistatin, n=12 for blebbistatin+CsA) or (E, F) tnnt2 MO (n=6 for tnnt2 MO, n=12 for tnnt2 MO+CsA). Examples of representative measured cells are shaded in white. * P < 0.01 vs paired control. Scale bar = 20 μM.
To complement the studies above, a genetic approach was employed to determine if downstream calcineurin activation was sufficient to induce cardiomyocyte hypertrophy in the absence of CaV1.2-mediated Ca2+ influx. Embryos were injected with cacna1c morpholinos alone (Figure 7, A) or co-injected with cacna1c morpholinos and a cRNA rescue construct encoding a constitutively active calcineurin (caCN) (Figure 7, B). Results showed that constitutively active calcineurin partially rescued cardiomyocyte hypertrophy during chamber formation in the absence of CaV1.2-mediated Ca2+ influx or contraction (Figure 7, C). Taken together with the prior experiments, these data suggest Ca2+ –mediated activation of calcineurin is a major stimulus for ventricular cardiomyocyte hypertrophy in the developing heart. Further, regulation of hypertrophy by calcineurin does not appear to be significantly influenced by or dependent upon the coincident effects of Ca2+ on the mechanical function of the heart (Table 1).
Figure 7.
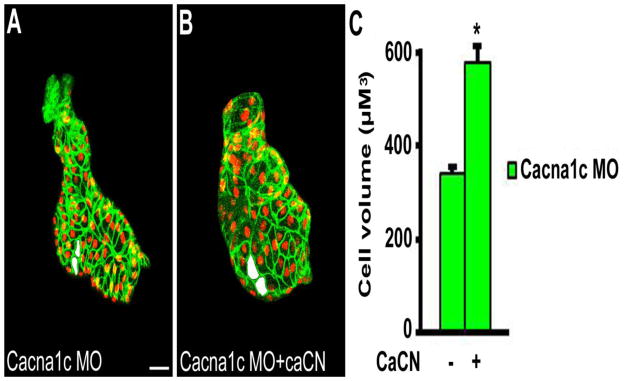
Constitutively active calcineurin rescues ventricular hypertrophy during chamber formation in the absence of L-type voltage-gated Ca2+ influx or contraction. Embryos were injected with (A) cacna1c morpholinos (MO) alone (n=19) or (B) co-injected with cacna1c MO and a cRNA rescue construct encoding a constitutively active calcineurin (caCN) (n=12). (C) Quantitation of outer curvature ventricular cardiomyocyte cell volume demonstrated that constitutively active calcineurin rescued cardiomyocyte growth during chamber formation in the absence of CaV1.2-mediated Ca2+ influx or contraction. Examples of representative measured cells are shaded in white. * P < 0.0001 vs cacna1c MO. Scale bar = 20 μM.
Table 1.
Summary of findings. Calcineurin activity correlates with hypertrophy, independent of contraction or blood flow. caCN = constitutively active calcineurin, CsA = cyclosporine A, MO = morpholino.
| Treatment | CaV1.2 | Calcineurin | Contraction/Blood Flow | Hypertrophy |
|---|---|---|---|---|
| Blebbistatin/tnnt2 MO | ↔ | ↔ | Absent | ↔ |
| Nisoldipine/cacna1c MO | ↓↓↓ | ↓↓↓ | Absent | ↓↓↓ |
| BayK/CaV1.2TS | ↑↑↑ | ↑↑↑ | ↔ | ↑↑↑ |
| BayK + Blebbistatin/tnnt2 MO | ↑↑↑ | ↑↑↑ | Absent | ↑↑↑ |
| CsA | ↔ | ↓↓↓ | ↔ | ↓↓↓ |
| CsA + Blebbistatin/tnnt2 MO | ↔ | ↓↓↓ | Absent | ↓↓↓ |
| Cacna1c MO + caCN | ↓↓↓ | ↔ | Absent | ↔ |
4. DISCUSSION
The present study examined the molecular mechanisms of ventricular cardiomyocyte hypertrophy during chamber expansion in attempt to address two fundamental questions of early cardiac development which may also have relevance to the HLHS debate: (1) Are heart function and blood flow required to stimulate myocardial hypertrophy during development? (2) Are Ca2+ signaling pathways that control adult cardiac hypertrophy also active during development? Using a zebrafish model, we first demonstrated that heart function and blood flow are not required to stimulate ventricular hypertrophy during early chamber formation. Next, we demonstrated that cardiomyocyte hypertrophy during development is at least partially regulated by conserved Ca2+ signaling pathways that function independent of hemodynamic forces. These findings provide new insights into the mechanisms of heart development and may inform treatments for HLHS or other heart malformations characterized by abnormal chamber growth.
