

Abstract
Stimulation of D1-like dopamine receptors (D1DRs) or D2-like dopamine receptors (D2DRs) in the nucleus accumbens (NAc) shell reinstates cocaine seeking in rats, an animal model of relapse. D2DRs and D1DRs activate protein kinase C (PKC) and recent studies indicate that activation of PKC in the NAc plays an important role in the reinstatement of drug seeking induced by a systemic cocaine priming injection. In the present study, pharmacological inhibition of PKC in the NAc shell attenuated cocaine seeking induced by intra-accumbens shell microinjection of a D2DR agonist, but not a D1DR agonist. D1DRs and D2DRs are primarily expressed on different accumbens medium spiny (MSN) neurons. Neuronal signaling and activity were assessed in these two populations of NAc neurons with transgenic mice expressing fluorescent labels under the control of D1DR and D2DR promoters. Following the extinction of cocaine self-administration, bath application of a PKC inhibitor produced similar effects on single evoked excitatory and inhibitory post-synaptic currents in D1DR- and D2DR-positive MSNs in the NAc shell. However, inhibition of PKC preferentially improved the ability of excitatory, but not inhibitory, synapses to sustain responding to brief train of stimuli specifically in D2DR-positive MSNs. This effect did not appear to involve modulation of presynaptic release mechanisms. Taken together, these findings indicate that the reinstatement of cocaine seeking is at least partially due to D2DR-dependent increases in PKC signaling in the NAc shell, which reduce excitatory synaptic efficacy in D2DR-expressing MSNs.
Introduction
Dopamine receptors are divided into two general categories, D1-like (D1 and D5) and D2-like (D2, D3 and D4), based on sequence homology and pharmacology (Lachowicz and Sibley, 1997). The two output pathways of the NAc selectively express D1-like dopamine receptors (D1DRs) and D2-like dopamine receptors (D2DRs), which project to the ventral tegmental area (VTA) and ventral pallidum, respectively (Gerfen, 1992; Lu et al., 1998). There are numerous examples of functional differences resulting from selective modulation of these accumbens afferents. For example, recent evidence indicated that preferential activation of D2DR-containing neurons in the NAc suppressed cocaine conditioned reward, whereas stimulation of D1DR-expressing neurons produced the opposite effect (Lobo et al., 2010). Similarly, activation of accumbens D2DR-positive neurons suppressed cocaine self-administration, whereas inhibition of these cells enhanced the motivation to obtain cocaine (Bock et al., 2013).
Increased dopamine transmission in the NAc shell is a major neurochemical trigger for cocaine priming-induced reinstatement of cocaine-seeking behavior, an animal model of relapse (Anderson et al., 2003; Anderson and Pierce, 2005; Bachtell et al., 2005). Thus, administration of D1DR agonists or D2DR agonists directly into the NAc shell promoted the reinstatement of cocaine seeking (Anderson et al., 2008; Schmidt et al., 2006; Schmidt and Pierce, 2006a). D1DRs and D2DRs have differential effects on adenylyl cyclase (AC)/protein kinase A (PKA) signaling with D1DRs activating the AC/PKA pathway via Gs and D2DRs producing the opposite effect through Gi (Missale et al., 1998). D2DRs also signal through Gβγ, which activates protein kinase C (PKC) via serial stimulation of phospholipase C (PLC), phosphatidylinositol biphosphate (PIP2) and diacetyl glycerol (DAG) (Beaulieu and Gainetdinov, 2011; Surmeier et al., 2007). Previous studies demonstrated a role for PKC in psychostimulant-mediated behaviors. For example, repeated cocaine administration resulted in the phosphorylation of some, but not all, isoforms of PKC in the NAc (Chen et al., 2007; Steketee et al., 1998) and PKC inhibitors suppress various psychostimulant-mediated behaviors (Aujla and Beninger, 2003; Cervo et al., 1997; Pierce et al., 1998). Moreover, stimuli associated with cocaine self-administration increased PKCγ mRNA in the NAc (Thomas and Everitt, 2001). We recently showed that phosphorylation of NAc PKCγ was increased during the reinstatement of cocaine seeking and intra-accumbens PKC inhibitors suppressed cocaine seeking (Schmidt et al., 2014; Schmidt et al., 2013).
These results, collectively, suggest that D2DR agonists promote cocaine reward, reinforcement and seeking by suppressing neuronal activity in D2DR-containing NAc MSNs projecting to the ventral pallidum. One way in which D2DR agonists may suppress excitatory transmission is through PKC-dependent regulation of AMPA receptors (AMPARs) (Anggono and Huganir, 2012; McCutcheon et al., 2011). Thus, we hypothesized that PKC inhibitors suppress cocaine seeking by reversing D2DR-induced inhibition of excitatory transmission. The present results indicate that pharmacological inhibition of PKC blocked cocaine seeking induced by intra-accumbal shell administration of a D2DR agonist and also triggered an activity-dependent increase of AMPAR-mediated excitatory postsynaptic currents specifically in NAc shell D2DR-containing MSNs.
Material and Methods
Animals and housing
Male Sprague-Dawley rats (Rattus norvegicus) weighing 250–275 g were obtained from Taconic Laboratories. Animals were individually housed with food and water available ad libitum in their home cages. Mice containing BAC Drd1a-tdTomato (Shuen et al., 2008) and Drd2-EGFP transgenes (Gong et al., 2003) were bred on a mixed B6SJLF1 x FVB/N background. Male mice (2–6 months old, 20–40g; age matched across group) were housed individually following stereotaxic surgery and during experimental paradigms. A 12/12 hr light/dark cycle was used with the lights on at 7:00 a.m. All experimental procedures were performed during the light cycle. The experimental protocols were all consistent with the guidelines issued by the U.S. National Institutes of Health and were approved by the University of Pennsylvania’s Institutional Animal Care and Use Committee.
Surgeries
Prior to surgery, rats and mice were anesthetized with 80 mg/kg ketamine and 12 mg/kg xylazine (Sigma-Aldrich, St. Louis, MO). An indwelling catheter (CamCath, Cambridge, UK) was inserted into the right jugular vein and sutured in place. The catheter was routed to a mesh backmount platform that was implanted subcutaneously dorsal to the shoulder blades. Catheters were flushed daily with 0.3 ml of antibiotic (Timentin, 0.93 mg/ml) dissolved in heparinized saline and sealed with plastic obturators when not in use. After catheter insertion, rats were immediately mounted in a stereotaxic apparatus (Kopf Instruments, CA). Guide cannulas (14 mm, 24 gauge) for microinjections were implanted bilaterally 2mm dorsal to the medial NAc shell. Guide cannulas were cemented in place by affixing dental acrylic to stainless steel screws secured in the skull. The coordinates for the ventral ends of the guide cannulas, relative to bregma according to the atlas of Paxinos and Watson (1997), were as follows: +1.0 mm A/P, ±1.0 mm M/L, and −5.0 mm D/V. An obturator (14mm, 33 gauge) was inserted into each guide cannula in order to prevent occlusion.
Rat cocaine self-administration, extinction and reinstatement of cocaine seeking
After surgery, rats were allowed 7 days to recover before behavioral testing commenced. Initially, rats were placed in operant chambers and allowed to lever-press for intravenous infusions of cocaine (0.25 mg cocaine/59 μl saline, infused over a 5 s period) on a fixed-ratio 1 (FR1) schedule of reinforcement. Each operant session began with the intravenous administration of 59 μl cocaine (0.25 mg) to fill the catheter. Rats were allowed to self-administer a maximum of 30 injections per 120 min operant session. Once an animal achieved at least 20 infusions of cocaine in a single daily operant session under the FR1 schedule, the subject was switched to a fixed-ratio 5 (FR5) schedule of reinforcement. The maximum number of injections was again limited to 30 per daily self-administration session under the FR5 schedule. For both FR1 and FR5 schedules, a 20 s time-out period followed each cocaine infusion, during which time active lever responses were tabulated but had no scheduled consequences. Responses made on the inactive lever, which had no scheduled consequences, were also recorded during both the FR1 and FR5 training sessions. Following 21 days of daily cocaine self-administration sessions, drug-seeking behavior was extinguished by replacing the cocaine with 0.9% saline. Daily extinction sessions continued until responding on the active lever was <15% of the total active lever responses completed on the last day of cocaine self-administration maintained on a FR5 schedule of reinforcement. Typically, it took ~7 days for rats to meet this criterion.
