

Abstract
Background
Mortality in sepsis is most often attributed to the development of multiple organ failure. In sepsis, inflammation-mediated endothelial activation, defined as a proinflammatory and procoagulant state of the endothelial cells, has been associated with severity of disease. Thus, the objective of this study was to test the hypothesis that AMPK activation limits inflammation and endothelium activation to protect against organ injury in sepsis. 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR), which is an AMP analogue, has been used to upregulate activity of AMPK. Compound C is a cell-permeable pyrrazolopyrimidine compound that inhibits AMPK activity.
Methods
Wild-type mice underwent CLP or Sham surgery. Mice were randomized to vehicle, AICAR, or Compound C. Mouse kidney endothelial cells were used for in vitro experiments. Renal and liver function, were determined by serum Cystatin C, BUN, creatinine, and ALT. Serum cytokines were measured by ELISA. Microvascular injury was determined using Evan’s blue dye and electron microscopy. Immunohistochemistry was used to measure protein levels of p-AMPK, LC3, and ICAM. LC3 levels were used as a measure of autophagosome formation.
Results
AICAR decreased liver, and kidney injury induced by CLP and minimized cytokine elevation, in vivo and in vitro. CLP increased renal and hepatic phosphorylation of AMPK and autophagic signaling as determined by LC3. Inhibition of AMPK with Compound C prevented CLP-induced autophagy and exacerbated tissue injury. Additionally, CLP led to endothelial injury as determined by electron microscopy and Evan’s blue dye extravasation, and AICAR limited this injury. Furthermore, AICAR limited CLP and LPS induced upregulation of ICAM in vivo and in vitro, and decreased LPS induced neutrophil adhesion in vitro.
Conclusion
In this model, activation of AMPK was protective and AICAR minimized organ injury by decreasing inflammatory cytokines and endothelial activation. These data suggest that AMPK signaling influences sepsis or LPS induced endothelial activation and organ injury.
Keywords: Sepsis, AMPK, energy, inflammation, endothelium, organ injury
Introduction
Sepsis is the leading cause of death in the critically ill patient population (1). Despite important efforts to understand the syndrome and multiple trials to test promising therapies, death rates have remained relatively stable for decades. Mortality by sepsis is directly related to the development of organ dysfunction (2), a process that remains incompletely understood. The pathogenesis of organ dysfunction is multifactorial, and includes direct cellular activation from circulating bacterial products, elaborated cytokines, as well as subsequent tissue hypoperfusion. Recent data has demonstrated that the cellular response to sepsis includes significant bioenergetic and metabolic regulation, including significant changes in mitochondrial responses (3–6).
Under normal physiologic conditions, cells maintain energy homeostasis through highly coordinated systems. Mitochondria have been shown to be central to these processes, not only in regards to production of ATP, but also as a critical signaling organelle that can sense changes in the metabolic environment and then signal to initiate adaptive responses. AMP-activated protein kinase (AMPK) is one of the most important energy regulators in the cell (7, 8). AMPK is a heterotrimeric kinase that fulfills a dual role. First, it is a very fine sensor of alterations in energy homeostasis as it monitors AMP to ATP ratio. Others and we have demonstrated increased AMP levels in the setting of sepsis (4, 5), suggesting an increment in ATP turnover, and perhaps a decrease in cellular energy charge. Second, its activation by relative increments of AMP to ATP modulates the activity and expression of key rate-limiting enzymes that control energy-consuming and energy-generating pathways (9, 10). In essence, AMPK regulates energy utilization and promotes energy homeostasis in the cell.
More recently, AMPK has been shown to regulate several additional important cellular pathways and processes, including transcription and protein synthesis, a number of membrane transport proteins in the kidney and other tissues (11), and autophagy (3, 12). These pleiotropic effects of AMPK are consonant with its role as a guardian of cellular energy homeostasis (7, 13).
Based on the fact that AMPK activation is part of the cellular response to stress (14), and based on the suggestion that such activation can protect against organ injury by decreasing inflammation in multiple animal models including hemorrhagic shock, ischemic preconditioning and ischemia/reperfusion (3, 15–21), these experiments were designed to test the hypothesis that AMPK protects against sepsis induced endothelial activation and injury, and that AMPK agonists would limit organ injury and inflammation.