Contrary to the classic “flow theory” of cardiac growth during development, we demonstrated that heart function and blood flow are not necessary stimuli for developmental hypertrophy as long as the Ca2+ conduction system remains intact. These findings mirror prior studies that demonstrated grossly normal ventricular morphology and two-dimensional cardiomyocyte cross-sectional area in non-contractile zebrafish hearts harboring the tnnt2 mutation [18, 25, 26] or atrial and ventricular myosin heavy chain deletions [21]. However, although overall cell size and volume were not affected in these non-contractile models, other experiments demonstrated that the absence of blood flow during chamber formation leads to subtle abnormalities in myocardial morphogenesis, such as changes in cell shape, alignment, and regional organization [21]. In addition, reduced or uncoordinated blood flow (as opposed to absent blood flow) can have various effects on cell size and myofibril maturation depending on the specific hemodynamic environment [21, 23, 30], and manipulation of fluid forces at later stages of development can alter cardiomyocyte proliferation and chamber maturation in higher order vertebrates [3, 31–36]. Beyond the myocardium, heart function and blood flow are known to be required for proper valve formation after chamber expansion [30, 37]. Thus, heart contraction and blood flow remain relevant to multiple other aspects of cardiac development and likely influence disease pathogenesis.
Although function and fluid forces are not obligatory stimuli for ventricular cell growth during early chamber formation, the signaling effects of Ca2+ entry are required. These findings were predicted given the body of literature supporting conservation of cardiac hypertrophy pathways between adult and fetal life [10] as well as the identification of impaired chamber formation in CaV1.2 mutant embryos [12] and reduced expression of calcineurin and NFAT transcripts in tissue from HLHS hearts [11]. While our data confirmed the requirement for CaV1.2-mediated Ca2+ entry and downstream calcineurin activity for developmental hypertrophy, other downstream Ca2+ sensitive mediators are likely involved given that constitutively active calcineurin expression did not achieve complete rescue of ventricular cell volume in the absence of CaV1.2 L-type Ca2+ channels.
These data shed light on the fundamental question of how chamber expansion is triggered during early heart development. The initiation of heart contraction at the linear heart tube stage immediately prior to chamber expansion led most to believe that mechanical activity or fluid forces triggered cellular growth. Our data instead suggest that chamber expansion is triggered by voltage-gated Ca2+ entry via downstream signaling mechanisms that are unaffected by the hemodynamic environment. However, given that the electromechanical and signaling functions of Ca2+ are intrinsically coupled in nature, it appears that contraction and hypertrophy are married events that occur simultaneously due to the onset of voltage-gated Ca2+ entry.
The laboratory findings reported here may have relevance to the etiology of HLHS and its potential treatments. It remains unresolved whether the myocardial defect in HLHS is caused by extrinsic flow-related phenomena or intrinsic abnormalities of myocardial cell maturation and growth. This controversy has gained additional scrutiny and importance as therapies for fetal intervention based on the former rationale have produced conflicting results [5, 38, 39]. The largest fetal intervention program for HLHS performed percutaneous in utero aortic valvuloplasty in highly selected fetuses with aortic stenosis who have been shown to almost uniformly progress to HLHS in prior studies [40]. These interventions led to significant improvements in the growth of the aortic valve, ascending aorta, and mitral valve and allowed for salvage of the left heart in 50% of patients [5, 38]. However, in comparison to historical controls, no change in the gestational growth of the left ventricle itself was observed after intervention [5].