Once cocaine self-administration was extinguished, animals entered the reinstatement phase of the experiment. During reinstatement test sessions, satisfaction of the response requirement (i.e., five presses on the active lever) resulted in an infusion of saline rather than cocaine. Using a between-sessions reinstatement paradigm, each reinstatement test session was followed by extinction sessions until responding was again <15% of the response rate maintained by cocaine self-administration. Generally, 1–2 days of extinction were necessary to reach extinction criterion between reinstatement test sessions. The FR5 schedule was used throughout the extinction and reinstatement phases of these experiments. Catheter patency was maintained in all rats throughout the reinstatement experiments. Since these experiments did not assess cocaine priming-induced reinstatement of drug seeking, no priming infusions of cocaine were administered prior to the reinstatement test sessions. Instead, a within-subjects design was used to study the effects of PKC inhibition in the shell on the ability of subsequent intra-accumbens shell microinjections of a D1-like or D2-like dopamine receptor agonist to reinstate cocaine-seeking behavior.
Rat microinjection procedures
Obturators were removed from the guide cannulas and 33 gauge, 16 mm stainless steel microinjectors were inserted. Bilateral infusions were performed simultaneously over 2 min in a total volume of 0.5 μl per hemisphere. The effect of intra-accumbal shell pretreatment with the PKC inhibitor Ro31-8220 or vehicle on D1-like and D2-like dopamine receptor agonist-induced reinstatement of cocaine seeking was assessed. Vehicle or 10.0 μM Ro 31-8220 were microinjected bilaterally into the NAc shell 10 min prior to infusion of the D1-like dopamine receptor agonist R(+)-SKF-81297 hydrobromide (0 and 1.0 μg/0.5μl) or the D2-like dopamine receptor agonist (−)-quinpirole hydrochloride (0 and 3.0 μg/0.5μl) directly into the NAc shell. Following infusion, microinjectors were left in place for an additional 1 min in order to allow for diffusion of the drug solution away from the tips of the microinjectors. To control for potential rank order effects of drug and vehicle administrations, all treatments were counterbalanced across reinstatement test sessions. Rats were placed immediately into operant chambers following infusions of a D1-like or D2-like dopamine receptor agonist into the accumbens shell.
Verification of cannula placements
After completion of all microinjection experiments, rats were given an overdose of pentobarbital (100 mg/kg) and perfused with 0.9% saline followed by 10% formalin. Brains were removed and coronal sections (100 μm) were taken at the level of the NAc with a Vibratome. The sections were mounted on gelatin-coated slides and stained with cresyl violet. An individual blind to behavioral responses determined cannula placements as well as excessive cannula-induced damage (defined as a cannula tract in excess of 500 μm) or drug-induced cell death (defined as neuronal death extending beyond 100 μm from the cannula tract) using light microscopy. Animals with cannula placements outside of the medial NAc shell, excessive mechanical damage or cell death were excluded from subsequent data analysis.
Mouse cocaine self-administration and extinction
Prior to catheterization, mice were trained to perform an operant response for sucrose pellets. The mice were placed in operant chambers (Med-Associates) and trained to spin a wheel manipulandum to receive a sucrose pellet on a FR1 schedule of reinforcement. A compound cue stimulus consisting of a cue light above the active lever, a 2,900 Hz tone, and house light off was delivered concurrently with each pellet administration. An 8 s time-out period, in which responding had no programmed consequences, followed each pellet delivery. During the food-training phase, mice were food restricted to approximately 90% of their free feeding weight. Following ten days of sucrose self-administration, mice were surgically implanted with jugular catheters. After surgery, mice were allowed 3–4 d to recover before beginning behavioral testing. Mice returned to ad libitum food access 3 days following the start of the cocaine self-administration phase. Mice were tested for cocaine self-administration behavior in 2-hour sessions (6 d per week) in the same chamber as used for sucrose pellet self-administration. During testing, responding on the wheel now delivered an intravenous cocaine injection (0.5 mg/kg/infusion), paired with the compound cue, under the same schedule as the food training. Mice self-administered an average of 23.56±0.62 infusions per day over the course of the 16-day self-administration phase. Following 16 days of cocaine self-administration, cocaine-seeking behavior was extinguished by replacing the cocaine with 0.9% saline. During this time the light and tone cues paired with cocaine delivery were not present. Daily 2-h extinction sessions continued until animals had met the extinction criterion of less than 25% of their self-administration responding. Mice reached the extinction criterion in an average of 6.9 ±0.68 days. Twenty-four hours following meeting the extinction criterion, brains were collected for physiological recordings.
Mouse NAc slices
Mice were decapitated following isoflurane anesthesia the day after the last extinction session. Brains were dissected and coronal slices (300 μm) containing the NAc were cut with a Vibratome (VT1000S, Leica Microsystems) in an ice-cold artificial cerebrospinal fluid solution (ACSF), in which NaCl was replaced by an equiosmolar concentration of sucrose. ACSF consisted of (in mM): 130 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, 1 MgCl2 and 2 CaCl2 (pH 7.2–7.4 when saturated with 95% O2/5% CO2). Slices were incubated in ACSF at 32–34 °C for 45 mi n and kept at 22–25 °C thereafter, until transfer t o the recording chamber. For all electrophysiology experiments, aCSF in the recording chamber was supplemented with 0.075 mM dopamine and 0.05 mM Na-metabisulfite to prevent dopamine oxidation (see Results). Slices were viewed using infrared differential interference contrast optics under an upright microscope (Eclipse FN1, Nikon Instruments Inc.) with a 40x water-immersion objective.
Electrophysiology
The recording chamber was continuously perfused (1–2 ml/min) with oxygenated ACSF heated to 32±1 °C using an automatic temperature co ntroller (Warner Instruments). Recording pipettes were pulled from borosilicate glass capillaries (World Precision Instruments) to a resistance of 4–7 MΩ when filled with the intracellular solution. The intracellular solution contained (in mM): 145 potassium gluconate, 2 MgCl2, 2.5 KCl, 2.5 NaCl, 0.1 BAPTA, 10 HEPES, 2 Mg-ATP, 0.5 GTP-Tris, 1 QX-314 (pH 7.2–7.3 with KOH, osmolarity 280–290 mOsm). QX-314 was omitted in experiments evaluating action potential firing. D1DR-positive and D2DR-positive MSNs in the NAc shell were identified by the presence of tdTomato and green fluorescent protein (GFP) luminescence, respectively. Evoked excitatory and inhibitory postsynaptic current (eEPSC and eIPSC, respectively) recordings were conducted in whole-cell voltage-clamp mode (Vh = −70 mV for eEPSC and Vh = 0 mV for eIPSCs). PKC inhibitors, chelerythrine (10 μM) or Ro 31-8220 (10 μM) were applied via the Y-tube perfusion system (Murase et al., 1989) for at least three minutes prior to any measurements. No differences were found between the effects of the two PKC inhibitors on synaptic activity in D1DR-positive or in D2DR-positive MSNs. Therefore, chelerythrine and Ro31-8220 results were pooled within each cell type. Action potential firing frequency was analyzed in current-clamp mode in response to a 500 ms depolarizing current step. Rheobase was determined as the amplitude of a minimum current step (advanced in 10 pA increments) to elicit an action potential response. All recordings were conducted with a MultiClamp700B amplifier (Molecular Devices). Currents were low-pass filtered at 2 kHz and digitized at 20 kHz using a Digidata 1440A digitizer and pClamp10 software (both from Molecular Devices). Access resistance (10–30 MΩ) was monitored throughout the recordings by injection of 10 mV hyperpolarizing pulses and data were discarded if access resistance changed by >25% over the course of data acquisition. Evoked responses were triggered by 100 μs constant-current pulses generated by an A310 Accupulser (World Precision Instruments) and delivered at 0.1 Hz via a bipolar tungsten stimulation electrode positioned within 100 μm of the recorded cell. The amplitude of the current pulses was controlled by a stimulus isolation unit (ISO-Flex, A.M.P.I.) and was adjusted to elicit monosynaptic responses in the range of 100–300 pA.