Materials and methods
Cecal ligation and puncture
Animal protocols were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Experiments were performed in adherence to the National Institutes of Health Guidelines on the Use of Laboratory Animals. Cecal ligation and puncture (CLP) was performed on male C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME; 8–10 weeks of age). These animals were anesthetized with pentobarbital (70 mg/kg, intraperitoneal [IP]). A 1- to 2-cm midline laparotomy was performed, and the cecum was identified. Stool was expressed to the tip of the cecum, and then the cecum was ligated at the level of the second cecal artery with 2-0 silk. The cecum was then perforated twice with a 22-gauge needle and returned into the abdomen. The muscle and skin were closed with a running 4-0 vicryl suture. Control animals underwent laparotomy and bowel manipulation without ligation or perforation. Animals were resuscitated with 1.0 mL of 0.9% normal saline, immediately after surgery via subcutaneous injection. Tissue and blood collection occurred at 8 or 24 hours post-CLP. No antibiotics were used, and animals had free access to food and water pre and postoperatively. In some experiments, mice were randomized to receive the AMPK agonist 5-Aminoimidazole-4-carboxamide ribonucleotide [AICAR (Biovision, San Francisco, CA); 100mg/kg; IP], or the AMPK inhibitor Compound C (Biovision) (30mg/kg; IP). Control mice received saline as vehicle only at the same volume (500 ml). Doses were selected based on previous reports from the literature.(20, 22–24)
Cell culture
Primary mouse peritoneal macrophages were harvested from male C57BL/6 mice by lavage of the peritoneal cavity with phosphate-buffered saline (PBS) (25). Macrophages were then plated on 6-well plates in RPMI 1640 medium supplemented with 10% FBS, 50 IU/ml penicillin, 50 μg/ml streptomycin and 2 mM L-glutamine. Four hours after plating, cells were washed 3 times with PBS to remove the non-adherent cells. Adherent cells were then incubated for an additional 24 h at 37°C before treatment. Primary male C57BL/6 mouse kidney glomerular endothelial cells (MKGECs) were purchased from Cell Biologics (Chicago, IL). They were cultured in cell culture medium (Cell Biologics) supplemented with MEM Non-essential amino acids solution (5.0 mL), L-Glutamine (5.0 mL), Penicillin-Streptomycin (5.0 mL) and 5% fetal bovine serum on coverslips for immunohistochemistry. Cells were used on day 2 of harvest. For in vitro experiments some cells were exposed to lipopolysaccharide (LPS 10–100ng/mL), AICAR (1mM), and/or Compound C (10μM).
Neutrophil adhesion assay
Mouse bone marrow neutrophils were prepared as described with some modifications (26). Briefly, PMNs were isolated from femurs and tibias flushed with Ca2+/Mg2+-free Hanks’ balanced salt solution (HBSS)-BSA. The obtained marrow was centrifuged at 300 g, 4°C for 10 min, and resuspended in 3 ml of HBSS. The suspension was subjected to a Percoll step gradient, the gradient was then centrifuged, and cells were removed from the neutrophil-enriched fraction. This procedure yielded >95% PMN purity and >95% viability, assessed by Trypan blue exclusion. Cells were washed with Ca2+/Mg2+-free HBSS (for calcein AM labeling). The assay for PMN adhesion to endothelial cells was performed as previously described (27). MKGECs were isolated and grown to confluence in 96-well gelatin-coated plates. Bone marrow PMNs loaded with calcein AM (Molecular Probes) at 2 μg/ml for 30 min at room temperature were added to MKGECs pretreated with AICAR (1 mM) for 1 h at 37°C. Cells were then treated with LPS (100 ng/mL and 1ug/mL) for 6 h. PMN adhesion was evaluated after treatment of PMNs with anti-CD11b mAb (M1/70) or anti-CD11a mAb (M17/4), each at a concentration of 10 μg/ml (BD Biosciences, San Diego, CA). The fluorescence readings were obtained with the Vactor II spectrofluorometer (Photon Technology International, Monmouth Junction, NJ) with detection at 485 and 535 nm, respectively. The percentage of adherent PMNs was calculated, and all assays were performed in duplicate.
Vascular leak assay
Eight hours post control or CLP surgery, animals were injected with 200 μL of 0.5% Evans blue dye (Sigma-Aldrich, St Louis, MO) via tail vein. The dye was allowed to percolate to the subendothelial spaces for 30 minutes, and then mice were sacrificed. Animals were perfused with cold PBS to wash away extra dye; whole kidneys were weighed and then dissociated with formamide (Sigma-Aldrich) for 48 h at 37° C. After 2 days, supernatants were spun down and read on a spectrophotometer at 620 nm.
Immunocyto/histochemistry
Cells were fixed on coverslips with paraformaldehyde for 15 minutes and then rinsed with cold PBS. Slides were then stained for intracellular adhesion molecule-1 (ICAM-1) (Santa Cruz Biotechnology, Dallas, TX) or vascular cell adhesion molecule-1 (VCAM-1) (Santa Cruz) to monitor endothelium activation. For immunohistochemistry, tissues were harvested, washed in cold PBS and then placed in paraformaldehyde (2%) for 1 hour, and then switched to 30% sucrose solution for 12 hours. The tissue was then slowly frozen in 2-methylbutane. Sections were stained against p-AMPK (Cell signaling, Beverly, MA), LC-3 (Cell signaling), or ICAM-1. Images were taken with an Olympus Provis Fluorescence microscope. Autophagy was determined as elevated LC3 levels in immunohistochemistry.