Interpretation of these results in the context of laboratory findings suggests intrinsic errors in myocardial growth may underlie some cases of HLHS. Although some have argued that the failure of HLHS myocardium to respond to improved in utero flow dynamics may be due to irreversible flow-mediated myocardial damage [38], an alternative explanation is that the chamber defect observed in some forms of HLHS is caused by a primary error in myocardial growth, possibly related to genetic or environmental disruption of Ca2+ signaling or other important pathways required for normal myocardial development [41]. In these instances, primary defects in myocardial hypertrophy and chamber formation may occur first, leading to secondary valvular defects as a consequence of impaired ventricular function and blood flow. Such a mechanism was suggested by an environmental toxin model of HLHS shown previously by our lab where exposure of zebrafish to a dioxin-like compound associated epidemiologically with HLHS [42] led to a phenotype resembling HLHS [20]. In this model, failure of cardiomyocyte hypertrophy occurred early in development prior to valve formation and was followed temporally by valve malformations and myocardial cell cycle arrest.
The therapeutic implications of this model are several-fold. First, while mechanical in utero intervention appears favorable to some HLHS patients [38], strategies to pharmacologically stimulate ventricular hypertrophy via systemic or catheter-based drug delivery may provide additional synergistic benefit or be of use in cases of HLHS not amenable to balloon valvuloplasty. Ca2+ signaling agonists appear to represent attractive pro-hypertrophic agents as Ca2+ -dependent hypertrophy pathways were able to stimulate or rescue ventricular cardiomyocyte hypertrophy even in perturbed hemodynamic environments. In addition, activation or over-activation of Ca2+ -dependent hypertrophy may be of benefit regardless of the precise cause of the hypoplastic phenotype, provided the block in cellular growth does not involve critical downstream Ca2+ sensitive machinery. Second, if some forms of HLHS are caused by primary myocardial growth defects beginning as early as chamber formation (3–4 weeks after fertilization), then treatments may need to be initiated significantly earlier then mid-gestation to have maximal effect. This would require novel avenues to identify and diagnose congenital heart defects prior to second trimester ultrasound which are presently unavailable. Lastly, given that Ca2+ signaling antagonists were also able to augment hypertrophy in the negative direction, inhibition of Ca2+ -dependent hypertrophy during development may also have utility in the reversal of hypertrophic heart defects.
However, several limitations to the extrapolation of these zebrafish results to the human condition should be acknowledged. First, the Ca2+ signaling pathways shown to regulate cardiomyocyte hypertrophy during early chamber development may not be active during later stages of heart development when HLHS is typically diagnosed and fetal intervention has been performed. Second, this study is limited to an analysis of hypertrophy and the effects of blood flow and Ca2+ signaling augmentation on cellular proliferation, heart function, and final heart patterning have not been evaluated. Lastly, the conservation of early developmental growth pathways between the two-chambered zebrafish heart and the four-chambered mammalian have not been tested, where left versus right-sided heart growth may depend on different molecular mechanisms. Thus, additional experimental analysis of the effects of Ca2+ signaling augmentation during various stages of heart development on hypertrophy, proliferation, heart function, and the final patterning and maturation of the 4-chambered mammalian heart are required to confirm relevance to humans. At present, the therapeutic implications of this study and relationship to human disease remain speculative and hypothesis-generating.
In summary, we demonstrated that ventricular cardiomyocyte hypertrophy during early heart development is at least partially regulated by conserved Ca2+ signaling pathways that function independent of heart function or hemodynamic forces. These findings may inform the understanding of developmental heart malformations characterized by abnormal chamber growth.
Supplementary Material
HIGHLIGHTS.