Drugs
Cocaine was obtained from the National Institute on Drug Abuse (Rockville, MD) and dissolved in bacteriostatic 0.9% saline. Ro 31-8220 mesylate (3-[3-[2, 5-Dihydro-4-(1-methyl-1H-indol-3-yl)-2, 5-dioxo-1H-pyrrol-3-yl]-1H-indol-1-yl]propyl carbamimidothioic acid ester mesylate) and chelerythrine chloride (1,2-Dimethoxy-12-methyl[1,3]benzodioxolo[5,6]phenanthridinium chloride) were purchased from Tocris (Minneapolis, MN). Chelerythrine chloride was dissolved in sterile 0.9% saline. Ro 31-8220 was dissolved in 100% DMSO to make stock solutions and then diluted in sterile 0.9% saline to required final working concentrations, resulting in final vehicle concentrations of 1% DMSO. R(+)-SKF-81297 hydrobromide (1.0 μg/0.5 μl) and (−)-quinpirole hydrochloride (3.0 μg/0.5 μl) were purchased from Sigma/RBI (St. Louis, MO) and dissolved in sterile 0.9% saline. Doses for each of the aforementioned pharmacological compounds were based on the following rat intra-cranial microinjection experiments: Ro 31-8220 mesylate (Loweth et al., 2009; Schmidt et al., 2014; Schmidt et al., 2013), chelerythrine chloride (Li et al., 2011; Narita et al., 2004; Schmidt et al., 2014; Schmidt et al., 2013), R(+)-SKF-81297 hydrobromide (Schmidt et al., 2006; Schmidt and Pierce, 2006a) and (−)-quinpirole hydrochloride(Schmidt et al., 2006; Schmidt and Pierce, 2006a). While the doses of chelerythrine chloride and Ro 31-8220 have been shown to have behavioral and biochemical effects specific to PKC inhibition in the rat brain, it is possible that these compounds could be influencing activity of other signaling molecules in our experiments.
Statistics
For the cocaine reinstatement experiments utilizing Ro 31-8220 and dopamine receptor agonists in the NAc shell, the total mean active and inactive lever responses were analyzed with separate one-way ANOVAs. Pairwise comparisons were made with Tukey’s HSD (P<0.05) following one-way ANOVAs. The mean±SEM of eEPSCs and eIPSCs were based on an average of 10 traces for single stimulations and on an average of 3 traces for stimulation trains. Electrophysiological experiments were analyzed with paired t-tests or ANOVAs with Tukey’s HSD post-hoc comparisons as indicated (P<0.05).
Results
Pharmacological inhibition of PKC in the NAc shell attenuates D2DR, but not D1DR, agonist-induced reinstatement of cocaine seeking
Microinjection sites for the rat behavioral experiments are shown in Figure 1A. Total lever responses (mean± SEM) following intra-accumbens shell administration of the D1DR agonist SKF-81297 are shown in Figure 1B. Active lever responses were analyzed with a one-way ANOVA, which revealed a significant main effect of treatment [F(2,23) = 26.11, p<0.0001]. Subsequent post hoc analyses showed a significant difference in total active lever responses between vehicle (n = 9) and 1.0 μg SKF-81297 (n = 8) and 10.0 μM Ro 31-8220 + 1.0 μg SKF-81297 (n = 9) treatments (Tukey’s HSD, p < 0.05). No significant differences on inactive lever responding were found between treatments [F(2,23) = 1.56, p < 0.24]. Total active lever responses (mean± SEM) following intra-accumbens shell administration of the D2DR agonist, quinpirole, are shown in Figure 1C. These data were analyzed with a one-way ANOVA, which revealed a significant main effect of treatment [F(2,26) = 24.01, p<0.0001]. Subsequent pairwise analyses revealed a significant difference in total active lever responses between 3.0 μg quinpirole (n = 10) and vehicle (n = 10) and 10.0 μM Ro 31-8220 + 3.0 μg quinpirole (n = 9) treatments (Tukey’s HSD, p<0.05). Inactive lever responses were analyzed with a one-way ANOVA, which revealed a significant main effect of treatment [F(2,26) = 3.52, p = 0.044]. However, further pairwise analyses did not reveal a significant difference in responding on the inactive lever among treatments (Tukey’s HSD, p > 0.05
Figure 1. Microinjection of a PKC inhibitor into the NAc shell blocks D2DR, but not D1DR, agonist-induced reinstatement of cocaine seeking.

(A) Coronal sections depicting microinjection sites, as indicated by closed circles, targeting the medial NAc shell in the dopamine receptor agonist experiments. Numbers on the left side of the coronal sections denote distance from bregma in the anteroposterior direction. (B) Total number of lever responses during the reinstatement test session following intra-accumbens shell administration of vehicle, 1.0 μg SKF-81297 or 10.0 μM Ro31-8220 + 1.0 μg SKF-81297. There was a significant increase in active lever responding in animals treated with 1.0 μg SKF-81297 or 10.0 μM Ro 31-8220 + 1.0 μg SKF-81297 when compared to vehicle treated controls (Tukey’s HSD, *p<0.05). (C) Total lever responses following intra-accumbens shell infusion of vehicle, 3.0 μg quinpirole or 10.0 μM Ro 31-8220 + 3.0 μg quinpirole. There was a significant increase in active lever responding in animals treated with 3.0 μg quinpirole when compared to animals treated with vehicle or 10.0 μM Ro 31-8220 + 3.0 μg quinpirole (Tukey’s HSD, *p<0.05). No significant differences in inactive lever responding were observed in either the SKF-81297 or quinpirole experiments.
PKC inhibition does not affect action potential firing in NAc shell D1DR-positive and D2DR-positive MSNs
To investigate the effects of PKC inhibitors on neuronal excitability, we made a simplifying assumption that D1DR agonist-induced reinstatement is mediated by the D1DR-positive MSNs in the NAc shell and D2DR agonist-induced reinstatement is mediated by D2DR-positive MSNs in the NAc shell. We utilized a transgenic mouse line, engineered to express tdTomato and GFP fluorescent tags in D1DR-positive and D2DR-positive neurons, respectively, to distinguish between the two cell populations (see Methods). Mice were trained to self-administer cocaine and underwent a period of extinction similar to cocaine-experienced rats and all electrophysiology experiments were conducted in mice. To approximate the conditions of elevated dopamine receptor signaling during reinstatement, an extracellular solution containing dopamine (75 μM) was applied to the slice. Dopamine at this concentration has been shown tomodulate both excitatory and inhibitory synaptic signaling in slice preparations (Hjelmstad, 2004; Nicola and Malenka, 1997). Exposure to dopamine increased the average firing frequency in response to depolarizing current steps to 121±4% in D1DR-positive and to 110±5% in D2DR positive MSNs, a smaller effect than that reported in NAc shell MSNs from younger animals (Hopf et al., 2003; Hopf et al., 2005). However, inhibition of PKC had little to no effect on action potential frequency across a range of injected currents (Figure 2) and did not change action potential rheobase (D1DR-positive MSNs: 70±28 pA and 70±23 pA without and with PKC antagonists, respectively; D2DR-positive MSNs: 84±15 pA and 84±18 pA without and with PKC antagonists, respectively). These results indicate that pharmacological inhibition of PKC does not markedly affect action potential firing in NAc shell D1DR-positive and D2DR-positive MSNs in mice whose cocaine self-administration had been extinguished.
Figure 2. Action potential firing in NAc shell D1DR- and D2DR-positive MSNs is not affected by PKC inhibition.
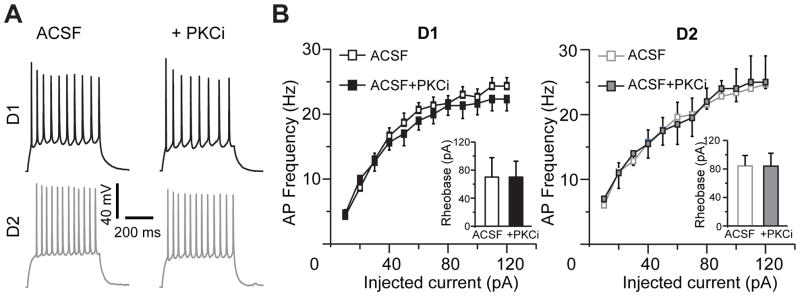
(A) Representative traces of action potentials elicited from the same D1DR- and D2DR-positive neuron in the NAc shell before and after PKC blockade. Responses to a current step of 60 pA above rheobase are shown in each cell. (B) The firing rate vs. input (f-I) curves illustrate minimal or no effect of PKC inhibition in D1DR-positive (left) or D2DR-positive (right) MSNs (n=5–7 cells from 2 animals). Insets, PKC blockade is likewise without effect on rheobase current in both cell types. PKCi, PKC inhibitor (chelerythrine (10 μM) or Ro 31-8220 (10 μM)).