Electron microscopy
Mice were perfused with cold PBS, then with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 mol/L phosphate buffer (pH 7.4) and processed for transmission electron microscopy (TEM) as described before (28). After dehydration, thin sections were stained with uranyl acetate and lead citrate for observation under a JEM 1011CX electron microscope (JEOL, Peabody, MA). Images were acquired digitally from a randomly selected pool of 10–15 fields under each condition.
Organ Injury measurement
Blood samples were obtained from cardiac puncture at 8 and 24 hours post-CLP. Cystatin C was determined from serum using a mouse Cystatin C kit according to the manufacturer’s instructions (R&D systems, Minneapolis, MN). Serum concentration of BUN, Creatinine, ALT and AST was determined with a HESKA DRI-CHEM 4000 chemistry analyzer (Loveland, CO).
Data analysis
Data is presented as mean ± standard error. One-way analysis of variance was used to determine differences between treatment groups. Statistical significance was determined as p<0.05.
Results
AMPK activation by AICAR minimizes sepsis-induced organ injury
The influence of AICAR on tissue injury was investigated in animals with CLP-induced sepsis. Sepsis-induced kidney failure was measured by serum Cystatin C (pg/mL), BUN, and Creatinine (mg/dL). As expected, CLP animals had a significant elevation of all Acute Kidney Injury (AKI) markers, compared to control animals (Cystatin C: 103.93 ng/mL ± 6.85 vs 29.18 ng/mL ± 2.42, p=0.0004; BUN: 62.18 mg/dL ± 1.2 vs 18 mg/dL ± 1.6, p<0.001; Creatinine 0.43 mg/dL ± 0.02 vs 0.16 mg/dL ± 0.01, p>0.001; Figure 1A–C). Administration of AICAR 24 hours before CLP protected against renal injury, with lower levels of AKI markers when compared to CLP mice (Cystatin C: 49.32 ng/mL ± 2.6, p=0.01; BUN: 43.71 ± 1.9, p=0.01; Creatinine: 0.20mg/dL ± 0.002, p=0.0003). There was a trend towards increased liver injury in CLP mice at the 8-hour time point (96.7 IU/L ± 9.93 vs 53.8 IU/L ± 11.8, p=0.3) and AICAR attenuated this injury (65 IU/L ± 3.6, p=0.3 versus CLP alone; Figure 1D).
Figure 1.

AICAR limited CLP-induced tissue injury. CLP resulted in acute kidney injury as determined by serum Cystatin C (A.), BUN (B.), and Creatinine (C.). CLP also resulted in liver injury as determined by serum ALT (D.). AICAR pretreatment protected against both acute kidney and liver injury. (*p<0.05 vs control; #p<0.05 vs CLP). Tissue was harvested 8h after CLP.
AMPK activation by AICAR decreased sepsis-induced cytokines
The activation of an inflammatory cascade with increased secretion of proinflammatory cytokines has been previously shown to contribute to the sepsis-induced organ dysfunction (29, 30). CLP induced increases in the serum level of multiple cytokines, including IL-1β, IL-6, IL-10, IL-17, RANTES, and TNF-α (*P<0.05 compared to control mice; Figure 2A–F). AICAR pretreatment limited these sepsis-induced changes (#P<0.05 compared to vehicle treated, CLP mice). The influence of AMPK activation on LPS-induced cytokines in cultured primary peritoneal macrophages was also determined. In vitro, AICAR limited LPS-induced increases in cell culture media TNF-α and IL-6 levels, and interestingly increased levels of the anti-inflammatory cytokine IL-10 (*P<0.05 compared to unstimulated controls; #P<0.05 compared to LPS alone; Figure 3A–C).
Figure 2.

AICAR pretreatment limited serum cytokine levels induced by CLP. Cytokine and chemokine levels (A. IL-6, B. IL-1b, C. IL-10, D. IL-17, E. RANTES, F. TNF-α) were measured in plasma by Luminex magpix assay 8 hours following CLP. *p<0.05 vs Control; #p<0.05 vs CLP.
Figure 3.