The mechanisms governing cardiomyocyte hypertrophy during development are unknown
We studied ventricular hypertrophy during chamber formation in a zebrafish model
Cardiomyocyte growth proceeded normally in the absence of contraction and blood flow
Cardiomyocyte growth was dependent upon fundamental Ca2+ signaling pathways
These findings may inform the understanding of developmental heart malformations
Acknowledgments
The authors acknowledge Nicholas Lent for technical assistance and Bijoy Thattaliyath for helpful discussion.
SOURCES OF FUNDING
The work was supported by NIH grants R01 HL084413 (MRH and MLK), R01 HL113136 (GSP), R01 HL071165 (GSP), The Hartwell Foundation (MRH, NDA, and GSP), the Thoracic Surgery Foundation for Research and Education (NDA), the March of Dimes grant 1-FY12-521 (GSP), the Duke Chancellor’s Science Council Pilot Funds program (GSP) and the George Brumley, Jr Neonatal Perinatal Research Institute at Duke University (MRH).
Footnotes
Presented at the American Heart Association Scientific Sessions 2013, November 16–20, 2013, Dallas, TX.
DISCLOSURES
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bailey LL, Gundry SR. Hypoplastic left heart syndrome. Pediatr Clin North Am. 1990;37:137–50. doi: 10.1016/s0031-3955(16)36836-5. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs JP, O’Brien SM, Chai PJ, Morell VO, Lindberg HL, Quintessenza JA. Management of 239 patients with hypoplastic left heart syndrome and related malformations from 1993 to 2007. Ann Thorac Surg. 2008;85:1691–6. doi: 10.1016/j.athoracsur.2008.01.057. discussion 7. [DOI] [PubMed] [Google Scholar]
- 3.Sedmera D, Hu N, Weiss KM, Keller BB, Denslow S, Thompson RP. Cellular changes in experimental left heart hypoplasia. Anat Rec. 2002;267:137–45. doi: 10.1002/ar.10098. [DOI] [PubMed] [Google Scholar]
- 4.Wilkins-Haug LE, Benson CB, Tworetzky W, Marshall AC, Jennings RW, Lock JE. In-utero intervention for hypoplastic left heart syndrome--a perinatologist’s perspective. Ultrasound Obstet Gynecol. 2005;26:481–6. doi: 10.1002/uog.2595. [DOI] [PubMed] [Google Scholar]
- 5.McElhinney DB, Marshall AC, Wilkins-Haug LE, Brown DW, Benson CB, Silva V, et al. Predictors of technical success and postnatal biventricular outcome after in utero aortic valvuloplasty for aortic stenosis with evolving hypoplastic left heart syndrome. Circulation. 2009;120:1482–90. doi: 10.1161/CIRCULATIONAHA.109.848994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soufan AT, van den Berg G, Ruijter JM, de Boer PA, van den Hoff MJ, Moorman AF. Regionalized sequence of myocardial cell growth and proliferation characterizes early chamber formation. Circ Res. 2006;99:545–52. doi: 10.1161/01.RES.0000239407.45137.97. [DOI] [PubMed] [Google Scholar]
- 7.Bohlmeyer TJ, Helmke S, Ge S, Lynch J, Brodsky G, Sederberg JH, et al. Hypoplastic left heart syndrome myocytes are differentiated but possess a unique phenotype. Cardiovasc Pathol. 2003;12:23–31. doi: 10.1016/s1054-8807(02)00127-8. [DOI] [PubMed] [Google Scholar]
- 8.Barry SP, Davidson SM, Townsend PA. Molecular regulation of cardiac hypertrophy. Int J Biochem Cell Biol. 2008;40:2023–39. doi: 10.1016/j.biocel.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 9.Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun. 2004;322:1178–91. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 10.Dirkx E, da Costa Martins PA, De Windt LJ. Regulation of fetal gene expression in heart failure. Biochim Biophys Acta. 2013;1832:2414–24. doi: 10.1016/j.bbadis.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 11.Gambetta K, Al-Ahdab MK, Ilbawi MN, Hassaniya N, Gupta M. Transcription repression and blocks in cell cycle progression in hypoplastic left heart syndrome. Am J Physiol Heart Circ Physiol. 2008;294:H2268–75. doi: 10.1152/ajpheart.91494.2007. [DOI] [PubMed] [Google Scholar]