PKC inhibition exerts similar effects on synaptic current amplitude in NAc shell D1DR-positive and D2DR-positive MSNs
We next focused on the effects of PKC inhibitors on synaptic signaling. AMPA receptor-mediated eEPSCs, triggered by single pulses of electrical stimulation, were isolated in the presence of dopamine (75 μM) by holding the cells close to the GABAA receptor reversal potential (Vh=−70 mV). Inhibition of PKC signaling attenuated the peak eEPSC amplitude to 75±6% of baseline in D1DR-positive MSNs (t(7)=3.49, p=0.01) and to 60.9±7% of baseline in D2DR-positive MSNs (t(6)=5.18, p=0.002) with no significant difference between the D1DR-positive and D2DR-positive MSNs (Figure 3). We then held the cells close to the reversal potential for eEPSC (Vh=0 mV) to isolate inhibitory postsynaptic responses mediated by the GABAA receptors. The effect of PKC blockade on eIPSCs was also not significantly different between the D1DR-positive and D2DR-positive MSNs although PKC antagonists slightly increased the mean eIPSC amplitude in the D1DR-positive neurons (to 112±10.9% of baseline) and slightly decreased the mean eIPSC amplitude in the D2DR-positive neurons (to 88.7±6.6% of baseline; Figure 4). We combined the effects of PKC inhibition on inhibitory and excitatory signaling by computing a ratio of eEPSC to eIPSC amplitudes. In the absence of PKC inhibition, eEPSC/eIPSC ratio was 0.64±0.1 in D1DR-positive MSNs and 0.67±0.15 in D2DR-positive MSNs. In the presence of PKC blockers, the eEPSC/eIPSC ratio was significantly reduced in both cell types to 0.54±0.12 in D1DR-positive MSNs (t(7)=2.78, p=0.027) and to 0.44±0.13 in D2DR-positive MSNs (t(7)=3.48, p=0.01). This corresponded to a 78.1±7.7% of baseline reduction in D1DR-positive MSNs and to a 66.4±7.4% of baseline reduction in D2DR-positive MSNs with no difference between the two cell types. We conclude that effects of PKC inhibition on excitatory and inhibitory synaptic activity evoked by single stimuli do not reliably discriminate between D1DR-positive and D2DR-positive MSNs in the NAc shell of mice whose cocaine self-administration had been extinguished.
Figure 3. PKC inhibition suppresses single stimulus eEPSCs in both D1DR- and D2DR-positive NAc shell MSNs.
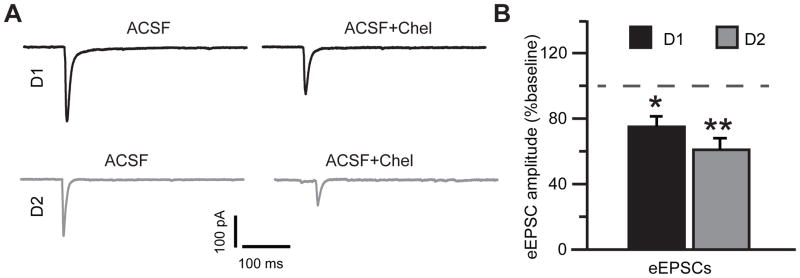
(A) eEPSC average traces from the same D1DR- and D2DR-positive cell in the absence and in the presence of PKC blockade. Stimulus artifacts were removed for illustration. (B) Peak amplitude of eEPSCs in D1DR- and D2DR-positive MSNs in the presence of PKC blockade is plotted as a percent of response recorded prior to PKC antagonist application. Note similar inhibition of eEPSC amplitude in both cell types (paired Student’s t-test, *, p<0.05; **, p<0.01; n=8–9 cells from 3 animals).
Figure 4. GABAA receptor-mediated single stimulus eIPSCs are not markedly affected by PKC inhibition.
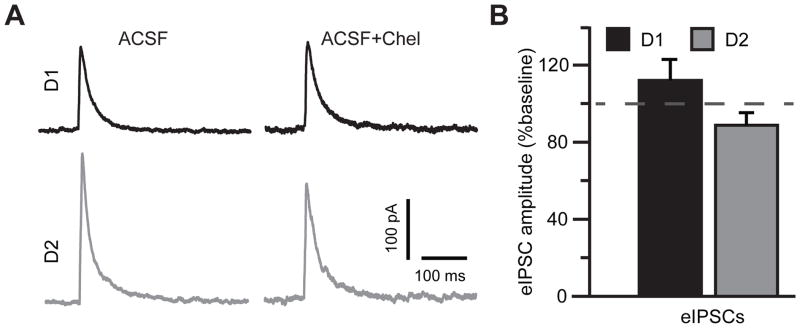
(A) eIPSC average traces from the same D1DR- and D2DR-positive cell in the absence and in the presence of PKC blockade. Stimulus artifacts were removed for illustration. (B) No significant differences in eIPSCs amplitude were observed in response to PKC antagonist application. Note, however, a trend toward PKCi-induced increase in D1DR-positive MSNs and PKC-induced decrease in D2DR-positive MSNs (n=8 cells from 3 animals).
Activity-dependent modulation of synaptic fidelity by PKC in D2DR, but not D1DR, -positive MSNs in the NAc shell
MSNs of the NAc fire across a broad frequency range (O’Donnell and Grace, 1993) and there are indications that the effect of dopamine on the ability of these cells to generate reliable postsynaptic responses varies across stimulation frequencies and is different for eEPSCs and eIPSCs (Hjelmstad, 2004). To test if D1DR-positive and D2DR-positive MSNs may respond differently to PKC blockade under conditions of continuous stimulation, we applied trains of 10 pulses at 1, 10, or 50 Hz and measured synaptic ability to sustain responding (i.e. synaptic fidelity) as a ratio of peak current amplitude generated by the last pulse to peak current amplitude generated by the first pulse. Under these conditions, PKC blockade did not affect fidelity of excitatory synaptic transmission in D1DR-positive MSNs: PKC inhibition changed the eEPSC10/eEPSC1 to 104.7±8.5%, 91.5±9.5%, and 103±10% of baseline at 1, 10, and 50 Hz, respectively (Figure 5A & B). However, there was a consistent increase in the ability of excitatory synapses to sustain responding to stimulus trains in D2DR-positive MSNs. In these neurons, PKC inhibition increased the eEPSC10/eEPSC1 to 116.1±6.8%, 124±6%, and 114.4±5.1% at 1, 10, and 50 Hz, respectively (Figure 5A & B). Two-way ANOVA of these data (with stimulation frequency and cell treatment as factors) revealed a significant effect of stimulation frequency (F(2,112)=25.99, p<0.001) and no significant effect of cell treatment (F(3,112)=2.35, p=0.08). Tukey’s post-hoc comparisons indicated a significant difference between D1+PKCi and D2+PKCi groups at 10 Hz (p<0.05). In the absence of PKC inhibition, there were no differences in eEPSC10/eEPSC1 between D1+ MSNs (1Hz: 0.94±0.08; 10 Hz: 0.67±0.05; 50 Hz: 0.68±0.11) and D2+ MSNs (1 Hz: 0.96±0.05; 10 Hz: 0.74±0.05; 50 Hz: 0.57±0.05). PKC blockade had a similar effect on eIPSC10/eIPSC1 across all treatments in both cell types (F(3, 108)=0.06, p=0.98; Figure 5A & C). In the absence of PKC inhibition, there were no differences in eIPSC10/eIPSC1 between D1+ MSNs (1 Hz: 0.77±0.05; 10 Hz: 0.62±0.05; 50 Hz: 1.04±0.15) and D2+ MSNs (1 Hz: 0.69±0.06; 10 Hz: 0.63±0.06; 50 Hz: 1.14±0.12).
Figure 5. Trains of stimuli differentially modulate synaptic excitation in NAc shell D1DR- and D2DR-positive MSNs following PKC blockade.

(A) Top, representative eEPSCs elicited by a 1 second train of stimuli at 10 Hz in D1DR- and D2DR-positive MSNs. Thick, black traces recorded in the presence of PKC inhibition are overlaid onto thin gray traces recorded in the absence of PKC inhibition. PKC inhibition induces a consistent increase in eEPSC amplitude throughout the stimulus train seen in D2DR-, but not D1DR-positive MSNs. Stimulus artifacts were removed for illustration. Responses in each overlaid set of traces are normalized to the peak amplitude of the first pulse in the absence of PKC inhibition for ease of comparison. Bottom, no consistent response to PKC inhibition could be observed for eIPSCs in D1DR- and D2DR-positive MSNs. (B) An activity-dependent effect on eEPSC trains is expressed as a ratio of the last (eEPSC10) to first (eEPSC1) current response at 3 frequencies of stimulation. The difference between D2DR+PKCi and D1DR+PKCi groups is significant (*, p<0.05, Tukey’s post-hoc; n=9–12 cells from 4–5 animals). (C) Same as (B), but for eIPSCs. No significant differences were found (n=8–12 cells from 3–5 animals).