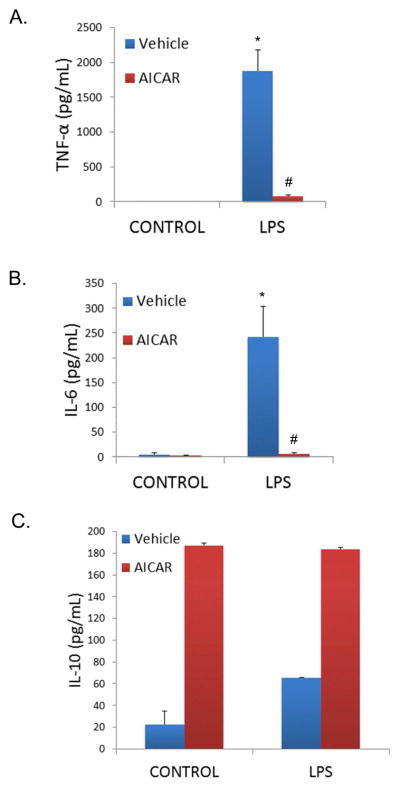
AICAR pretreatment minimized cytokine level expression in LPS stimulated macrophages (A. TNF-α, B. IL-6, C. IL-10). Peritoneal macrophages were pretreated with or without AICAR for 1 hour, and LPS (10ng/mL) was added to the cell culture. Cytokine levels were measured in cell culture media after 4 hours by ELISA. *p<0.05 vs Control; #p<0.05 vs LPS.
Inhibition of AMPK prevents autophagy and increases tissue injury
The activation of AMPK has been shown to be associated with increased adaptive cell signaling responses, including autophagy (3, 12, 31, 32). Additionally, autophagic signaling has been shown to be a protective response in the setting of sepsis (4, 12, 33–35). The influence of CLP on phosphorylation of AMPK was confirmed (Figure 4A demonstrates increases in hepatic phospho-AMPK, which is inhibited by Compound C). Compound C also inhibited CLP-induced autophagic signaling as determined by hepatic and renal LC3 staining (Figures 4B, C). Inhibition of AMPK activation with compound C also led to increased CLP-induced cell death as determined by TUNEL staining at 24 hours (Figure 4D). Moreover, inhibition of AMPK by compound C led to exacerbated IL-6 levels, liver, and kidney injury at a 24 hour time point after CLP compared to vehicle treated, CLP controls (Figures 4E–H).
Figure 4.

Inhibition of AMPK signaling exacerbated injury and inflammation. A. CLP increased phosphorylation of AMPK as demonstrated by immunohistochemistry in liver tissue. Compound C prevented CLP-induced AMPK phosphorylation [Green=actin; blue=dapi; red=p-AMPK]. B, C. Autophagy as determined by LC3 protein levels was increased in liver (B.) and Kidney (C.) following CLP [Green=actin; blue=dapi; red=LC3]. Compound C limited these increases in LC3 levels. D. Minimal apoptotic cell death is seen following CLP, however, there is increased apoptotic cell death with inhibition of AMPK signaling as determined by TUNEL staining in kidney tissue [Green=actin; blue=dapi; red=TUNEL]. E–H. Inhibition of AMPK signaling resulted in worse inflammation and tissue injury at 24 hours. Serum IL-6 levels normalized by 24 hours following CLP, however, Compound C treated animals continued to show elevated levels at this time point (*P<0.05 compared to vehicle, CLP mice; E.). Furthermore, there was no significant difference in organ injury in vehicle treated CLP mice compared to control mice at 24 hours following CLP. Compound C pretreatment led to exacerbated injury at this timepoint (*P<0.05 compared to vehicle, CLP mice; F–H.).
AICAR limits CLP-induced endothelial activation and injury
One of the hallmarks of sepsis is the up regulation of adhesion molecules and endothelium activation. This is a normal defense mechanism that allows neutrophils to transmigrate from the circulation to the affected region and fight infection. However, this effect is also partly responsible of the capillary leak and microvascular disruption that characterizes the septic clinical syndrome and likely one of the pathways to explain organ dysfunction. The potential protective effect of AMPK signaling in sepsis on endothelial activation and injury was investigated. CLP increased both renal and hepatic up regulation of the adhesion molecule ICAM-1 (Figure 5A, B). AICAR pretreatment limited this increase in ICAM-1 protein levels. Furthermore, microvascular endothelial injury was also determined by transmission electron microscopy ultra structural changes. CLP leads to renal microvascular injury demonstrated by increased endothelial fenestrations. AICAR pretreatment prevented these changes (Figure 6A). Moreover, AICAR limited CLP-induced vascular leakage as determined by Evan’s blue renal tissue extravasation (Figure 6B).
Figure 5.

AICAR pretreatment minimized endothelial cell activation induced by CLP. A. CLP increased endothelial activation in kidney as demonstrated by immunohistochemistry staining of ICAM-1 (red) in kidney tissue, at 8 hours following CLP. Pharmacological activation of AMPK by AICAR given 24 hours before CLP minimized this upregulation. B. CLP also caused an increase in endothelial activation in liver, measured by ICAM-1 (red), which was minimized by AICAR pretreatment.