- 12.Rottbauer W, Baker K, Wo ZG, Mohideen MA, Cantiello HF, Fishman MC. Growth and function of the embryonic heart depend upon the cardiac-specific L-type calcium channel alpha1 subunit. Dev Cell. 2001;1:265–75. doi: 10.1016/s1534-5807(01)00023-5. [DOI] [PubMed] [Google Scholar]
- 13.Asnani A, Peterson RT. The zebrafish as a tool to identify novel therapies for human cardiovascular disease. Dis Model Mech. 2014;7:763–7. doi: 10.1242/dmm.016170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mably JD, Mohideen MA, Burns CG, Chen JN, Fishman MC. heart of glass regulates the concentric growth of the heart in zebrafish. Curr Biol. 2003;13:2138–47. doi: 10.1016/j.cub.2003.11.055. [DOI] [PubMed] [Google Scholar]
- 15.Huang CJ, Tu CT, Hsiao CD, Hsieh FJ, Tsai HJ. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev Dyn. 2003;228:30–40. doi: 10.1002/dvdy.10356. [DOI] [PubMed] [Google Scholar]
- 16.Jin SW, Beis D, Mitchell T, Chen JN, Stainier DY. Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development. 2005;132:5199–209. doi: 10.1242/dev.02087. [DOI] [PubMed] [Google Scholar]
- 17.Nüsslein-Volhard C, Dahm R. Zebrafish: A Practical Approach. New York, New York, USA: Oxford University Press; 2002. [Google Scholar]
- 18.Sehnert AJ, Huq A, Weinstein BM, Walker C, Fishman M, Stainier DY. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat Genet. 2002;31:106–10. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- 19.Ramachandran KV, Hennessey JA, Barnett AS, Yin X, Stadt HA, Foster E, et al. Calcium influx through L-type CaV1.2 Ca2+ channels regulates mandibular development. J Clin Invest. 2013;123:1638–46. doi: 10.1172/JCI66903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grimes AC, Erwin KN, Stadt HA, Hunter GL, Gefroh HA, Tsai HJ, et al. PCB126 exposure disrupts zebrafish ventricular and branchial but not early neural crest development. Toxicol Sci. 2008;106:193–205. doi: 10.1093/toxsci/kfn154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auman HJ, Coleman H, Riley HE, Olale F, Tsai HJ, Yelon D. Functional modulation of cardiac form through regionally confined cell shape changes. PLoS Biol. 2007;5:e53. doi: 10.1371/journal.pbio.0050053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirschy A, Schatzmann F, Ehler E, Perriard JC. Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev Biol. 2006;289:430–41. doi: 10.1016/j.ydbio.2005.10.046. [DOI] [PubMed] [Google Scholar]
- 23.Lin YF, Swinburne I, Yelon D. Multiple influences of blood flow on cardiomyocyte hypertrophy in the embryonic zebrafish heart. Dev Biol. 2012;362:242–53. doi: 10.1016/j.ydbio.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jou CJ, Spitzer KW, Tristani-Firouzi M. Blebbistatin effectively uncouples the excitation-contraction process in zebrafish embryonic heart. Cell Physiol Biochem. 2010;25:419–24. doi: 10.1159/000303046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chi NC, Bussen M, Brand-Arzamendi K, Ding C, Olgin JE, Shaw RM, et al. Cardiac conduction is required to preserve cardiac chamber morphology. Proc Natl Acad Sci U S A. 2010;107:14662–7. doi: 10.1073/pnas.0909432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang W, Zhang R, Xu X. Myofibrillogenesis in the developing zebrafish heart: A functional study of tnnt2. Dev Biol. 2009;331:237–49. doi: 10.1016/j.ydbio.2009.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goonasekera SA, Hammer K, Auger-Messier M, Bodi I, Chen X, Zhang H, et al. Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J Clin Invest. 2012;122:280–90. doi: 10.1172/JCI58227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 29.Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, et al. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–63. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 30.Hove JR, Koster RW, Forouhar AS, Acevedo-Bolton G, Fraser SE, Gharib M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature. 2003;421:172–7. doi: 10.1038/nature01282. [DOI] [PubMed] [Google Scholar]
- 31.Fishman NH, Hof RB, Rudolph AM, Heymann MA. Models of congenital heart disease in fetal lambs. Circulation. 1978;58:354–64. doi: 10.1161/01.cir.58.2.354. [DOI] [PubMed] [Google Scholar]
- 32.Clark EB, Hu N, Frommelt P, Vandekieft GK, Dummett JL, Tomanek RJ. Effect of increased pressure on ventricular growth in stage 21 chick embryos. Am J Physiol. 1989;257:H55–61. doi: 10.1152/ajpheart.1989.257.1.H55. [DOI] [PubMed] [Google Scholar]
- 33.Saiki Y, Konig A, Waddell J, Rebeyka IM. Hemodynamic alteration by fetal surgery accelerates myocyte proliferation in fetal guinea pig hearts. Surgery. 1997;122:412–9. doi: 10.1016/s0039-6060(97)90034-9. [DOI] [PubMed] [Google Scholar]
- 34.Eghtesady P, Michelfelder E, Altaye M, Ballard E, Hirsh R, Beekman RH., 3rd Revisiting animal models of aortic stenosis in the early gestation fetus. Ann Thorac Surg. 2007;83:631–9. doi: 10.1016/j.athoracsur.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 35.Sedmera D, Pexieder T, Rychterova V, Hu N, Clark EB. Remodeling of chick embryonic ventricular myoarchitecture under experimentally changed loading conditions. Anat Rec. 1999;254:238–52. doi: 10.1002/(SICI)1097-0185(19990201)254:2<238::AID-AR10>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 36.Kowalski WJ, Teslovich NC, Menon PG, Tinney JP, Keller BB, Pekkan K. Left atrial ligation alters intracardiac flow patterns and the biomechanical landscape in the chick embryo. Dev Dyn. 2014;243:652–62. doi: 10.1002/dvdy.24107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartman T, Walsh EC, Wen KK, McKane M, Ren J, Alexander J, et al. Early myocardial function affects endocardial cushion development in zebrafish. PLoS Biol. 2004;2:E129. doi: 10.1371/journal.pbio.0020129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freud LR, McElhinney DB, Marshall AC, Marx GR, Friedman KG, Del Nido PJ, et al. Fetal aortic valvuloplasty for evolving hypoplastic left heart syndrome: postnatal outcomes of the first 100 patients. Circulation. 2014;130:638–45. doi: 10.1161/CIRCULATIONAHA.114.009032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rychik J. Hypoplastic left heart syndrome: can we change the rules of the game? Circulation. 2014;130:629–31. doi: 10.1161/CIRCULATIONAHA.114.011728. [DOI] [PubMed] [Google Scholar]
- 40.Makikallio K, McElhinney DB, Levine JC, Marx GR, Colan SD, Marshall AC, et al. Fetal aortic valve stenosis and the evolution of hypoplastic left heart syndrome: patient selection for fetal intervention. Circulation. 2006;113:1401–5. doi: 10.1161/CIRCULATIONAHA.105.588194. [DOI] [PubMed] [Google Scholar]
- 41.Singh MK, Li Y, Li S, Cobb RM, Zhou D, Lu MM, et al. Gata4 and Gata5 cooperatively regulate cardiac myocyte proliferation in mice. J Biol Chem. 2010;285:1765–72. doi: 10.1074/jbc.M109.038539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuehl KS, Loffredo CA. A cluster of hypoplastic left heart malformation in Baltimore, Maryland. Pediatr Cardiol. 2006;27:25–31. doi: 10.1007/s00246-005-0859-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.