Dopamine is known to modulate both excitatory and inhibitory synaptic signaling at concentrations similar to those used in extracellular solutions in our study (Hjelmstad, 2004; Nicola and Malenka, 1997). Therefore, we evaluated whether dopamine receptor activation was necessary for discrete effects of PKC at D1DR- and D2DR-positive MSNs in the accumbens shell. In the absence of bath-applied dopamine, PKC inhibition decreased the amplitude of eEPSCs evoked by single stimuli to 79.1±8.9% in 4 out of 6 D1DR-positive MSNs, similar to that observed in the presence of dopamine (Figure 3). In 2 out of 6 D1DR-positive MSNs, PKC blockade increased the eEPSC amplitude to 180.4% and 137.1%. In D1DR-positive MSN, PKC blockade reduced single-pulse eEPSCs to 70.5±8.9% in 4 out of 5 cells and increased the eEPSC amplitude to 107.2% in 1 cell. There were no significant differences when results from all D1DR-positive cells were compared to all D2DR-positive cells (D1DR-positive:105.6±18.6%; D2DR-positive: 77.8±9.9%; t(9)=1.24, p=0.24). PKC blockade reduced the amplitude of single-pulse eIPSCs in D1DR-positive MSN (to 83.8±4.2%, n=6) and in D2DR-positive MSNs (to 95±8.7%, n=5, including two cells with an increase to 115% and 110%). These mean differences were not significant between cell types (t(9)=1.23, p=0.25). Note, however that PKC antagonists tended to potentiate single-pulse eIPSCs in D1DR-positive MSNs, in the presence of bath-applied dopamine (Figure 3). For synaptic currents triggered by stimulus trains, two-way ANOVAs revealed a significant effect of stimulation frequency for both eEPSCs (F(2,119)=11.39, p<0.01) and eIPSCs (F(2,116)=11.18, p<0.01), but no other significant differences (Figure 6). These data indicate that while effects of PKC blockade on eEPSCs triggered by single stimuli do not require elevated levels of dopamine receptor activation, the ability of PKC to influence sustained synaptic responding during stimulation trains does.
Figure 6. Stimulus trains do not affect fidelity of synaptic signaling in the absence of extracellular dopamine.

(A) Top, representative eEPSCs elicited by a 1 second train of stimuli at 10 Hz in D1DR- and D2DR-positive MSNs. Thick, black traces recorded in the presence of PKC inhibition are overlaid onto thin gray traces recorded in the absence of PKC inhibition. PKC inhibition has similar effects on synaptic responding in D1DR- and D2DR-positive MSNs. Stimulus artifacts were removed for illustration. Responses in each overlaid set of traces are normalized to the peak amplitude of the first pulse in the absence of PKC inhibition for ease of comparison. Bottom, eIPSC responses to PKC inhibition are also similar in D1DR- and D2DR-positive MSNs. (B) Ratios of the last (eEPSC10) to first (eEPSC1) current response are plotted at 3 frequencies of stimulation. There are no significant differences between any of the groups (n=9–12 cells from 6 animals). (C) Same as (B), but for eIPSCs. No significant differences were found between groups (n=10–12 cells from 6 animals).
To investigate whether the observed frequency-dependence of synaptic transmission in the presence of bath-applied dopamine could be explained by differential modulation of presynaptic glutamate release probability by PKC blockers in the D2DR-positive MSNs, we measured the paired-pulse ratio (PPR) of eEPSCs and eIPSCs. We found no differences in the PPR between the D1DR-positive and D2DR-positive MSNs following PKC inhibition on either the excitatory or the inhibitory postsynaptic currents (Figure 7). Overall, these results indicate that PKC inhibition following extinction preferentially increases the fidelity of excitatory synaptic transmission in D2DR-positive MSNs of the NAc shell, an effect that is particularly prominent for moderate (10 Hz) intensity stimuli. Furthermore, this effect cannot be attributed to PKC modulation of presynaptic probability of glutamate release as measured by the paired-pulse ratio.
Figure 7. PKC inhibition does not affect the paired-pulse ratio of excitatory and inhibitory PSCs in MSNs of the NAc shell.
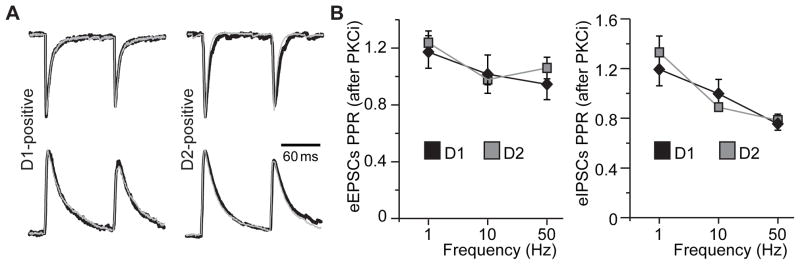
(A) eEPSCs (top traces) or eIPSCs (bottom traces) evoked by a 10 Hz paired-pulse stimulus. Overlays of traces in the absence (thick black traces) and in the presence (thin gray traces) of PKC inhibition are pictured. Responses in each overlaid set of traces are normalized to the peak amplitude of the first pulse in the absence of PKC inhibition for ease of comparison. Stimulus artifacts were removed for illustration. (B) Paired-pulse ratios of eEPSCs (left) and eIPSCs (right) after PKC inhibition are plotted for D1DR- and D2DR-positive MSNs at indicated paired-pulse frequencies. No differences were found between any of the groups (n=8–9 cells from 3 animals).
Discussion
The present study represents the first investigation of the role of PKC-mediated signaling in expression of behavioral and neuronal responses to D1-like and D2-like dopamine receptor stimulation in the NAc shell following extinction of cocaine self-administration. Here, we demonstrate for the first time that (1) stimulation of D2DRs in the NAc shell reinstates cocaine seeking, at least in part, through activation of PKC, while D1DR agonist-mediated reinstatement is PKC independent, (2) pharmacological inhibition of PKC exerts similar modulatory effects on excitatory and inhibitory synaptic activity evoked by single electrical pulses in NAc shell D1DR-positive and D2DR-positive MSNs during extinction, and (3) blocking PKC signaling preferentially facilitates excitatory, but not inhibitory, activity evoked by synaptic train stimulation in D2DR-, but not D1DR-, positive MSNs in the NAc shell of mice whose cocaine self-administration was extinguished. Taken together, these results suggest that stimulation of NAc shell D2DRs activates PKC to promote cocaine seeking via an activity-dependent decrease in the fidelity of excitatory synaptic signaling in D2DR-positive MSNs. These data support the hypothesis that the net effect of activating D2DR-PKC signaling in the NAc shell is to suppress D2DR-mediated output to the ventral pallidum thereby biasing MSN signaling in the NAc toward D1DR-mediated excitation. Future studies necessary to directly evaluate the role of such bias in the reinstatement of cocaine-seeking behavior and will require manipulation of synaptic activity in vivo.
Our results contribute to and expand upon previous studies demonstrating a role for NAc PKC in cocaine seeking. There is clear evidence that both D1DRs and D2DRs in the NAc shell play critical roles in the reinstatement of cocaine seeking (Anderson et al., 2003; Anderson et al., 2006; Bachtell et al., 2005; Schmidt et al., 2006; Schmidt and Pierce, 2006a, b). Consistent with these studies, we show that administration of a D1DR or D2DR agonist into the NAc shell is sufficient to reinstate cocaine seeking. While it is clear that stimulation of D1DRs and D2DRs in the NAc shell promotes cocaine seeking, the role of non-traditional secondary effectors in this behavioral response is largely unknown. D1DRs and D2DRs are traditionally thought to exert opposing influences on 3′, 5′-cyclic AMP (cAMP)-dependent signaling through their associated heterotrimeric GTP-binding proteins (G proteins)(Missale et al., 1998). D1DRs are coupled to stimulatory G proteins (Gsα or Golfα) that activate adenylyl cyclase and increase the hydrolysis of ATP to cAMP (Sibley et al., 1993). In contrast, D2DRs are coupled to inhibitory G proteins (Giα or Goα) that inhibit adenylyl cyclase, thus decreasing or having no effect on cAMP (Sibley et al., 1993). D1DR and D2DR signaling, however, is more complex in that other signaling cascades can be influenced in the same way following stimulation of D1DRs and D2DRs. For example, stimulation of D1DRs and D2DRs also activates phospholipase C (PLC) and PKC (Lee et al., 2004; Neve et al., 2004; Pollack, 2004), which regulate excitation of MSNs in the NAc shell (Hopf et al., 2005). While recent evidence indicates an important role for NAc PKC in cocaine seeking (Schmidt et al., 2014; Schmidt et al., 2013), no studies have examined dopamine receptor-PKC signaling in reinstatement of cocaine seeking. Here, we show that pharmacological inhibition of PKC in the NAc shell markedly attenuates cocaine seeking induced by intra-accumbens shell administration of a D2DR, but not D1DR, agonist. These findings indicate that D2DR-facilitated cocaine seeking is mediated by PKC activation in the NAc shell and are consistent with studies demonstrating that D2DRs stimulate PKC signaling via activation of Gi protein-coupled receptor βγ subunits (Choi et al., 1999; Liu et al., 1992; Morris et al., 2007). Our recent studies indicate that cocaine priming-induced reinstatement of drug seeking is associated with activation of PKCγ in the NAc and intra-accumbens PKC inhibitors suppressed cocaine seeking (Schmidt et al., 2014; Schmidt et al., 2013). Together with the present findings, these data suggest that cocaine seeking is mediated, in part, through D2DR-mediated activation of PKCγ in the NAc shell.