Figure 6.

AICAR minimized morphological changes and microvascular injury induced by CLP. A. Transmission Electron Microscopy of kidney tissue 8h following CLP, demonstrates an increase in the diameter and number of fenestrations induced by CLP. AICAR pretreatment minimized these morphological changes. Podocyte structure and number were not affected by CLP. Arrow demonstrates endothelial fenestrations in kidney tissue. CL= capillary lumen, P= Podocytes, RBC= Red blood cell. B. Microvascular injury was measured by Evan’s blue dye extravasation in kidney injury at 8h following CLP with or without AICAR pretreatment. AICAR pretreatment minimized microvascular injury as measured by Evan’s blue dye extravasation in kidney (#p<0.05 compared to vehicle treated CLP animals).
AMPK activation by AICAR minimizes in vitro endothelium activation, and leucocyte adhesion
The influence of AICAR on LPS-induced endothelial activation was determined. Primary mouse kidney glomerular endothelial cells (MKGECs) were utilized for these experiments and were treated with or without LPS for 4 hours, and with or without AICAR. Similar to that seen in vivo, LPS increased protein levels of ICAM-1 and VCAM-1 (Figure 7A, B). These changes were prevented by AICAR treatment. As a functional marker of endothelial activation, neutrophil adhesion to MKGECs was determined. LPS treatment of MKGEC increased neutrophil adhesion and this was limited by AICAR (*P<0.05 compared to control; #P<0.05 compared to vehicle treated, LPS activated cells; Figure 7C).
Figure 7.

AICAR minimized endothelial cell activation, and neutrophil adhesion. A, B. Mouse Kidney Glomerular Endothelial Cells were pretreated with or without AICAR for 1 h, and LPS (10ng/mL) was added to the cell culture. LPS induced upregulated expression of ICAM-1(A.), and VCAM-1 (B.). AICAR pretreatment minimized the upregulation of ICAM-1 and VCAM-1. (Nucleus=blue, ICAM-1, VCAM-1=red). C. Bone marrow derived neutrophils were added to Mouse Kidney Glomerular Endothelial Cell culture with or without AICAR pretreatment, and stimulated with LPS for 6 hours. Neutrophil adhesion was increased in vehicle treated cells (*P<0.05 vs control). AICAR pretreatment minimized the number of adhered neutrophils when compared to LPS stimulated cells (#p<0.05 vs LPS alone).
Discussion
The release of PAMPs and DAMPs locally and systemically leads to the activation of endothelial cells, circulating and tissue based immune cells, as well as parenchymal cells. Many inflammatory pathways are activated by such signaling, and subsequent expression of proinflammatory cytokines such as interleukin 1β (IL-1β) and TNF-α (36) follows. This enhanced pro-inflammatory state has been associated with several effects that characterize the clinical phenotype of sepsis, including oxidative stress in parenchymal cells of diverse organs, as well as endothelial activation with increased production of reactive oxygen and nitrogen intermediates, such as superoxide and nitric oxide (NO). This excess in NO production is a major contributor to the vasodilatation and vascular hyporeactivity seen in septic shock. Inflammation has been shown to cause an alteration to endothelial lining of organs, and there is recent evidence of TNF induced glomerular damage in the kidney with TLR 4 activation by LPS (37).
The results of this study suggest that activation of the AMPK signaling pathway serves to limit inflammation, endothelial activation and organ dysfunction in experimental sepsis. Importantly, these results illustrate that this signaling pathway can be activated exogenously, suggesting that the system can be harnessed to limit injury. Although causality is not hereby established, these data suggest that the protective effects of AICAR may be at least in part explained by modulation of the inflammatory response and protection of the endothelium. Furthermore, inhibition of AMPK activation by Compund C exacerbated injury supporting the hypothesis that the AMPK pathway may be part of the adaptive response to sepsis. These findings are in agreement with previous studies suggesting a link between metabolic homeostasis regulation and the immune response (19, 38). As Sag et al. has suggested (38), our data suggest that AMPK activation through AICAR produces a significant decline in cytokine release and an association with maintenance of renal function early after the septic insult. This is important, because decreasing inflammation in the vicinity of the tubular epithelium may help decrease overall cellular injury, allowing the tubular epithelial cells to maintain important processes for the organ as a whole (i.e. tubular transport) and thus sustain renal function. Our experiments showed a discrepancy between the in vivo and in vitro release of IL-10 to inflammatory stimuli. However, our data agrees with that of other groups that have found similar elevations of IL-10 after stimulation of peritoneal macrophages in vitro.(39) The difference with the results in the in vivo experiments may have several explanations. First, the inflammatory injury caused by LPS vs. CLP is not the same, with LPS exerting a more specific TLR-4 mediated response, whereas CLP with multi-organism peritonitis exerts a more diffuse compromise in terms of cellular signaling; second, the cytokine release in the whole animal may be very different to that in cell culture, as there are many other mechanisms in play that can modify the response to inflammation and treatment; third, AICAR and AMPK activation are known to improve neutrophil chemotaxis, and bacterial clearance, suggesting that pre-treated animals with AICAR could have less inflammatory response (and this is inflammatory and anti-inflammatory), possibly due to better control of the infectious source. However, our data does not provide enough information as to describe the mechanism by which AICAR exerts its protective effects on renal function. An important question to answer is what is the contribution of AMPK activation to energy regulatory processes in parenchymal cells, such as the tubular epithelial cell, and what is the effect of this on organ function not only early after the septic insult, but also during convalescence.