In addition to PKC, D2DRs modulate activity of ion channels via G protein-coupled βγ subunits. Stimulation of D2DRs activates G protein-coupled inward rectifying potassium (GIRK) channels (Greif et al., 1995; Lacey et al., 1987; Petit-Jacques et al., 1999; Pillai et al., 1998) and inhibits several types of calcium channels (Okada et al., 2003; Ramanathan et al., 2008; Zamponi and Snutch, 1998). All of these ion channels are important elements controlling cellular excitability that can modulate action potential generation and influence behavior. Indeed, similar to previous reports (Hopf et al., 2003; Hopf et al., 2005) we find that extracellular dopamine increases the MSN firing rate in response to depolarizing stimuli. However, in contrast to Hopf et al. (2005) our data indicate that inhibition of PKC did not affect the f-I curve of action potential responses or the action potential rheobase in either D1DR-positive or D2DR-positive MSNs in the NAc shell of animals whose cocaine taking had been extinguished. This discrepancy could be due to the effects of cocaine self-administration and extinction training on mechanisms coupling dopamine receptor activation to action potential generation or on mechanisms involved in dopamine receptor interaction with PKC. Compensatory interactions between multiple targets of intracellular signaling cascades engaged by D2DR activation (Neve et al., 2004; Nicola et al., 2000) could also contribute to the behavioral consequences of PKC blockade on reinstatement of cocaine seeking.
Multiple reports find that PKC activation by diacylglycerol or phorbol esters increases the amplitude of spontaneous and evoked EPSCs (Francis et al., 2002; Korogod et al., 2007; Lou et al., 2008; Wierda et al., 2007; however see, Chen and Roper, 2003). Consistent with these findings, pharmacological inhibition of PKC in the NAc shell both in the absence and in the presence of dopamine receptor stimulation by extracellular dopamine reduced the amplitude of AMPA receptor-mediated evoked excitatory currents in D1DR-positive and the majority of D2DR-positive MSNs. In contrast, GABAA receptor-mediated evoked inhibitory currents were not significantly affected by PKC inhibition, although there was a non-significant trend toward increased eIPSC amplitude in D1DR-positive MSNs, but decreased eIPSC amplitude in D2DR-positive MSNs. These results suggest that PKC may differently modulate gating of AMPA and GABAA receptors in NAc shell MSNs following cocaine self-administration. Alternatively, the difference in PKC effects on the amplitude of evoked excitatory and inhibitory responses could be attributed to distinct pre-synaptic modulation of excitatory versus inhibitory afferents to NAc shell D1DR-positive and D2DR-positive MSNs.
Modulation of pre-synaptic release by PKC has been repeatedly demonstrated at a number of synapses with some disagreement over whether PKC affects the probabilities of vesicular fusion and release or the size of the readily releasable pool of vesicles (Basu et al., 2007; Chu et al., 2012; Lou et al., 2008; Stevens and Sullivan, 1998; Wierda et al., 2007). Using train stimulations, our data indicate activity-dependent differences in AMPAR-mediated eEPSCs at excitatory afferents onto D1DR-positive and D2DR-positive MSNs in the NAc shell of cocaine-experienced animals. Inhibition of PKC improves the ability of NAc shell D2DR-positive MSNs to sustain excitatory synaptic responses, most prominently in the 10 Hz frequency range. In contrast, there was no effect of PKC inhibition on eEPSCs evoked by stimulus trains at D1DR-positive MSNs. Additionally, PKC inhibition did not affect the paired-pulse ratio, a measure of presynaptic probability of release, in D1DR-positive or D2DR-positive MSNs. The latter finding does not agree with more sophisticated analyses postulating a role of PKC in modulating pre-synaptic pools of releasable neurotransmitter vesicles (see Stevens and Sullivan, 1998). This discrepancy may reflect limitations of the paired-pulse ratio as an independent measure of pre-synaptic release mechanisms, in particular, given the complicating factors such as fluctuations in vesicular pool size, rapid vesicle refilling, activity-dependent decreases of presynaptic Ca2+ transients, and others (Kirischuk et al., 2002; Zucker and Regehr, 2002). Regardless of the involvement of pre-synaptic mechanisms, activity-dependent regulation of AMPAR-mediated eEPSCs by PKC suggests that modulation of neuronal activity levels in the NAc associated with psychostimulant exposure (Carelli and Ijames, 2000; Peoples and Cavanaugh, 2003; Peoples et al., 2007) may determine the impact of PKC signaling on cocaine-mediated behavioral plasticity.
Collectively, these results suggest that D2DR agonists promote cocaine seeking by suppressing neuronal activity in D2DR-expressing NAc shell MSNs. Although our report establishes only an indirect link between PKC modulation of synaptic activity and PKC modulation of cocaine reinstatement, a number of previous findings support this hypothesis. First, both in vitro and in vivo studies demonstrate suppression of excitatory signaling in the NAc by D2DR agonists (Benoit-Marand and O’Donnell, 2008; Brady and O’Donnell, 2004). Second, D2DR agonists have been shown to suppress excitatory transmission through PKC-dependent regulation of AMPA receptors (Anggono and Huganir, 2012; McCutcheon et al., 2011). PKC phosphorylates GluA2 subunits at Ser880, which influences trafficking of GluA2-containing AMPA receptors (Shepherd and Huganir, 2007; Song and Huganir, 2002). Third, increased phosphorylation of GluA2 at Ser880 (Famous et al., 2008) and decreased surface expression of GluA2 (Wiggins et al., 2011) in the NAc is associated with cocaine priming-induced reinstatement of drug seeking. In contrast, increased surface expression of GluA1-containing AMPA receptors in NAc MSNs promotes cocaine seeking (Anderson et al., 2008; Loweth et al., 2013; Schmidt and Pierce, 2010). Since the vast majority of AMPA receptors in the NAc contain both GluA1 and GluA2 subunits (Boudreau et al., 2007), it is possible that cocaine seeking is driven by increased surface expression of GluA1-containing AMPA receptors in D1DR-positive MSNs and endocytosis of GluA2-containing AMPA receptors primarily in NAc outputs that express D2DRs. Our findings suggest that phosphorylation events associated with D2DR-induced activation of PKC may alter the dynamics of AMPAR signaling to decrease the fidelity of AMPA receptor-mediated eEPSCs in D2DR-positive MSNs of the NAc shell and to promote cocaine seeking.
Conclusion
Our experiments identify a role for NAc shell PKC signaling in D2DR, but not D1DR, agonist-induced reinstatement of cocaine seeking. These results support the hypothesis that cocaine seeking may result from differential effects of cocaine on the major output pathways of the NAc, which express D1DRs and D2DRs relatively selectively (Gerfen et al., 1990; Lu et al., 1998). Furthermore, we show that activation of PKC associated with D2DR stimulation reduces excitatory synaptic signaling in D2DR-positive MSNs of the NAc shell. Excitation of D2DRs inhibits NAc efferents to the ventral pallidum (indirect pathway), whereas stimulation of D1DRs activates NAc outputs to the VTA (direct pathway) (Albin et al., 1995; Surmeier et al., 2007). It is possible that cell type-specific effect of PKC observed in our study developed as a result of particular behavioral history of cocaine exposure rather than reflecting a more general impact of PKC signaling on excitatory neurotransmission at D1DR-positive and D2DR-positive neurons. For example, cocaine self-administration alone or subsequent cocaine abstinence during extinction may have altered the strength of PKC regulation of excitatory neurotransmission in D2DR-positive cells. Our results indicate, however, that following extinction of cocaine taking, increased PKC signaling in D2DR-positive MSNs in the NAc shell may reduce information flow through the D2DR-mediated indirect pathway thereby biasing NAc signaling in favor of D1DR-mediated, direct pathway output to promote reinstatement behavior.
Blockade of PKC signaling attenuates reinstatement of cocaine-seeking
PKC inhibition has distinct effects on excitatory signaling in D1DR and D2DR MSNs
Reinstatement of cocaine seeking partly relies on D2DR increases in PKC signaling
Acknowledgments
This work was supported by NIDA grants DA031747 (P.I.O.), DA033372 (L.A.B.), DA22339 and DA18678 (R.C.P.), DA30445 (H.D.S.).
Footnotes
Conflict of Interest
The authors declare no potential conflict of interest relating to this study.
Authors’ Contributions
P.I.O. was responsible for designing, performing and analyzing the electrophysiology data as well as drafting the manuscript. L.A.B. contributed to the acquisition of the animal data and editing of the manuscript. R.C.P. was also responsible for the study design and provided critical revisions of the manuscript. H.D.S was responsible for the study concept and design, supervised and contributed to the acquisition of behavioral data, analyzed the data and drafted the manuscript. All authors reviewed content and approved the final version for publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albin RL, Young AB, Penney JB. The functional anatomy of disorders of the basal ganglia. Trends Neurosci. 1995;18:63–64. [PubMed] [Google Scholar]
- Anderson SM, Bari AA, Pierce RC. Administration of the D1-like dopamine receptor antagonist SCH-23390 into the medial nucleus accumbens shell attenuates cocaine priming-induced reinstatement of drug-seeking behavior in rats. Psychopharmacology (Berl) 2003;168:132–138. doi: 10.1007/s00213-002-1298-5. [DOI] [PubMed] [Google Scholar]
- Anderson SM, Famous KR, Sadri-Vakili G, Kumaresan V, Schmidt HD, Bass CE, Terwilliger EF, Cha JH, Pierce RC. CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat Neurosci. 2008;11:344–353. doi: 10.1038/nn2054. [DOI] [PubMed] [Google Scholar]
- Anderson SM, Pierce RC. Cocaine-induced alterations in dopamine receptor signaling: implications for reinforcement and reinstatement. Pharmacol Ther. 2005;106:389–403. doi: 10.1016/j.pharmthera.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Anderson SM, Schmidt HD, Pierce RC. Administration of the D2 dopamine receptor antagonist sulpiride into the shell, but not the core, of the nucleus accumbens attenuates cocaine priming-induced reinstatement of drug seeking. Neuropsychopharmacology. 2006;31:1452–1461. doi: 10.1038/sj.npp.1300922. [DOI] [PubMed] [Google Scholar]
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012 doi: 10.1016/j.conb.2011.12.006. Epub 2 Jan 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aujla H, Beninger RJ. Intra-accumbens protein kinase C inhibitor NPC 15437 blocks amphetamine-produced conditioned place preference in rats. Behav Brain Res. 2003;147:41–48. doi: 10.1016/s0166-4328(03)00136-0. [DOI] [PubMed] [Google Scholar]
- Bachtell RK, Whisler K, Karanian D, Self DW. Effects of intra-nucleus accumbens shell administration of dopamine agonists and antagonists on cocaine-taking and cocaine-seeking behaviors in the rat. Psychopharmacology (Berl) 2005;183:41–53. doi: 10.1007/s00213-005-0133-1. [DOI] [PubMed] [Google Scholar]
- Basu J, Betz A, Brose N, Rosenmund C. Munc13-1 C1 domain activation lowers the energy barrier for synaptic vesicle fusion. J Neurosci. 2007;27:1200–1210. doi: 10.1523/JNEUROSCI.4908-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Benoit-Marand M, O’Donnell P. D2 dopamine modulation of corticoaccumbens synaptic responses changes during adolescence. Eur J Neurosci. 2008;27:1364–1372. doi: 10.1111/j.1460-9568.2008.06107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock R, Shin JH, Kaplan AR, Dobi A, Markey E, Kramer PF, Gremel CM, Christensen CH, Adrover MF, Alvarez VA. Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat Neurosci. 2013;16:632–638. doi: 10.1038/nn.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady AM, O’Donnell P. Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo. J Neurosci. 2004;24:1040–1049. doi: 10.1523/JNEUROSCI.4178-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carelli RM, Ijames SG. Nucleus accumbens cell firing during maintenance, extinction, and reinstatement of cocaine self-administration behavior in rats. Brain Res. 2000;866:44–54. doi: 10.1016/s0006-8993(00)02217-4. [DOI] [PubMed] [Google Scholar]
- Cervo L, Mukherjee S, Bertaglia A, Samanin R. Protein kinases A and C are involved in the mechanisms underlying consolidation of cocaine place conditioning. Brain Res. 1997;775:30–36. doi: 10.1016/s0006-8993(97)00866-4. [DOI] [PubMed] [Google Scholar]
- Chen HX, Roper SN. PKA and PKC enhance excitatory synaptic transmission in human dentate gyrus. J Neurophysiol. 2003;89:2482–2488. doi: 10.1152/jn.01031.2002. [DOI] [PubMed] [Google Scholar]
- Chen Q, Lee TH, Wetsel WC, Sun QA, Liu Y, Davidson C, Xiong X, Ellinwood EH, Zhang X. Reversal of cocaine sensitization-induced behavioral sensitization normalizes GAD67 and GABAA receptor alpha2 subunit expression, and PKC zeta activity. Biochem Biophys Res Commun. 2007;356:733–738. doi: 10.1016/j.bbrc.2007.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi EY, Jeong D, Park KW, Baik JH. G protein-mediated mitogen-activated protein kinase activation by two dopamine D2 receptors. Biochem Biophys Res Commun. 1999;256:33–40. doi: 10.1006/bbrc.1999.0286. [DOI] [PubMed] [Google Scholar]
- Chu Y, Fioravante D, Thanawala M, Leitges M, Regehr WG. Calcium-dependent isoforms of protein kinase C mediate glycine-induced synaptic enhancement at the calyx of Held. J Neurosci. 2012;32:13796–13804. doi: 10.1523/JNEUROSCI.2158-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng L-J, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famous KR, Kumaresan V, Sadri-Vakili G, Schmidt HD, Mierke DF, Cha JH, Pierce RC. Phosphorylation-dependent trafficking of GluR2-containing AMPA receptors in the nucleus accumbens plays a critical role in the reinstatement of cocaine seeking. J Neurosci. 2008;28:11061–11070. doi: 10.1523/JNEUROSCI.1221-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis HW, Scott JC, Manis PB. Protein kinase C mediates potentiation of synaptic transmission by phorbol ester at parallel fibers in the dorsal cochlear nucleus. Brain Res. 2002;951:9–22. doi: 10.1016/s0006-8993(02)03095-0. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Greif GJ, Lin YJ, Liu JC, Freedman JE. Dopamine-modulated potassium channels on rat striatal neurons: specific activation and cellular expression. J Neurosci. 1995;15:4533–4544. doi: 10.1523/JNEUROSCI.15-06-04533.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmstad GO. Dopamine Excites Nucleus Accumbens Neurons through the Differential Modulation of Glutamate and GABA Release. Journal of Neuroscience. 2004;24:8621–8628. doi: 10.1523/JNEUROSCI.3280-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf FW, Cascini MG, Gordon AS, Diamond I, Bonci A. Cooperative activation of dopamine D1 and D2 receptors increases spike firing of nucleus accumbens neurons via G-protein betagamma subunits. J Neurosci. 2003;23:5079–5087. doi: 10.1523/JNEUROSCI.23-12-05079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf FW, Mailliard WS, Gonzalez GF, Diamond I, Bonci A. Atypical protein kinase C is a novel mediator of dopamine-enhanced firing in nucleus accumbens neurons. J Neurosci. 2005;25:985–989. doi: 10.1523/JNEUROSCI.3099-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk S, Clements JD, Grantyn R. Presynaptic and postsynaptic mechanisms underlie paired pulse depression at single GABAergic boutons in rat collicular cultures. J Physiol. 2002;543:99–116. doi: 10.1113/jphysiol.2002.021576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Posttetanic potentiation critically depends on an enhanced Ca(2+) sensitivity of vesicle fusion mediated by presynaptic PKC. Proc Natl Acad Sci U S A. 2007;104:15923–15928. doi: 10.1073/pnas.0704603104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachowicz JE, Sibley DR. Molecular characteristics of mammalian dopamine receptors. Pharmacol Toxicol. 1997;81:105–113. doi: 10.1111/j.1600-0773.1997.tb00039.x. [DOI] [PubMed] [Google Scholar]
- Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lanca AJ, O’Dowd BF, George SR. Dopamine D1 and D2 receptor Co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem. 2004;279:35671–35678. doi: 10.1074/jbc.M401923200. [DOI] [PubMed] [Google Scholar]