We have also demonstrated that pre-emptive AMPK activation decreases global endothelial activation as measured by a decrease in endothelial expression of adhesion molecules (specifically ICAM-1), a decrease in vascular leak (Evan’s blue) and a decrease in neutrophil adhesion, as well as minimizing morphological changes induced by sepsis (TEM). We acknowledge that these effects may be secondary to the effects of AICAR on the inflammatory response described above. In the same way, these results are in agreement with data by other groups that have shown that AMPK activation in the vascular endothelium may confer protection via decreased cytokine-induced NF-kB activation (40). Other studies have demonstrated suppression of oxidative stress by induction of MnSOD and PGC-1α-dependent mitochondrial biogenesis (41, 42), as well as decreased mitochondrial production of radical oxygen species (ROS) (41) and inhibition of apoptosis (43), as pathways to enhance organ protection via AMPK upregulation. In addition, the data cannot clarify whether this endothelial protection actually preceded the decrease in cytokine outflow, and thus whether protection came from the action of AICAR on the endothelium, or instead on circulating activated leukocytes.
In conclusion, this study demonstrated that AICAR-induced AMPK activation has a measurable effect on cytokine release, on disease-relevant markers of endothelial activation, and ultimately on renal function. This data suggest that AMPK activation protects against sepsis-induced renal dysfunction, and that this protection is associated with AMPK-induced decrease in circulating cytokines and endothelial activation. Whether re-prioritization of energy utilization within the renal tubular cell has a role in such a protective signal remains unknown. Finally, these data suggest that this protection may not be organ-specific, given that we also found a signal suggesting liver protection.
Highlights.
AICAR induced AMPK activation was associated with a decrease in sepsis-induced acute kidney and hepatic injury markers early after induction of CLP.
AMPK activation limited sepsis-induced inflammatory response as measured by plasma cytokines.
Activation of AMPK decreased endothelial activation and vascular leak in vivo, and neutrophil adhesion in vitro.
Acknowledgments
Source of funding
This work is supported by National Institutes of Health grants R01 GM082830 (BSZ), 1K12HL109068-02 (HG), Veterans Affairs Merit Award 1I01BX000566 (BSZ), and Department of Defense DM102439 (BSZ).
List of Abbreviations
- AKI
Acute Kidney Injury
- AMPK
AMP activated protein kinase
- CLP
Cecal Ligation and Puncture
- DAMPs
Damage Associated Molecular Patterns
- ICAM-1
Intracellular adhesion molecule-1
- LPS
Lipopolysaccharide
- MKGECs
Mouse Kidney Glomerular Endothelial Cells
- mTOR
mammalian Target of Rapamycin
- NO
Nitric Oxide
- PAMPs
Pathogen Associated Molecular Patterns
- PBS
Phosphate-buffered Saline
- PMNs
Polymorphonuclear lymphocytes
- ROS
Reactive Oxygen Species
- TEM
Transmission Electron Microscopy
- VCAM-1
Vascular adhesion molecule-1
Footnotes
Conflicts of interest: No conflict of interest declared.