- Li YQ, Xue YX, He YY, Li FQ, Xue LF, Xu CM, Sacktor TC, Shaham Y, Lu L. Inhibition of PKMzeta in nucleus accumbens core abolishes long-term drug reward memory. J Neurosci. 2011;31:5436–5446. doi: 10.1523/JNEUROSCI.5884-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YF, Civelli O, Grandy DK, Albert PR. Differential sensitivity of the short and long human dopamine D2 receptor subtypes to protein kinase C. J Neurochem. 1992;59:2311–2317. doi: 10.1111/j.1471-4159.1992.tb10125.x. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Covington HE, 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han MH, Nestler EJ. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Korogod N, Brose N, Schneggenburger R. Phorbol esters modulate spontaneous and Ca2+-evoked transmitter release via acting on both Munc13 and protein kinase C. J Neurosci. 2008;28:8257–8267. doi: 10.1523/JNEUROSCI.0550-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loweth JA, Svoboda R, Austin JD, Guillory AM, Vezina P. The PKC inhibitor Ro31-8220 blocks acute amphetamine-induced dopamine overflow in the nucleus accumbens. Neurosci Lett. 2009;455:88–92. doi: 10.1016/j.neulet.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loweth JA, Tseng KY, Wolf ME. Adaptations in AMPA receptor transmission in the nucleus accumbens contributing to incubation of cocaine craving. Neuropharmacology. 2013 doi: 10.1016/j.neuropharm.2013.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XY, Ghasemzadeh MB, Kalivas PW. Expression of D1 receptor, D2 receptor, substance P and enkephalin messenger RNAs in the neurons projecting from the nucleus accumbens. Neuroscience. 1998;82:767–780. doi: 10.1016/s0306-4522(97)00327-8. [DOI] [PubMed] [Google Scholar]
- McCutcheon JE, Loweth JA, Ford KA, Marinelli M, Wolf ME, Tseng KY. Group I mGluR activation reverses cocaine-induced accumulation of calcium-permeable AMPA receptors in nucleus accumbens synapses via a protein kinase C-dependent mechanism. J Neurosci. 2011;31:14536–14541. doi: 10.1523/JNEUROSCI.3625-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- Morris SJ, Van H, II, Daigle M, Robillard L, Sajedi N, Albert PR. Differential desensitization of dopamine D2 receptor isoforms by protein kinase C: the importance of receptor phosphorylation and pseudosubstrate sites. Eur J Pharmacol. 2007;577:44–53. doi: 10.1016/j.ejphar.2007.08.027. [DOI] [PubMed] [Google Scholar]
- Narita M, Akai H, Nagumo Y, Sunagawa N, Hasebe K, Nagase H, Kita T, Hara C, Suzuki T. Implications of protein kinase C in the nucleus accumbens in the development of sensitization to methamphetamine in rats. Neuroscience. 2004;127:941–948. doi: 10.1016/j.neuroscience.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Malenka RC. Dopamine depresses excitatory and inhibitory synaptic transmission by distinct mechanisms in the nucleus accumbens. J Neurosci. 1997;17:5697–5710. doi: 10.1523/JNEUROSCI.17-15-05697.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- O’Donnell P, Grace AA. Physiological and morphological properties of accumbens core and shell neurons recorded in vitro. Synapse. 1993;13:135–160. doi: 10.1002/syn.890130206. [DOI] [PubMed] [Google Scholar]
- Okada Y, Miyamoto T, Toda K. Dopamine modulates a voltage-gated calcium channel in rat olfactory receptor neurons. Brain Res. 2003;968:248–255. doi: 10.1016/s0006-8993(03)02267-4. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press; New York: 1997. [DOI] [PubMed] [Google Scholar]
- Peoples LL, Cavanaugh D. Differential changes in signal and background firing of accumbal neurons during cocaine self-administration. J Neurophysiol. 2003;90:993–1010. doi: 10.1152/jn.00849.2002. [DOI] [PubMed] [Google Scholar]
- Peoples LL, Kravitz AV, Lynch KG, Cavanaugh DJ. Accumbal neurons that are activated during cocaine self-administration are spared from inhibitory effects of repeated cocaine self-administration. Neuropsychopharmacology. 2007;32:1141–1158. doi: 10.1038/sj.npp.1301203. [DOI] [PubMed] [Google Scholar]
- Petit-Jacques J, Sui JL, Logothetis DE. Synergistic activation of G protein-gated inwardly rectifying potassium channels by the betagamma subunits of G proteins and Na(+) and Mg(2+) ions. J Gen Physiol. 1999;114:673–684. doi: 10.1085/jgp.114.5.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Quick EA, Reeder DC, Morgan ZR, Kalivas PW. Calcium-mediated second messengers modulate the expression of behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1998;286:1171–1176. [PubMed] [Google Scholar]
- Pillai G, Brown NA, McAllister G, Milligan G, Seabrook GR. Human D2 and D4 dopamine receptors couple through betagamma G-protein subunits to inwardly rectifying K+ channels (GIRK1) in a Xenopus oocyte expression system: selective antagonism by L-741,626 and L-745,870 respectively. Neuropharmacology. 1998;37:983–987. doi: 10.1016/s0028-3908(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Pollack A. Coactivation of D1 and D2 dopamine receptors: in marriage, a case of his, hers, and theirs. Sci STKE 2004. 2004:pe50. doi: 10.1126/stke.2552004pe50. [DOI] [PubMed] [Google Scholar]
- Ramanathan S, Tkatch T, Atherton JF, Wilson CJ, Bevan MD. D2-like dopamine receptors modulate SKCa channel function in subthalamic nucleus neurons through inhibition of Cav2.2 channels. J Neurophysiol. 2008;99:442–459. doi: 10.1152/jn.00998.2007. [DOI] [PubMed] [Google Scholar]
- Schmidt HD, Anderson SM, Pierce RC. Stimulation of D1-like or D2 Dopamine Receptors in the Shell, but Not the Core, of the Nucleus Accumbens Reinstates Cocaine-Seeking Behavior in the Rat. European Journal of Neuroscience. 2006;23:219–228. doi: 10.1111/j.1460-9568.2005.04524.x. [DOI] [PubMed] [Google Scholar]
- Schmidt HD, Kimmey BA, Arreola AC, Pierce RC. Group I metabotropic glutamate receptor-mediated activation of PKC gamma in the nucleus accumbens core promotes the reinstatement of cocaine seeking. Addict Biol. 2014 doi: 10.1111/adb.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Pierce RC. Cooperative activation of D1-like and D2-like dopamine receptors in the nucleus accumbens shell is required for the reinstatement of cocaine-seeking behavior in the rat. Neuroscience. 2006a;142:451–461. doi: 10.1016/j.neuroscience.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Schmidt HD, Pierce RC. Systemic administration of a dopamine, but not a serotonin or norepinephrine, transporter inhibitor reinstates cocaine seeking in the rat. Behav Brain Res. 2006b;175:189–194. doi: 10.1016/j.bbr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Schmidt HD, Pierce RC. Cocaine-induced neuroadaptations in glutamate transmission: potential therapeutic targets for craving and addiction. Ann N Y Acad Sci. 2010;1187:35–75. doi: 10.1111/j.1749-6632.2009.05144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Schassburger RL, Guercio LA, Pierce RC. Stimulation of mGluR5 in the Accumbens Shell Promotes Cocaine Seeking by Activating PKC Gamma. J Neurosci. 2013;33:14160–14169. doi: 10.1523/JNEUROSCI.2284-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shuen JA, Chen M, Gloss B, Calakos N. Drd1a-tdTomato BAC transgenic mice for simultaneous visualization of medium spiny neurons in the direct and indirect pathways of the basal ganglia. J Neurosci. 2008;28:2681–2685. doi: 10.1523/JNEUROSCI.5492-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley DR, Monsma FJ, Jr, Shen Y. Molecular neurobiology of dopaminergic receptors. Int Rev Neurobiol. 1993;35:391–415. doi: 10.1016/s0074-7742(08)60573-5. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Steketee JD, Rowe LA, Chandler LJ. The effects of acute and repeated cocaine injections on protein kinase C activity and isoform levels in dopaminergic brain regions. Neuropharmacology. 1998;37:339–347. doi: 10.1016/s0028-3908(98)00022-7. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Thomas KL, Everitt BJ. Limbic-cortical-ventral striatal activation during retrieval of a discrete cocaine-associated stimulus: a cellular imaging study with gamma protein kinase C expression. J Neurosci. 2001;21:2526–2535. doi: 10.1523/JNEUROSCI.21-07-02526.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron. 2007;54:275–290. doi: 10.1016/j.neuron.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Wiggins A, Smith RJ, Shen HW, Kalivas PW. Integrins modulate relapse to cocaine-seeking. J Neurosci. 2011;31:16177–16184. doi: 10.1523/JNEUROSCI.3816-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP. Decay of prepulse facilitation of N type calcium channels during G protein inhibition is consistent with binding of a single Gbeta subunit. Proc Natl Acad Sci U S A. 1998;95:4035–4039. doi: 10.1073/pnas.95.7.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]