Author contribution: Conception of experimental design (HG, BSZ, DE, MRR), animal experimentation (DE, AMBQ, BCK, JL, PL, SD), Collection of data (DE, AMBQ, BCK, JL, PL, SD), Manuscript elaboration (DE, HG, BSZ, MRR), Review of final manuscript (DE, AMBQ, BCK, JL, PL, SD, HG, BSZ, MRR)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Critical care medicine. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Marshall JC, Cook DJ, Christou NV, Bernard GR, Sprung CL, et al. Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Critical care medicine. 1995;23:1638–1652. doi: 10.1097/00003246-199510000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Howell GM, Gomez H, Collage RD, Loughran P, Zhang X, et al. Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PloS one. 2013;8:e69520. doi: 10.1371/journal.pone.0069520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology. 2011;53:2053–2062. doi: 10.1002/hep.24324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whelan SP, Carchman EH, Kautza B, Nassour I, Mollen K, et al. Polymicrobial sepsis is associated with decreased hepatic oxidative phosphorylation and an altered metabolic profile. The Journal of surgical research. 2014;186:297–303. doi: 10.1016/j.jss.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SJ, Zhang J, Choi AM, Kim HP. Mitochondrial dysfunction induces formation of lipid droplets as a generalized response to stress. Oxidative medicine and cellular longevity. 2013;2013:327167. doi: 10.1155/2013/327167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes & development. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hallows KR, Mount PF, Pastor-Soler NM, Power DA. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. American journal of physiology. Renal physiology. 2010;298:F1067–1077. doi: 10.1152/ajprenal.00005.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pastor-Soler NM, Hallows KR. AMP-activated protein kinase regulation of kidney tubular transport. Current opinion in nephrology and hypertension. 2012;21:523–533. doi: 10.1097/MNH.0b013e3283562390. [DOI] [PubMed] [Google Scholar]
- 10.Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annual review of biochemistry. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 11.Hallows KR. Emerging role of AMP-activated protein kinase in coupling membrane transport to cellular metabolism. Current opinion in nephrology and hypertension. 2005;14:464–471. doi: 10.1097/01.mnh.0000174145.14798.64. [DOI] [PubMed] [Google Scholar]
- 12.Guo L, Stripay JL, Zhang X, Collage RD, Hulver M, et al. CaMKIalpha regulates AMP kinase-dependent, TORC-1-independent autophagy during lipopolysaccharide-induced acute lung neutrophilic inflammation. Journal of immunology. 2013;190:3620–3628. doi: 10.4049/jimmunol.1102975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiological reviews. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 14.Alexander A, Walker CL. The role of LKB1 and AMPK in cellular responses to stress and damage. FEBS letters. 2011;585:952–957. doi: 10.1016/j.febslet.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Alkhulaifi AM, Pugsley WB. Role of acadesine in clinical myocardial protection. British heart journal. 1995;73:304–305. doi: 10.1136/hrt.73.4.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galinanes M, Zhai X, Bullough D, Mullane KM, Hearse DJ. Protection against injury during ischemia and reperfusion by acadesine derivatives GP-1-468 and GP-1-668. Studies in the transplanted rat heart. The Journal of thoracic and cardiovascular surgery. 1995;110:752–761. doi: 10.1016/S0022-5223(95)70108-7. [DOI] [PubMed] [Google Scholar]
- 17.Bullough DA, Zhang C, Montag A, Mullane KM, Young MA. Adenosine-mediated inhibition of platelet aggregation by acadesine. A novel antithrombotic mechanism in vitro and in vivo. The Journal of clinical investigation. 1994;94:1524–1532. doi: 10.1172/JCI117493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peralta C, Bartrons R, Serafin A, Blazquez C, Guzman M, et al. Adenosine monophosphate-activated protein kinase mediates the protective effects of ischemic preconditioning on hepatic ischemia-reperfusion injury in the rat. Hepatology. 2001;34:1164–1173. doi: 10.1053/jhep.2001.29197. [DOI] [PubMed] [Google Scholar]
- 19.Zhao X, Zmijewski JW, Lorne E, Liu G, Park YJ, et al. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. American journal of physiology Lung cellular and molecular physiology. 2008;295:L497–504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaskin FS, Kamada K, Yusof M, Durante W, Gross G, et al. AICAR preconditioning prevents postischemic leukocyte rolling and adhesion: role of K(ATP) channels and heme oxygenase. Microcirculation. 2009;16:167–176. doi: 10.1080/10739680802355897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathew JP, Rinder CS, Tracey JB, Auszura LA, O’Connor T, et al. Acadesine inhibits neutrophil CD11b up-regulation in vitro and during in vivo cardiopulmonary bypass. The Journal of thoracic and cardiovascular surgery. 1995;109:448–456. doi: 10.1016/S0022-5223(95)70275-X. [DOI] [PubMed] [Google Scholar]
- 22.Gaskin FS, Kamada K, Yusof M, Korthuis RJ. 5′-AMP-activated protein kinase activation prevents postischemic leukocyte-endothelial cell adhesive interactions. American journal of physiology. Heart and circulatory physiology. 2007;292:H326–332. doi: 10.1152/ajpheart.00744.2006. [DOI] [PubMed] [Google Scholar]
- 23.Gross ER, Nithipatikom K, Hsu AK, Peart JN, Falck JR, et al. Cytochrome P450 omega-hydroxylase inhibition reduces infarct size during reperfusion via the sarcolemmal KATP channel. Journal of molecular and cellular cardiology. 2004;37:1245–1249. doi: 10.1016/j.yjmcc.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 24.Shigematsu S, Ishida S, Gute DC, Korthuis RJ. Postischemic anti-inflammatory effects of bradykinin preconditioning. American journal of physiology. Heart and circulatory physiology. 2001;280:H441–454. doi: 10.1152/ajpheart.2001.280.1.H441. [DOI] [PubMed] [Google Scholar]
- 25.Saraswathi V, Hasty AH. The role of lipolysis in mediating the proinflammatory effects of very low density lipoproteins in mouse peritoneal macrophages. Journal of lipid research. 2006;47:1406–1415. doi: 10.1194/jlr.M600159-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, et al. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 27.Rahman A, Anwar KN, Uddin S, Xu N, Ye RD, et al. Protein kinase C-delta regulates thrombin-induced ICAM-1 gene expression in endothelial cells via activation of p38 mitogen-activated protein kinase. Molecular and cellular biology. 2001;21:5554–5565. doi: 10.1128/MCB.21.16.5554-5565.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stolz DB, Ross MA, Salem HM, Mars WM, Michalopoulos GK, et al. Cationic colloidal silica membrane perturbation as a means of examining changes at the sinusoidal surface during liver regeneration. The American journal of pathology. 1999;155:1487–1498. doi: 10.1016/S0002-9440(10)65464-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodrigues CE, Sanches TR, Volpini RA, Shimizu MH, Kuriki PS, et al. Effects of continuous erythropoietin receptor activator in sepsis-induced acute kidney injury and multi-organ dysfunction. PloS one. 2012;7:e29893. doi: 10.1371/journal.pone.0029893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. American journal of physiology. Lung cellular and molecular physiology. 2006;290:L622–L645. doi: 10.1152/ajplung.00477.2005. [DOI] [PubMed] [Google Scholar]
- 31.Wang LT, Chen BL, Wu CT, Huang KH, Chiang CK, et al. Protective Role of AMP-Activated Protein Kinase-Evoked Autophagy on an In Vitro Model of Ischemia/Reperfusion-Induced Renal Tubular Cell Injury. PloS one. 2013;8:e79814. doi: 10.1371/journal.pone.0079814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Guan KL. AMPK connects energy stress to PIK3C3/VPS34 regulation. Autophagy. 2013;9:1110–1111. doi: 10.4161/auto.24877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujishima Y, Nishiumi S, Masuda A, Inoue J, Nguyen NM, et al. Autophagy in the intestinal epithelium reduces endotoxin-induced inflammatory responses by inhibiting NF-kappaB activation. Archives of biochemistry and biophysics. 2011;506:223–235. doi: 10.1016/j.abb.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 34.Carchman EH, Whelan S, Loughran P, Mollen K, Stratamirovic S, et al. Experimental sepsis-induced mitochondrial biogenesis is dependent on autophagy, TLR4, and TLR9 signaling in liver. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27:4703–4711. doi: 10.1096/fj.13-229476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe E, Muenzer JT, Hawkins WG, Davis CG, Dixon DJ, et al. Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study. Laboratory investigation; a journal of technical methods and pathology. 2009;89:549–561. doi: 10.1038/labinvest.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carre JE, Singer M. Cellular energetic metabolism in sepsis: the need for a systems approach. Biochimica et biophysica acta. 2008;1777:763–771. doi: 10.1016/j.bbabio.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 37.Xu C, Chang A, Hack BK, Eadon MT, Alper SL, et al. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney international. 2013 doi: 10.1038/ki.2013.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. Journal of immunology. 2008;181:8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nath N, Giri S, Prasad R, Salem ML, Singh AK, et al. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. Journal of immunology. 2005;175:566–574. doi: 10.4049/jimmunol.175.1.566. [DOI] [PubMed] [Google Scholar]
- 40.Katerelos M, Mudge SJ, Stapleton D, Auwardt RB, Fraser SA, et al. 5-aminoimidazole-4-carboxamide ribonucleoside and AMP-activated protein kinase inhibit signalling through NF-kappaB. Immunology and cell biology. 2010;88:754–760. doi: 10.1038/icb.2010.44. [DOI] [PubMed] [Google Scholar]
- 41.Ouslimani N, Peynet J, Bonnefont-Rousselot D, Therond P, Legrand A, et al. Metformin decreases intracellular production of reactive oxygen species in aortic endothelial cells. Metabolism: clinical and experimental. 2005;54:829–834. doi: 10.1016/j.metabol.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 42.Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55:120–127. [PubMed] [Google Scholar]
- 43.Ido Y, Carling D, Ruderman N. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes. 2002;51:159–167. doi: 10.2337/diabetes.51.1.159. [DOI] [PubMed] [Google Scholar]